
Anti-Inflammatory and/or Anti-Fibrotic Treatment of MPO-ANCA-Positive Interstitial Lung Disease: A Short Review

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Prevalence and Clinical Manifestations
3. Morphological Domain
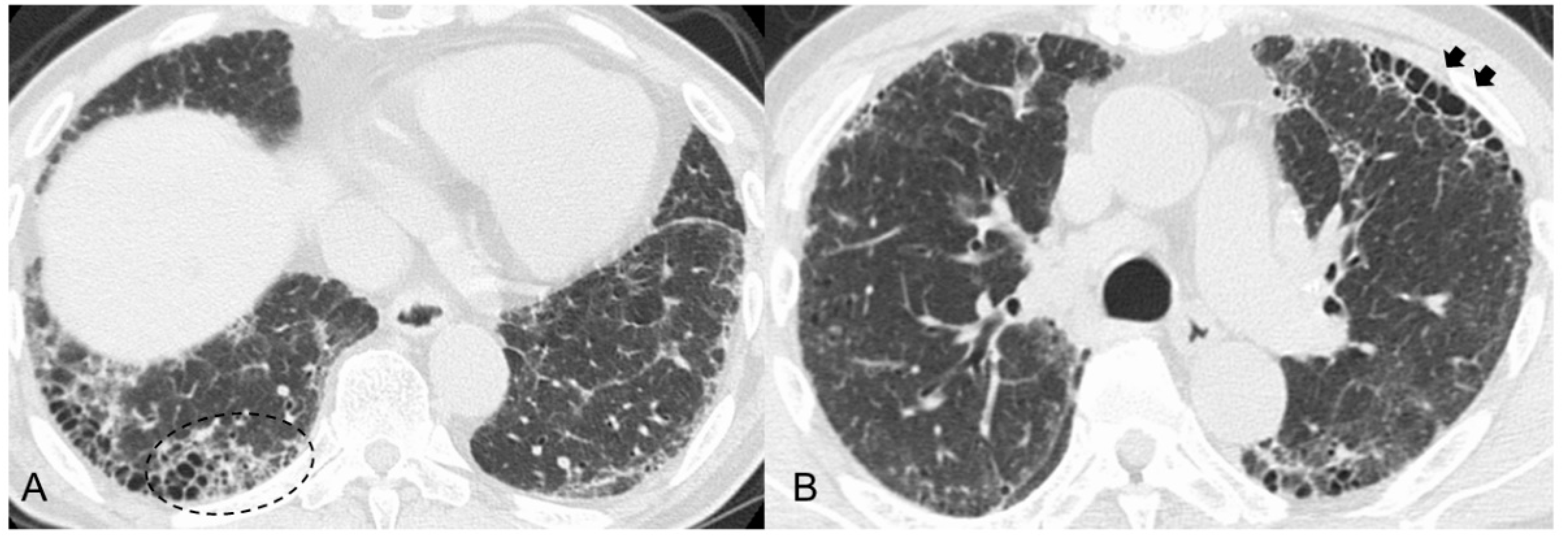
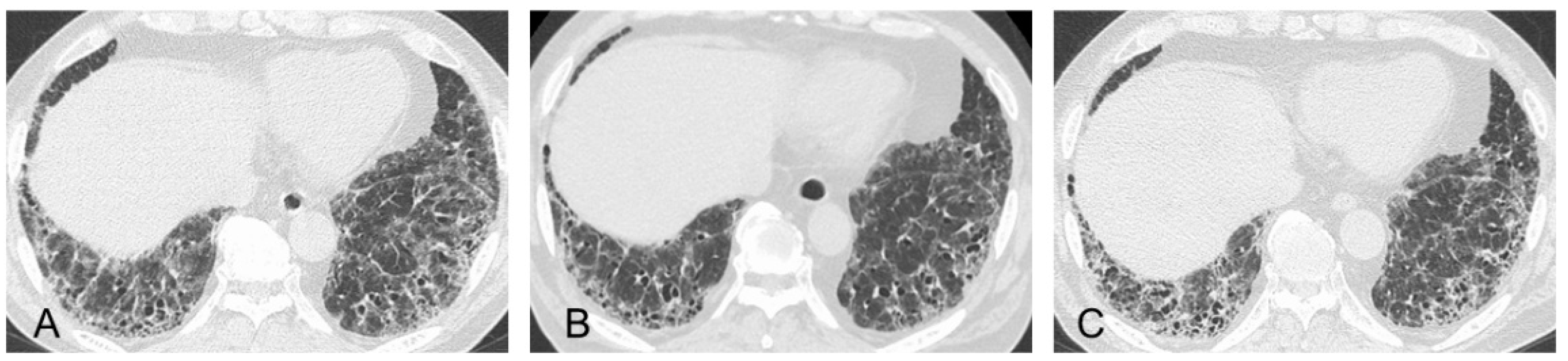
3.1. Radiological Findings
3.2. Pathological Findings
3.3. Differences between MPO-ANCA-Positive ILD and UIP/IPF
4. Prognosis and Causes of Death
5. Therapeutic Assessment of MPO-ANCA-Positive ILD
5.1. ILD Patients with MPA
5.2. MPO-ANCA-Positive ILD Patients without Systemic Vasculitis as Idiopathic ILD
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Xiao, H.; Heeringa, P.; Hu, P.; Liu, Z.; Zhao, M.; Aratani, Y.; Maeda, N.; Falk, R.J.; Jennette, J.C. Antineutrophil cytoplasmic autoantibodies specific for myeloperoxidase cause glomerulonephritis and vasculitis in mice. J. Clin. Investig. 2002, 110, 955–963. [Google Scholar] [CrossRef]
- Jennette, J.C.; Falk, R.J.; Bacon, P.A.; Basu, N.; Cid, M.C.; Ferrario, F.; Flores-Suarez, L.F.; Gross, W.L.; Guillevin, L.; Hagen, E.C.; et al. 2012 revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheumatol. 2013, 65, 1–11. [Google Scholar] [CrossRef]
- Kallenberg, C.G. The diagnosis and classification of microscopic polyangiitis. J. Autoimmun. 2014, 48–49, 90–93. [Google Scholar] [CrossRef]
- Suppiah, R.; Robson, J.C.; Grayson, P.C.; Ponte, C.; Craven, A.; Khalid, S.; Judge, A.; Hutchings, A.; Merkel, P.A.; Luqmani, R.A.; et al. 2022 American College of Rheumatology/European Alliance of Associations for Rheumatology classification criteria for microscopic polyangiitis. Ann. Rheum. Dis. 2022, 81, 321–326. [Google Scholar] [CrossRef]
- Maillet, T.; Goletto, T.; Beltramo, G.; Dupuy, H.; Jouneau, S.; Borie, R.; Crestani, B.; Cottin, V.; Blockmans, D.; Lazaro, E.; et al. Usual interstitial pneumonia in ANCA-associated vasculitis: A poor prognostic factor. J. Autoimmun. 2020, 106, 102338. [Google Scholar] [CrossRef]
- Katsumata, Y.; Kawaguchi, Y.; Yamanaka, H. Interstitial lung disease with ANCA-associated vasculitis. Clin. Med. Insights Circ. Respir. Pulm. Med. 2015, 9 (Suppl. 1), 51–56. [Google Scholar] [CrossRef]
- Kishaba, T. Current perspective of progressive-fibrosing interstitial lung disease. Respir. Investig. 2022. [Google Scholar] [CrossRef] [PubMed]
- Bando, M.; Homma, S.; Harigai, M. MPO-ANCA positive interstitial pneumonia: Current knowledge and future perspectives. Sarcoidosis Vasc. Diffus. Lung Dis. 2022, 38, e2021045. [Google Scholar]
- Kwon, M.; Lee, A.S.; Mira-Avendano, I.; Rojas, C.A.; Grage, R.; Abril, A. Interstitial lung disease in antineutrophil cytoplasmic antibody-associated vasculitis patients: Comparison with idiopathic pulmonary fibrosis. J. Clin. Rheumatol. 2021, 27, 324–330. [Google Scholar] [CrossRef]
- Hozumi, H.; Kono, M.; Hasegawa, H.; Yasui, H.; Suzuki, Y.; Karayama, M.; Furuhashi, K.; Enomoto, N.; Fujisawa, T.; Inui, N.; et al. Clinical significance of interstitial lung disease and its acute exacerbation in microscopic polyangiitis. Chest 2021, 159, 2334–2345. [Google Scholar] [CrossRef]
