
COVID-19-Related ARDS: Key Mechanistic Features and Treatments

 , ,
, ,  and
and
Abstract
:1. Introduction
2. “Typical” ARDS
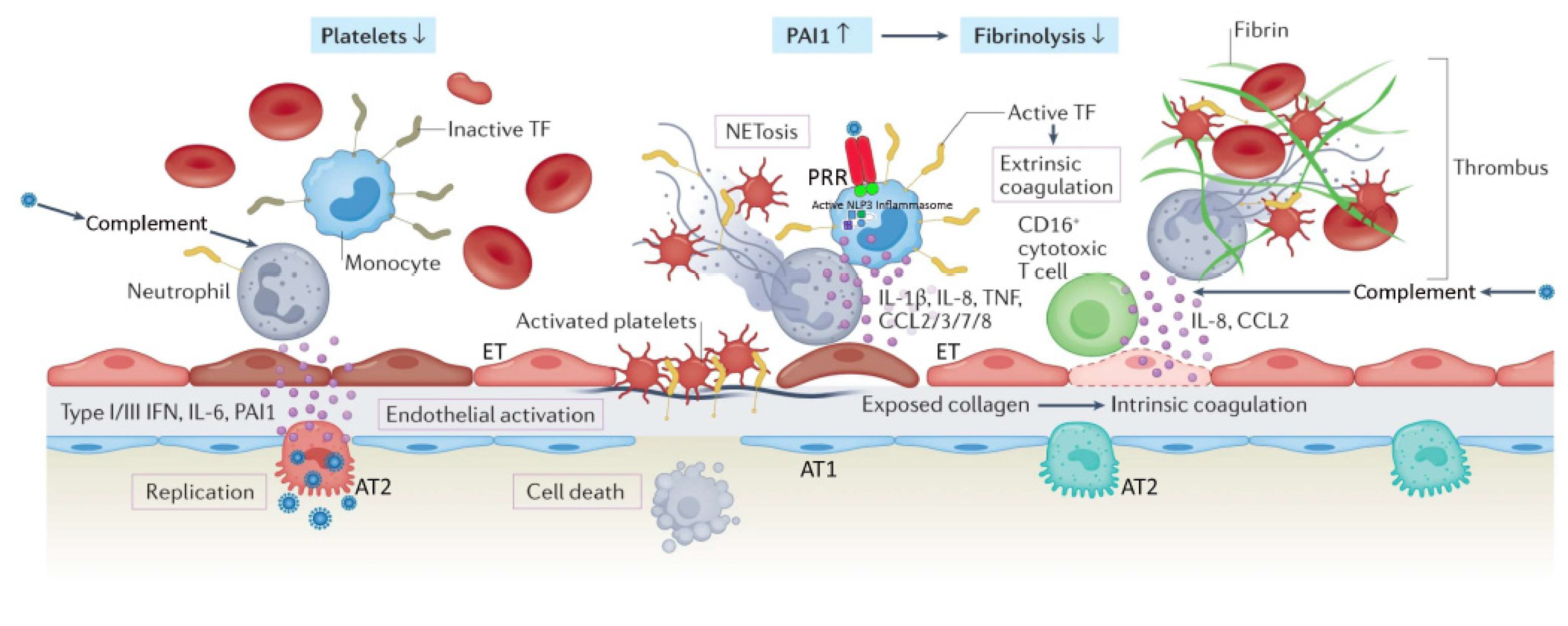
3. Viral Pathogenesis of SARS-CoV-2
4. Pharmacologic Interventions
5. Blood Purification Interventions
6. Distinct Pathologic Features of C-ARDS
7. Respiratory Mechanics and Gas Exchange in C-ARDS
8. Mechanical Ventilation in C-ARDS
8.1. Tidal Volume in C-ARDS
- (1)
- Data from the ARMA trial, derived primarily from patients with ARDS secondary to bacterial pneumonia and sepsis, may not be wholly translatable to patients with ARDS secondary to novel forms of viral pneumonia with unique pathologic features, such as C-ARDS.
- (2)
- Even in the ARMA trial, tidal volumes could be liberalized if necessary to facilitate patient comfort and adequate ventilation.