- Matsuda, S.; Kotani, T.; Suzuka, T.; Kiboshi, T.; Fukui, K.; Wakama, M.; Ishida, T.; Fujiki, Y.; Shiba, H.; Nagai, K.; et al. Evaluation of poor prognostic factors of respiratory related death in microscopic polyangiitis complicated by interstitial lung disease. Sci. Rep. 2021, 11, 1490. [Google Scholar] [CrossRef] [PubMed]
- Ozaki, S.; Atsumi, T.; Hayashi, T.; Ishizu, A.; Kobayashi, S.; Kumagai, S.; Kurihara, Y.; Kurokawa, M.S.; Makino, H.; Nagafuchi, H.; et al. Severity-based treatment for Japanese patients with MPO-ANCA-associated vasculitis: The JMAAV study. Mod. Rheumatol. 2012, 22, 394–404. [Google Scholar] [CrossRef] [PubMed]
- Kagiyama, N.; Takayanagi, N.; Kanauchi, T.; Ishiguro, T.; Yanagisawa, T.; Sugita, Y. Antineutrophil cytoplasmic antibody-positive conversion and microscopic polyangiitis development in patients with idiopathic pulmonary fibrosis. BMJ Open Respir. Res. 2015, 2, e000058. [Google Scholar] [CrossRef] [PubMed]
- Kadura, S.; Raghu, G. Antineutrophil cytoplasmic antibody-associated interstitial lung disease: A review. Eur. Respir. Rev. 2021, 30, 210123. [Google Scholar] [CrossRef]
- Hozumi, H.; Oyama, Y.; Yasui, H.; Suzuki, Y.; Kono, M.; Karayama, M.; Furuhashi, K.; Enomoto, N.; Fujisawa, T.; Inui, N.; et al. Clinical significance of myeloperoxidase-anti-neutrophil cytoplasmic antibody in idiopathic interstitial pneumonias. PLoS ONE 2018, 13, e0199659. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.Y.; Ventura, I.B.; Achtar-Zadeh, N.; Elicker, B.M.; Jones, K.D.; Wolters, P.J.; Collard, H.R.; Adegunsoye, A.; Strek, M.E.; Ley, B. Prevalence and clinical significance of antineutrophil cytoplasmic antibodies in North American patients with idiopathic pulmonary fibrosis. Chest 2019, 156, 715–723. [Google Scholar] [CrossRef] [PubMed]
- Alba, M.A.; Flores-Suárez, L.F.; Henderson, A.G.; Xiao, H.; Hu, P.; Nachman, P.H.; Falk, R.J.; Charles Jennette, J. Interstital lung disease in ANCA vasculitis. Autoimmun. Rev. 2017, 16, 722–729. [Google Scholar] [CrossRef] [PubMed]
- Homma, S.; Suzuki, A.; Sato, K. Pulmonary involvement in ANCA-associated vasculitis from the view of the pulmonologist. Clin. Exp. Nephrol. 2013, 17, 667–671. [Google Scholar] [CrossRef] [PubMed]
- Sada, K.E.; Yamamura, M.; Harigai, M.; Fujii, T.; Dobashi, H.; Takasaki, Y.; Ito, S.; Yamada, H.; Wada, T.; Hirahashi, J.; et al. Classification and characteristics of Japanese patients with antineutrophil cytoplasmic antibody-associated vasculitis in a nationwide, prospective, inception cohort study. Arthritis Res. Ther. 2014, 16, R101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tzelepis, G.E.; Kokosi, M.; Tzioufas, A.; Toya, S.P.; Boki, K.A.; Zormpala, A.; Moutsopoulos, H.M. Prevalence and outcome of pulmonary fibrosis in microscopic polyangiitis. Eur. Respir. J. 2010, 36, 116–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arulkumaran, N.; Periselneris, N.; Gaskin, G.; Strickland, N.; Ind, P.W.; Pusey, C.D.; Salama, A.D. Interstitial lung disease and ANCA-associated vasculitis: A retrospective observational cohort study. Rheumatology 2011, 50, 2035–2043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohammad, A.J.; Mortensen, K.H.; Babar, J.; Smith, R.; Jones, R.B.; Nakagomi, D.; Sivasothy, P.; Jayne, D.R.W. Pulmonary involvement in antineutrophil cytoplasmic antibodies (ANCA)-associated vasculitis: The influence of ANCA subtype. J. Rheumatol. 2017, 44, 1458–1467. [Google Scholar] [CrossRef] [PubMed]
- Schirmer, J.H.; Wright, M.N.; Vonthein, R.; Herrmann, K.; Nölle, B.; Both, M.; Henes, F.O.; Arlt, A.; Gross, W.L.; Schinke, S.; et al. Clinical presentation and long-term outcome of 144 patients with microscopic polyangiitis in a monocentric German cohort. Rheumatology 2016, 55, 71–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furuta, S.; Chaudhry, A.N.; Hamano, Y.; Fujimoto, S.; Nagafuchi, H.; Makino, H.; Matsuo, S.; Ozaki, S.; Endo, T.; Muso, E.; et al. Comparison of phenotype and outcome in microscopic polyangiitis between Europe and Japan. J. Rheumatol. 2014, 41, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Fernandez Casares, M.; Gonzalez, A.; Fielli, M.; Caputo, F.; Bottinelli, Y.; Zamboni, M. Microscopic polyangiitis associated with pulmonary fibrosis. Clin. Rheumatol. 2015, 34, 1273–1277. [Google Scholar] [CrossRef]
- Katsuyama, T.; Sada, K.E.; Makino, H. Current concept and epidemiology of systemic vasculitides. Allergol. Int. 2014, 63, 505–513. [Google Scholar] [CrossRef] [Green Version]
- Lynch, D.A.; Sverzellati, N.; Travis, W.D.; Brown, K.K.; Colby, T.V.; Galvin, J.R.; Goldin, J.G.; Hansell, D.M.; Inoue, Y.; Johkoh, T.; et al. Diagnostic criteria for idiopathic pulmonary fibrosis: A Fleischner Society White Paper. Lancet Respir. Med. 2018, 6, 138–153. [Google Scholar] [CrossRef]
- Raghu, G.; Remy-Jardin, M.; Myers, J.L.; Richeldi, L.; Ryerson, C.J.; Lederer, D.J.; Behr, J.; Cottin, V.; Danoff, S.K.; Morell, F.; et al. Diagnosis of Idiopathic Pulmonary Fibrosis. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2018, 198, e44–e68. [Google Scholar] [CrossRef]
- Palmucci, S.; Inì, C.; Cosentino, S.; Fanzone, L.; Di Pietro, S.; Di Mari, A.; Galioto, F.; Tiralongo, F.; Vignigni, G.; Toscano, S.; et al. Pulmonary vasculitides: A radiological review emphasizing parenchymal HRCT features. Diagnostics 2021, 11, 2318. [Google Scholar] [CrossRef]
- Suzuki, A.; Sakamoto, S.; Kurosaki, A.; Kurihara, Y.; Satoh, K.; Usui, Y.; Nanki, T.; Arimura, Y.; Makino, H.; Okada, Y.; et al. Chest high-resolution CT findings of microscopic polyangiitis: A Japanese first nationwide prospective cohort study. AJR Am. J. Roentgenol. 2019, 213, 104–114. [Google Scholar] [CrossRef]