- (3)
8.2. Application of PEEP in C-ARDS
8.3. Body Positioning
9. Extracorporeal Life Support for C-ARDS
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ashbaugh, D.G.; Bigelow, D.B.; Petty, T.L.; Levine, B.E. Acute respiratory distress in adults. Lancet 1967, 2, 319–323. [Google Scholar] [CrossRef]
- Gattinoni, L.; Marini, J.J. Isn’t it time to abandon ARDS? The COVID-19 lesson. Crit. Care 2021, 25, 326. [Google Scholar] [CrossRef] [PubMed]
- Bernard, G.R. Acute respiratory distress syndrome: A historical perspective. Am. J. Respir. Crit. Care Med. 2005, 172, 798–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thille, A.W.; Esteban, A.; Fernandez-Segoviano, P.; Rodriguez, J.M.; Aramburu, J.A.; Penuelas, O.; Cortes-Puch, I.; Cardinal-Fernandez, P.; Lorente, J.A.; Frutos-Vivar, F. Comparison of the Berlin definition for acute respiratory distress syndrome with autopsy. Am. J. Respir. Crit. Care Med. 2013, 187, 761–767. [Google Scholar] [CrossRef] [Green Version]
- Marini, J.J. Limitations of clinical trials in acute lung injury and acute respiratory distress syndrome. Curr. Opin. Crit. Care 2006, 12, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Force, A.D.T.; Ranieri, V.M.; Rubenfeld, G.D.; Thompson, B.T.; Ferguson, N.D.; Caldwell, E.; Fan, E.; Camporota, L.; Slutsky, A.S. Acute respiratory distress syndrome: The Berlin Definition. JAMA 2012, 307, 2526–2533. [Google Scholar] [CrossRef]
- Iuliano, A.D.; Brunkard, J.M.; Boehmer, T.K.; Peterson, E.; Adjei, S.; Binder, A.M.; Cobb, S.; Graff, P.; Hidalgo, P.; Panaggio, M.J.; et al. Trends in Disease Severity and Health Care Utilization During the Early Omicron Variant Period Compared with Previous SARS-CoV-2 High Transmission Periods—United States, December 2020–January 2022. MMWR Morb. Mortal. Wkly. Rep. 2022, 71, 146–152. [Google Scholar] [CrossRef]
- Lim, Z.J.; Subramaniam, A.; Ponnapa Reddy, M.; Blecher, G.; Kadam, U.; Afroz, A.; Billah, B.; Ashwin, S.; Kubicki, M.; Bilotta, F.; et al. Case Fatality Rates for Patients with COVID-19 Requiring Invasive Mechanical Ventilation. A Meta-analysis. Am. J. Respir. Crit. Care Med. 2021, 203, 54–66. [Google Scholar] [CrossRef]
- Acute Respiratory Distress Syndrome Network; Brower, R.G.; Matthay, M.A.; Morris, A.; Schoenfeld, D.; Thompson, B.T.; Wheeler, A. Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. N. Engl. J. Med. 2000, 342, 1301–1308. [Google Scholar] [CrossRef] [Green Version]
- Thompson, B.T.; Chambers, R.C.; Liu, K.D. Acute Respiratory Distress Syndrome. N. Engl. J. Med. 2017, 377, 1904–1905. [Google Scholar] [CrossRef]
- Holm, B.A.; Matalon, S. Role of pulmonary surfactant in the development and treatment of adult respiratory distress syndrome. Anesth. Analg. 1989, 69, 805–818. [Google Scholar] [CrossRef] [PubMed]
- Niklason, L.; Eckerstrom, J.; Jonson, B. The influence of venous admixture on alveolar dead space and carbon dioxide exchange in acute respiratory distress syndrome: Computer modelling. Crit. Care 2008, 12, R53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radermacher, P.; Maggiore, S.M.; Mercat, A. Fifty Years of Research in ARDS. Gas Exchange in Acute Respiratory Distress Syndrome. Am. J. Respir. Crit. Care Med. 2017, 196, 964–984. [Google Scholar] [CrossRef] [PubMed]
- Robertson, H.T.; Swenson, E.R. What do dead-space measurements tell us about the lung with acute respiratory distress syndrome? Respir. Care 2004, 49, 1006–1007. [Google Scholar]
- Katzenstein, A.L.; Bloor, C.M.; Leibow, A.A. Diffuse alveolar damage—The role of oxygen, shock, and related factors. A review. Am. J. Pathol. 1976, 85, 209–228. [Google Scholar]
- Gattinoni, L.; Mascheroni, D.; Torresin, A.; Marcolin, R.; Fumagalli, R.; Vesconi, S.; Rossi, G.P.; Rossi, F.; Baglioni, S.; Bassi, F.; et al. Morphological response to positive end expiratory pressure in acute respiratory failure. Computerized tomography study. Intensive Care Med. 1986, 12, 137–142. [Google Scholar] [CrossRef]
- Gattinoni, L.; Pelosi, P.; Vitale, G.; Pesenti, A.; D’Andrea, L.; Mascheroni, D. Body position changes redistribute lung computed-tomographic density in patients with acute respiratory failure. Anesthesiology 1991, 74, 15–23. [Google Scholar] [CrossRef]
- Gattinoni, L.; Pesenti, A.; Avalli, L.; Rossi, F.; Bombino, M. Pressure-volume curve of total respiratory system in acute respiratory failure. Computed tomographic scan study. Am. Rev. Respir. Dis. 1987, 136, 730–736. [Google Scholar] [CrossRef]
- Gattinoni, L.; Marini, J.J.; Pesenti, A.; Quintel, M.; Mancebo, J.; Brochard, L. The “baby lung” became an adult. Intensive Care Med. 2016, 42, 663–673. [Google Scholar] [CrossRef]
- Gattinoni, L.; Pesenti, A. The concept of “baby lung”. Intensive Care Med. 2005, 31, 776–784. [Google Scholar] [CrossRef]
- Gattinoni, L.; Pesenti, A.; Bombino, M.; Baglioni, S.; Rivolta, M.; Rossi, F.; Rossi, G.; Fumagalli, R.; Marcolin, R.; Mascheroni, D.; et al. Relationships between lung computed tomographic density, gas exchange, and PEEP in acute respiratory failure. Anesthesiology 1988, 69, 824–832. [Google Scholar] [CrossRef] [PubMed]
- Marini, J.J.; Gattinoni, L. Time Course of Evolving Ventilator-Induced Lung Injury: The “Shrinking Baby Lung”. Crit. Care Med. 2020, 48, 1203–1209. [Google Scholar] [CrossRef] [PubMed]
- Wiersinga, W.J.; Rhodes, A.; Cheng, A.C.; Peacock, S.J.; Prescott, H.C. Pathophysiology, Transmission, Diagnosis, and Treatment of Coronavirus Disease 2019 (COVID-19): A Review. JAMA 2020, 324, 782–793. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Kruger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e278. [Google Scholar] [CrossRef]
- Lamers, M.M.; Haagmans, B.L. SARS-CoV-2 pathogenesis. Nat. Rev. Microbiol. 2022, 20, 270–284. [Google Scholar] [CrossRef]
- Ogando, N.S.; Dalebout, T.J.; Zevenhoven-Dobbe, J.C.; Limpens, R.; van der Meer, Y.; Caly, L.; Druce, J.; de Vries, J.J.C.; Kikkert, M.; Barcena, M.; et al. SARS-coronavirus-2 replication in Vero E6 cells: Replication kinetics, rapid adaptation and cytopathology. J. Gen. Virol. 2020, 101, 925–940. [Google Scholar] [CrossRef]
- Khan, M.; Yoo, S.J.; Clijsters, M.; Backaert, W.; Vanstapel, A.; Speleman, K.; Lietaer, C.; Choi, S.; Hether, T.D.; Marcelis, L.; et al. Visualizing in deceased COVID-19 patients how SARS-CoV-2 attacks the respiratory and olfactory mucosae but spares the olfactory bulb. Cell 2021, 184, 5932–5949.e5915. [Google Scholar] [CrossRef]