- Yamakawa, H.; Sato, S.; Nakamura, T.; Nishizawa, T.; Kawabe, R.; Oba, T.; Horikoshi, M.; Akasaka, K.; Amano, M.; Kuwano, K.; et al. Prognostic value of radiological findings indeterminate for UIP pattern and anterior upper lobe honeycomb-like lesion in chronic fibrosing interstitial lung disease associated with MPO-ANCA. BMC Pulm. Med. 2021, 21, 346. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Peng, M.; Zhang, T.; Li, Z.; Song, L.; Li, M.; Shi, J. Clinical features and long-term outcomes of interstitial lung disease with anti-neutrophil cytoplasmic antibody. BMC Pulm. Med. 2021, 21, 88. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Otani, K.; Egashira, R.; Kashima, Y.; Taniguchi, H.; Kondoh, Y.; Kataoka, K.; Shiraki, A.; Kitasato, Y.; Leslie, K.O.; et al. Interstitial pneumonia associated with MPO-ANCA: Clinicopathological features of nine patients. Respir. Med. 2012, 106, 1765–1770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosoda, C.; Baba, T.; Hagiwara, E.; Ito, H.; Matsuo, N.; Kitamura, H.; Iwasawa, T.; Okudela, K.; Takemura, T.; Ogura, T. Clinical features of usual interstitial pneumonia with anti-neutrophil cytoplasmic antibody in comparison with idiopathic pulmonary fibrosis. Respirology 2016, 21, 920–926. [Google Scholar] [CrossRef]
- Baqir, M.; Yi, E.E.; Colby, T.V.; Cox, C.W.; Ryu, J.H.; Specks, U. Radiologic and pathologic characteristics of myeloperoxidase-antineutrophil cytoplasmic antibody-associated interstitial lung disease: A retrospective analysis. Sarcoidosis Vasc. Diffus. Lung Dis. 2019, 36, 195–201. [Google Scholar] [CrossRef] [Green Version]
- Yamakawa, H.; Ogura, T.; Kameda, H.; Kishaba, T.; Iwasawa, T.; Takemura, T.; Kuwano, K. Decision-making strategy for the treatment of rheumatoid arthritis-associated interstitial lung disease (RA-ILD). J. Clin. Med. 2021, 10, 3806. [Google Scholar] [CrossRef]
- Juge, P.A.; Lee, J.S.; Ebstein, E.; Furukawa, H.; Dobrinskikh, E.; Gazal, S.; Kannengiesser, C.; Ottaviani, S.; Oka, S.; Tohma, S.; et al. MUC5B promoter variant and rheumatoid arthritis with interstitial lung disease. N. Engl. J. Med. 2018, 379, 2209–2219. [Google Scholar] [CrossRef]
- Putman, R.K.; Gudmundsson, G.; Araki, T.; Nishino, M.; Sigurdsson, S.; Gudmundsson, E.F.; Eiríksdottír, G.; Aspelund, T.; Ross, J.C.; San José Estépar, R.; et al. The MUC5B promoter polymorphism is associated with specific interstitial lung abnormality subtypes. Eur. Respir. J. 2017, 50, 1700537. [Google Scholar] [CrossRef] [Green Version]
- Seibold, M.A.; Wise, A.L.; Speer, M.C.; Steele, M.P.; Brown, K.K.; Loyd, J.E.; Fingerlin, T.E.; Zhang, W.; Gudmundsson, G.; Groshong, S.D.; et al. A common MUC5B promoter polymorphism and pulmonary fibrosis. N. Engl. J. Med. 2011, 364, 1503–1512. [Google Scholar] [CrossRef] [Green Version]
- Akiyama, M.; Kaneko, Y. Pathogenesis, clinical features, and treatment strategy for rheumatoid arthritis-associated interstitial lung disease. Autoimmun. Rev. 2022, 21, 103056. [Google Scholar] [CrossRef]
- Namba, N.; Kawasaki, A.; Sada, K.E.; Hirano, F.; Kobayashi, S.; Yamada, H.; Furukawa, H.; Shimada, K.; Hashimoto, A.; Matsui, T.; et al. Association of MUC5B promoter polymorphism with interstitial lung disease in myeloperoxidase-antineutrophil cytoplasmic antibody-associated vasculitis. Ann. Rheum. Dis. 2019, 78, 1144–1146. [Google Scholar] [CrossRef] [PubMed]