- Hou, Y.J.; Okuda, K.; Edwards, C.E.; Martinez, D.R.; Asakura, T.; Dinnon, K.H., 3rd; Kato, T.; Lee, R.E.; Yount, B.L.; Mascenik, T.M.; et al. SARS-CoV-2 Reverse Genetics Reveals a Variable Infection Gradient in the Respiratory Tract. Cell 2020, 182, 429–446.e414. [Google Scholar] [CrossRef]
- Osuchowski, M.F.; Winkler, M.S.; Skirecki, T.; Cajander, S.; Shankar-Hari, M.; Lachmann, G.; Monneret, G.; Venet, F.; Bauer, M.; Brunkhorst, F.M.; et al. The COVID-19 puzzle: Deciphering pathophysiology and phenotypes of a new disease entity. Lancet Respir. Med. 2021, 9, 622–642. [Google Scholar] [CrossRef]
- Barkauskas, C.E.; Cronce, M.J.; Rackley, C.R.; Bowie, E.J.; Keene, D.R.; Stripp, B.R.; Randell, S.H.; Noble, P.W.; Hogan, B.L. Type 2 alveolar cells are stem cells in adult lung. J. Clin. Investig. 2013, 123, 3025–3036. [Google Scholar] [CrossRef] [PubMed]
- Carty, M.; Guy, C.; Bowie, A.G. Detection of Viral Infections by Innate Immunity. Biochem. Pharmacol. 2021, 183, 114316. [Google Scholar] [CrossRef] [PubMed]
- Land, W.G. Role of DAMPs in respiratory virus-induced acute respiratory distress syndrome-with a preliminary reference to SARS-CoV-2 pneumonia. Genes Immun. 2021, 22, 141–160. [Google Scholar] [CrossRef]
- Hernandez Acosta, R.A.; Esquer Garrigos, Z.; Marcelin, J.R.; Vijayvargiya, P. COVID-19 Pathogenesis and Clinical Manifestations. Infect. Dis. Clin. N. Am. 2022, 36, 231–249. [Google Scholar] [CrossRef] [PubMed]
- Sebag, S.C.; Bastarache, J.A.; Ware, L.B. Therapeutic modulation of coagulation and fibrinolysis in acute lung injury and the acute respiratory distress syndrome. Curr. Pharm. Biotechnol. 2011, 12, 1481–1496. [Google Scholar] [CrossRef]
- Iba, T.; Levy, J.H.; Levi, M.; Thachil, J. Coagulopathy in COVID-19. J. Thromb. Haemost. 2020, 18, 2103–2109. [Google Scholar] [CrossRef]
- Varga, Z.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.S.; Mehra, M.R.; Schuepbach, R.A.; Ruschitzka, F.; Moch, H. Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020, 395, 1417–1418. [Google Scholar] [CrossRef]
- Ackermann, M.; Verleden, S.E.; Kuehnel, M.; Haverich, A.; Welte, T.; Laenger, F.; Vanstapel, A.; Werlein, C.; Stark, H.; Tzankov, A.; et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in Covid-19. N. Engl. J. Med. 2020, 383, 120–128. [Google Scholar] [CrossRef]
- Owens, A.P., 3rd; Mackman, N. Tissue factor and thrombosis: The clot starts here. Thromb. Haemost. 2010, 104, 432–439. [Google Scholar] [CrossRef]
- Kenawy, H.I.; Boral, I.; Bevington, A. Complement-Coagulation Cross-Talk: A Potential Mediator of the Physiological Activation of Complement by Low pH. Front. Immunol. 2015, 6, 215. [Google Scholar] [CrossRef] [Green Version]
- Swieringa, F.; Spronk, H.M.H.; Heemskerk, J.W.M.; van der Meijden, P.E.J. Integrating platelet and coagulation activation in fibrin clot formation. Res. Pract. Thromb. Haemost. 2018, 2, 450–460. [Google Scholar] [CrossRef] [PubMed]
- Bonaventura, A.; Vecchie, A.; Dagna, L.; Martinod, K.; Dixon, D.L.; Van Tassell, B.W.; Dentali, F.; Montecucco, F.; Massberg, S.; Levi, M.; et al. Endothelial dysfunction and immunothrombosis as key pathogenic mechanisms in COVID-19. Nat. Rev. Immunol. 2021, 21, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, T.S.; de Sa, K.S.G.; Ishimoto, A.Y.; Becerra, A.; Oliveira, S.; Almeida, L.; Goncalves, A.V.; Perucello, D.B.; Andrade, W.A.; Castro, R.; et al. Inflammasomes are activated in response to SARS-CoV-2 infection and are associated with COVID-19 severity in patients. J. Exp. Med. 2021, 218, e20201707. [Google Scholar] [CrossRef]