- Yamakawa, H.; Ogura, T.; Sato, S.; Nishizawa, T.; Kawabe, R.; Oba, T.; Kato, A.; Horikoshi, M.; Akasaka, K.; Amano, M.; et al. The potential utility of anterior upper lobe honeycomb-like lesion in interstitial lung disease associated with connective tissue disease. Respir. Med. 2020, 172, 106125. [Google Scholar] [CrossRef] [PubMed]
- Hirayama, K.; Kobayashi, M.; Usui, J.; Arimura, Y.; Sugiyama, H.; Nitta, K.; Muso, E.; Wada, T.; Matsuo, S.; Yamagata, K. Pulmonary involvements of anti-neutrophil cytoplasmic autoantibody-associated renal vasculitis in Japan. Nephrol. Dial. Transplant. 2015, 30 (Suppl. 1), i83–i93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foulon, G.; Delaval, P.; Valeyre, D.; Wallaert, B.; Debray, M.P.; Brauner, M.; Nicaise, P.; Cadranel, J.; Cottin, V.; Tazi, A.; et al. ANCA-associated lung fibrosis: Analysis of 17 patients. Respir. Med. 2008, 102, 1392–1398. [Google Scholar] [CrossRef]
- Sebastiani, M.; Luppi, F.; Sambataro, G.; Castillo Villegas, D.; Cerri, S.; Tomietto, P.; Cassone, G.; Bocchino, M.; Atienza-Mateo, B.; Cameli, P.; et al. Interstitial lung disease and anti-myeloperoxidase antibodies: Not a simple association. J. Clin. Med. 2021, 10, 2548. [Google Scholar] [CrossRef]
- Wijsenbeek, M.; Cottin, V. Spectrum of fibrotic lung diseases. N. Engl. J. Med. 2020, 383, 958–968. [Google Scholar] [CrossRef]
- Terrier, B.; Saadoun, D.; Sène, D.; Ghillani, P.; Amoura, Z.; Deray, G.; Fautrel, B.; Piette, J.C.; Cacoub, P. Antimyeloperoxidase antibodies are a useful marker of disease activity in antineutrophil cytoplasmic antibody-associated vasculitides. Ann. Rheum. Dis. 2009, 68, 1564–1571. [Google Scholar] [CrossRef]
- Kondoh, Y.; Makino, S.; Ogura, T.; Suda, T.; Tomioka, H.; Amano, H.; Anraku, M.; Enomoto, N.; Fujii, T.; Fujisawa, T.; et al. 2020 guide for the diagnosis and treatment of interstitial lung disease associated with connective tissue disease. Respir. Investig. 2021, 59, 709–740. [Google Scholar] [CrossRef]
- Ravaglia, C.; Wells, A.U.; Tomassetti, S.; Dubini, A.; Cavazza, A.; Piciucchi, S.; Sverzellati, N.; Gurioli, C.; Gurioli, C.; Costabel, U.; et al. Transbronchial lung cryobiopsy in diffuse parenchymal lung disease: Comparison between biopsy from 1 segment and biopsy from 2 segments-diagnostic yield and complications. Respiration 2017, 93, 285–292. [Google Scholar] [CrossRef]
- Enomoto, N.; Suda, T.; Kato, M.; Kaida, Y.; Nakamura, Y.; Imokawa, S.; Ida, M.; Chida, K. Quantitative analysis of fibroblastic foci in usual interstitial pneumonia. Chest 2006, 130, 22–29. [Google Scholar] [CrossRef]
- Elicker, B.M.; Kallianos, K.G.; Henry, T.S. The role of high-resolution computed tomography in the follow-up of diffuse lung disease. Eur. Respir. Rev. 2017, 26, 170008. [Google Scholar] [CrossRef] [PubMed]
- Bulpa, P.A.; Dive, A.M.; Mertens, L.; Delos, M.A.; Jamart, J.; Evrard, P.A.; Gonzalez, M.R.; Installé, E.J. Combined bronchoalveolar lavage and transbronchial lung biopsy: Safety and yield in ventilated patients. Eur. Respir. J. 2003, 21, 489–494. [Google Scholar] [CrossRef] [PubMed]
- Makino, S. Progressive fibrosing interstitial lung diseases: A new concept and indication of nintedanib. Mod. Rheumatol. 2021, 31, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Yamakawa, H.; Kitamura, H.; Takemura, T.; Ikeda, S.; Sekine, A.; Baba, T.; Iwasawa, T.; Hagiwara, E.; Sato, S.; Ogura, T. Prognostic factors and disease behaviour of pathologically proven fibrotic non-specific interstitial pneumonia. Respirology 2018, 23, 1032–1040. [Google Scholar] [CrossRef] [Green Version]
- Gibson, C.D.; Kugler, M.C.; Deshwal, H.; Munger, J.S.; Condos, R. Advances in targeted therapy for progressive fibrosing interstitial lung disease. Lung 2020, 198, 597–608. [Google Scholar] [CrossRef] [PubMed]
- Silva, C.I.; Müller, N.L.; Hansell, D.M.; Lee, K.S.; Nicholson, A.G.; Wells, A.U. Nonspecific interstitial pneumonia and idiopathic pulmonary fibrosis: Changes in pattern and distribution of disease over time. Radiology 2008, 247, 251–259. [Google Scholar] [CrossRef]
- Adegunsoye, A.; Oldham, J.M.; Bellam, S.K.; Montner, S.; Churpek, M.M.; Noth, I.; Vij, R.; Strek, M.E.; Chung, J.H. Computed tomography honeycombing identifies a progressive fibrotic phenotype with increased mortality across diverse interstitial lung diseases. Ann. Am. Thorac. Soc. 2019, 16, 580–588. [Google Scholar] [CrossRef]
- Leuschner, G.; Behr, J. Acute exacerbation in interstitial lung disease. Front. Med. 2017, 4, 176. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, A.; Kondoh, Y.; Brown, K.K.; Johkoh, T.; Kataoka, K.; Fukuoka, J.; Kimura, T.; Matsuda, T.; Yokoyama, T.; Fukihara, J.; et al. Acute exacerbations of fibrotic interstitial lung diseases. Respirology 2020, 25, 525–534. [Google Scholar] [CrossRef]
- Richeldi, L.; du Bois, R.M.; Raghu, G.; Azuma, A.; Brown, K.K.; Costabel, U.; Cottin, V.; Flaherty, K.R.; Hansell, D.M.; Inoue, Y.; et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N. Engl. J. Med. 2014, 370, 2071–2082. [Google Scholar] [CrossRef] [Green Version]
- Fletcher, S.V.; Jones, M.G.; Renzoni, E.A.; Parfrey, H.; Hoyles, R.K.; Spinks, K.; Kokosi, M.; Kwok, A.; Warburton, C.; Titmuss, V.; et al. Safety and tolerability of nintedanib for the treatment of idiopathic pulmonary fibrosis in routine UK clinical practice. ERJ Open Res. 2018, 4, 00049–02018. [Google Scholar] [CrossRef] [PubMed]