- Kambas, K.; Markiewski, M.M.; Pneumatikos, I.A.; Rafail, S.S.; Theodorou, V.; Konstantonis, D.; Kourtzelis, I.; Doumas, M.N.; Magotti, P.; Deangelis, R.A.; et al. C5a and TNF-alpha up-regulate the expression of tissue factor in intra-alveolar neutrophils of patients with the acute respiratory distress syndrome. J. Immunol. 2008, 180, 7368–7375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Georg, P.; Astaburuaga-Garcia, R.; Bonaguro, L.; Brumhard, S.; Michalick, L.; Lippert, L.J.; Kostevc, T.; Gabel, C.; Schneider, M.; Streitz, M.; et al. Complement activation induces excessive T cell cytotoxicity in severe COVID-19. Cell 2022, 185, 493–512.e425. [Google Scholar] [CrossRef]
- Ebeyer-Masotta, M.; Eichhorn, T.; Weiss, R.; Laukova, L.; Weber, V. Activated Platelets and Platelet-Derived Extracellular Vesicles Mediate COVID-19-Associated Immunothrombosis. Front. Cell Dev. Biol. 2022, 10, 914891. [Google Scholar] [CrossRef]
- Mackman, N.; Antoniak, S.; Wolberg, A.S.; Kasthuri, R.; Key, N.S. Coagulation Abnormalities and Thrombosis in Patients Infected With SARS-CoV-2 and Other Pandemic Viruses. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 2033–2044. [Google Scholar] [CrossRef]
- Williamson, E.J.; Walker, A.J.; Bhaskaran, K.; Bacon, S.; Bates, C.; Morton, C.E.; Curtis, H.J.; Mehrkar, A.; Evans, D.; Inglesby, P.; et al. Factors associated with COVID-19-related death using OpenSAFELY. Nature 2020, 584, 430–436. [Google Scholar] [CrossRef]
- O’Driscoll, M.; Ribeiro Dos Santos, G.; Wang, L.; Cummings, D.A.T.; Azman, A.S.; Paireau, J.; Fontanet, A.; Cauchemez, S.; Salje, H. Age-specific mortality and immunity patterns of SARS-CoV-2. Nature 2021, 590, 140–145. [Google Scholar] [CrossRef]
- Zhang, Q.; Bastard, P.; Liu, Z.; Le Pen, J.; Moncada-Velez, M.; Chen, J.; Ogishi, M.; Sabli, I.K.D.; Hodeib, S.; Korol, C.; et al. Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science 2020, 370, eabd4570. [Google Scholar] [CrossRef]
- Severe Covid, G.G.; Ellinghaus, D.; Degenhardt, F.; Bujanda, L.; Buti, M.; Albillos, A.; Invernizzi, P.; Fernandez, J.; Prati, D.; Baselli, G.; et al. Genomewide Association Study of Severe Covid-19 with Respiratory Failure. N. Engl. J. Med. 2020, 383, 1522–1534. [Google Scholar] [CrossRef]
- Pairo-Castineira, E.; Clohisey, S.; Klaric, L.; Bretherick, A.D.; Rawlik, K.; Pasko, D.; Walker, S.; Parkinson, N.; Fourman, M.H.; Russell, C.D.; et al. Genetic mechanisms of critical illness in COVID-19. Nature 2021, 591, 92–98. [Google Scholar] [CrossRef]
- Initiative, C.-H.G. Mapping the human genetic architecture of COVID-19. Nature 2021, 600, 472–477. [Google Scholar] [CrossRef] [PubMed]
- Asano, T.; Boisson, B.; Onodi, F.; Matuozzo, D.; Moncada-Velez, M.; Maglorius Renkilaraj, M.R.L.; Zhang, P.; Meertens, L.; Bolze, A.; Materna, M.; et al. X-linked recessive TLR7 deficiency in ~1% of men under 60 years old with life-threatening COVID-19. Sci. Immunol. 2021, 6, eabl4348. [Google Scholar] [CrossRef] [PubMed]