- King, T.E., Jr.; Bradford, W.Z.; Castro-Bernardini, S.; Fagan, E.A.; Glaspole, I.; Glassberg, M.K.; Gorina, E.; Hopkins, P.M.; Kardatzke, D.; Lancaster, L.; et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N. Engl. J. Med. 2014, 370, 2083–2092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flaherty, K.R.; Wells, A.U.; Cottin, V.; Devaraj, A.; Walsh, S.L.F.; Inoue, Y.; Richeldi, L.; Kolb, M.; Tetzlaff, K.; Stowasser, S.; et al. Nintedanib in progressive fibrosing interstitial lung diseases. N. Engl. J. Med. 2019, 381, 1718–1727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wells, A.U.; Flaherty, K.R.; Brown, K.K.; Inoue, Y.; Devaraj, A.; Richeldi, L.; Moua, T.; Crestani, B.; Wuyts, W.A.; Stowasser, S.; et al. Nintedanib in patients with progressive fibrosing interstitial lung diseases-subgroup analyses by interstitial lung disease diagnosis in the INBUILD trial: A randomised, double-blind, placebo-controlled, parallel-group trial. Lancet Respir. Med. 2020, 8, 453–460. [Google Scholar] [CrossRef]
- Behr, J.; Prasse, A.; Kreuter, M.; Johow, J.; Rabe, K.F.; Bonella, F.; Bonnet, R.; Grohe, C.; Held, M.; Wilkens, H.; et al. Pirfenidone in patients with progressive fibrotic interstitial lung diseases other than idiopathic pulmonary fibrosis (RELIEF): A double-blind, randomised, placebo-controlled, phase 2b trial. Lancet Respir. Med. 2021, 9, 476–486. [Google Scholar] [CrossRef]
- Raghu, G.; Collard, H.R.; Egan, J.J.; Martinez, F.J.; Behr, J.; Brown, K.K.; Colby, T.V.; Cordier, J.F.; Flaherty, K.R.; Lasky, J.A.; et al. An official ATS/ERS/JRS/ALAT statement: Idiopathic pulmonary fibrosis: Evidence-based guidelines for diagnosis and management. Am. J. Respir. Crit. Care Med. 2011, 183, 788–824. [Google Scholar] [CrossRef] [Green Version]
- Naccache, J.M.; Jouneau, S.; Didier, M.; Borie, R.; Cachanado, M.; Bourdin, A.; Reynaud-Gaubert, M.; Bonniaud, P.; Israël-Biet, D.; Prévot, G.; et al. Cyclophosphamide added to glucocorticoids in acute exacerbation of idiopathic pulmonary fibrosis (EXAFIP): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Respir. Med. 2022, 10, 26–34. [Google Scholar] [CrossRef]
- Chiu, Y.H.; Spierings, J.; de Jong, P.A.; Hoesein, F.M.; Grutters, J.C.; van Laar, J.M.; Voortman, M. Predictors for progressive fibrosis in patients with connective tissue disease associated interstitial lung diseases. Respir. Med. 2021, 187, 106579. [Google Scholar] [CrossRef]
- Zamora-Legoff, J.A.; Krause, M.L.; Crowson, C.S.; Ryu, J.H.; Matteson, E.L. Risk of serious infection in patients with rheumatoid arthritis-associated interstitial lung disease. Clin. Rheumatol. 2016, 35, 2585–2589. [Google Scholar] [CrossRef]
- Yamaguchi, K.; Yamaguchi, A.; Itai, M.; Onuki, Y.; Shin, Y.; Uno, S.; Hanazato, C.; Taguchi, K.; Umetsu, K.; Aikawa, M.; et al. Interstitial lung disease with myeloperoxidase-antineutrophil cytoplasmic antibody-associated vasculitis in elderly patients. Rheumatol. Int. 2021, 41, 1641–1650. [Google Scholar] [CrossRef]
- Denning, D.W.; Cadranel, J.; Beigelman-Aubry, C.; Ader, F.; Chakrabarti, A.; Blot, S.; Ullmann, A.J.; Dimopoulos, G.; Lange, C. Chronic pulmonary aspergillosis: Rationale and clinical guidelines for diagnosis and management. Eur. Respir. J. 2016, 47, 45–68. [Google Scholar] [CrossRef] [PubMed]
- Yamakawa, H.; Takayanagi, N.; Miyahara, Y.; Ishiguro, T.; Kanauchi, T.; Hoshi, T.; Yanagisawa, T.; Sugita, Y. Prognostic factors and radiographic outcomes of nontuberculous mycobacterial lung disease in rheumatoid arthritis. J. Rheumatol. 2013, 40, 1307–1315. [Google Scholar] [CrossRef] [PubMed]
- Kurosaki, F.; Bando, M.; Nakayama, M.; Mato, N.; Nakaya, T.; Yamasawa, H.; Yoshimoto, T.; Fukushima, N.; Sugiyama, Y. Clinical features of pulmonary aspergillosis associated with interstitial pneumonia. Intern. Med. 2014, 53, 1299–1306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Odashima, K.; Kagiyama, N.; Kanauchi, T.; Ishiguro, T.; Takayanagi, N. Incidence and etiology of chronic pulmonary infections in patients with idiopathic pulmonary fibrosis. PLoS ONE 2020, 15, e0230746. [Google Scholar] [CrossRef] [PubMed]
- Kawamura, K.; Ichikado, K.; Ichiyasu, H.; Anan, K.; Yasuda, Y.; Suga, M.; Sakagami, T. Acute exacerbation of chronic fibrosing interstitial pneumonia in patients receiving antifibrotic agents: Incidence and risk factors from real-world experience. BMC Pulm. Med. 2019, 19, 113. [Google Scholar] [CrossRef]
- Sebastiani, M.; Manfredi, A.; Vacchi, C.; Cassone, G.; Faverio, P.; Cavazza, A.; Sverzellati, N.; Salvarani, C.; Luppi, F. Epidemiology and management of interstitial lung disease in ANCA-associated vasculitis. Clin. Exp. Rheumatol. 2020, 38 (Suppl. 124), 221–231. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MPO-ANCA-Positive ILD/MPA-ILD | IPF | |
|---|---|---|
| Age | Commonly older than 65 years old (patients younger than 50 years old are rare) | |
| Smoking | The majority of patients have a history of past cigarette smoking | |
| Sex | Male (45.2–66.7%) ≒ Female (33.3–54.7%) | Male (>70%) > Female |
| HRCT findings | Framework: Subpleural and basal predominant distribution is often heterogeneous | |
| Increased attenuation around honeycombing and traction bronchiectasis (19–39%) | ― | |
| Pathological findings | Framework: Dense fibrosis with architectural distortion, predominant subpleural and/or paraseptal distribution of fibrosis | |
| More prominent inflammatory cell infiltration and cellular bronchiolitis compared with IPF | ― | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yamakawa, H.; Toyoda, Y.; Baba, T.; Kishaba, T.; Fukuda, T.; Takemura, T.; Kuwano, K. Anti-Inflammatory and/or Anti-Fibrotic Treatment of MPO-ANCA-Positive Interstitial Lung Disease: A Short Review. J. Clin. Med. 2022, 11, 3835. https://doi.org/10.3390/jcm11133835
Yamakawa H, Toyoda Y, Baba T, Kishaba T, Fukuda T, Takemura T, Kuwano K. Anti-Inflammatory and/or Anti-Fibrotic Treatment of MPO-ANCA-Positive Interstitial Lung Disease: A Short Review. Journal of Clinical Medicine. 2022; 11(13):3835. https://doi.org/10.3390/jcm11133835
Chicago/Turabian StyleYamakawa, Hideaki, Yuko Toyoda, Tomohisa Baba, Tomoo Kishaba, Taiki Fukuda, Tamiko Takemura, and Kazuyoshi Kuwano. 2022. "Anti-Inflammatory and/or Anti-Fibrotic Treatment of MPO-ANCA-Positive Interstitial Lung Disease: A Short Review" Journal of Clinical Medicine 11, no. 13: 3835. https://doi.org/10.3390/jcm11133835