- Koning, R.; Bastard, P.; Casanova, J.L.; Brouwer, M.C.; van de Beek, D.; Amsterdam, U.M.C. COVID-19 Biobank Investigators. Autoantibodies against type I interferons are associated with multi-organ failure in COVID-19 patients. Intensive Care Med. 2021, 47, 704–706. [Google Scholar] [CrossRef]
- Bastard, P.; Rosen, L.B.; Zhang, Q.; Michailidis, E.; Hoffmann, H.H.; Zhang, Y.; Dorgham, K.; Philippot, Q.; Rosain, J.; Beziat, V.; et al. Autoantibodies against type I IFNs in patients with life-threatening COVID-19. Science 2020, 370, eabd4585. [Google Scholar] [CrossRef]
- Berlin, D.A.; Gulick, R.M.; Martinez, F.J. Severe Covid-19. N. Engl. J. Med. 2020, 383, 2451–2460. [Google Scholar] [CrossRef]
- Kox, M.; Waalders, N.J.B.; Kooistra, E.J.; Gerretsen, J.; Pickkers, P. Cytokine Levels in Critically Ill Patients With COVID-19 and Other Conditions. JAMA 2020, 234, 1565–1567. [Google Scholar] [CrossRef]
- Leisman, D.E.; Ronner, L.; Pinotti, R.; Taylor, M.D.; Sinha, P.; Calfee, C.S.; Hirayama, A.V.; Mastroiani, F.; Turtle, C.J.; Harhay, M.O.; et al. Cytokine elevation in severe and critical COVID-19: A rapid systematic review, meta-analysis, and comparison with other inflammatory syndromes. Lancet Respir. Med. 2020, 8, 1233–1244. [Google Scholar] [CrossRef]
- The REMAP-CAP; ACTIV-4a; ATTACC Investigators; Goligher, E.C.; Bradbury, C.A.; McVerry, B.J.; Lawler, P.R.; Berger, J.S.; Gong, M.N.; Carrier, M.; et al. Therapeutic Anticoagulation with Heparin in Critically Ill Patients with COVID-19. N. Engl. J. Med. 2021, 385, 777–789. [Google Scholar] [CrossRef]
- Group, R.C.; Horby, P.; Lim, W.S.; Emberson, J.R.; Mafham, M.; Bell, J.L.; Linsell, L.; Staplin, N.; Brightling, C.; Ustianowski, A.; et al. Dexamethasone in Hospitalized Patients with COVID-19. N. Engl. J. Med. 2021, 384, 693–704. [Google Scholar] [CrossRef]
- The WHO Rapid Evidence Appraisal for COVID-19 Therapies (REACT) Working Group; Sterne, J.A.C.; Murthy, S.; Diaz, J.V.; Slutsky, A.S.; Villar, J.; Angus, D.C.; Annane, D.; Azevedo, L.C.P.; Berwanger, O.; et al. Association Between Administration of Systemic Corticosteroids and Mortality Among Critically Ill Patients With COVID-19: A Meta-analysis. JAMA 2020, 324, 1330–1341. [Google Scholar] [CrossRef] [PubMed]
- Stone, J.H.; Frigault, M.J.; Serling-Boyd, N.J.; Fernandes, A.D.; Harvey, L.; Foulkes, A.S.; Horick, N.K.; Healy, B.C.; Shah, R.; Bensaci, A.M.; et al. Efficacy of Tocilizumab in Patients Hospitalized with Covid-19. N. Engl. J. Med. 2020, 383, 2333–2344. [Google Scholar] [CrossRef] [PubMed]
- The WHO Rapid Evidence Appraisal for COVID-19 Therapies (REACT) Working Group; Shankar-Hari, M.; Vale, C.L.; Godolphin, P.J.; Fisher, D.; Higgins, J.P.T.; Spiga, F.; Savovic, J.; Tierney, J.; Baron, G.; et al. Association Between Administration of IL-6 Antagonists and Mortality Among Patients Hospitalized for COVID-19: A Meta-analysis. JAMA 2021, 326, 499–518. [Google Scholar] [CrossRef]
- Kalil, A.C.; Patterson, T.F.; Mehta, A.K.; Tomashek, K.M.; Wolfe, C.R.; Ghazaryan, V.; Marconi, V.C.; Ruiz-Palacios, G.M.; Hsieh, L.; Kline, S.; et al. Baricitinib plus Remdesivir for Hospitalized Adults with Covid-19. N. Engl. J. Med. 2021, 384, 795–807. [Google Scholar] [CrossRef]
- Marconi, V.C.; Ramanan, A.V.; de Bono, S.; Kartman, C.E.; Krishnan, V.; Liao, R.; Piruzeli, M.L.B.; Goldman, J.D.; Alatorre-Alexander, J.; de Cassia Pellegrini, R.; et al. Efficacy and safety of baricitinib for the treatment of hospitalised adults with COVID-19 (COV-BARRIER): A randomised, double-blind, parallel-group, placebo-controlled phase 3 trial. Lancet Respir. Med. 2021, 9, 1407–1418. [Google Scholar] [CrossRef]
- Wolfe, C.R.; Tomashek, K.M.; Patterson, T.F.; Gomez, C.A.; Marconi, V.C.; Jain, M.K.; Yang, O.O.; Paules, C.I.; Palacios, G.M.R.; Grossberg, R.; et al. Baricitinib versus dexamethasone for adults hospitalised with COVID-19 (ACTT-4): A randomised, double-blind, double placebo-controlled trial. Lancet Respir. Med. 2022. [Google Scholar] [CrossRef]
- Selvaraj, V.; Finn, A.; Lal, A.; Khan, M.S.; Dapaah-Afriyie, K.; Carino, G.P. Baricitinib in hospitalised patients with COVID-19: A meta-analysis of randomised controlled trials. EClinicalMedicine 2022, 49, 101489. [Google Scholar] [CrossRef]
- Kyriazopoulou, E.; Poulakou, G.; Milionis, H.; Metallidis, S.; Adamis, G.; Tsiakos, K.; Fragkou, A.; Rapti, A.; Damoulari, C.; Fantoni, M.; et al. Early treatment of COVID-19 with anakinra guided by soluble urokinase plasminogen receptor plasma levels: A double-blind, randomized controlled phase 3 trial. Nat. Med. 2021, 27, 1752–1760. [Google Scholar] [CrossRef]
- Friesecke, S.; Trager, K.; Schittek, G.A.; Molnar, Z.; Bach, F.; Kogelmann, K.; Bogdanski, R.; Weyland, A.; Nierhaus, A.; Nestler, F.; et al. International registry on the use of the CytoSorb(R) adsorber in ICU patients: Study protocol and preliminary results. Med. Klin. Intensivmed. Notfmed. 2019, 114, 699–707. [Google Scholar] [CrossRef]
- Hawchar, F.; Laszlo, I.; Oveges, N.; Trasy, D.; Ondrik, Z.; Molnar, Z. Extracorporeal cytokine adsorption in septic shock: A proof of concept randomized, controlled pilot study. J. Crit. Care 2019, 49, 172–178. [Google Scholar] [CrossRef] [Green Version]
- Brouwer, W.P.; Duran, S.; Kuijper, M.; Ince, C. Hemoadsorption with CytoSorb shows a decreased observed versus expected 28-day all-cause mortality in ICU patients with septic shock: A propensity-score-weighted retrospective study. Crit. Care 2019, 23, 317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rieder, M.; Wengenmayer, T.; Staudacher, D.; Duerschmied, D.; Supady, A. Cytokine adsorption in patients with severe COVID-19 pneumonia requiring extracorporeal membrane oxygenation. Crit. Care 2020, 24, 435. [Google Scholar] [CrossRef] [PubMed]
- Alharthy, A.; Faqihi, F.; Memish, Z.A.; Balhamar, A.; Nasim, N.; Shahzad, A.; Tamim, H.; Alqahtani, S.A.; Brindley, P.G.; Karakitsos, D. Continuous renal replacement therapy with the addition of CytoSorb cartridge in critically ill patients with COVID-19 plus acute kidney injury: A case-series. Artif. Organs 2021, 45, E101–E112. [Google Scholar] [CrossRef] [PubMed]
- Stockmann, H.; Thelen, P.; Stroben, F.; Pigorsch, M.; Keller, T.; Krannich, A.; Spies, C.; Treskatsch, S.; Ocken, M.; Kunz, J.V.; et al. CytoSorb Rescue for COVID-19 Patients with Vasoplegic Shock and Multiple Organ Failure: A Prospective, Open-Label, Randomized Controlled Pilot Study. Crit. Care Med. 2022, 50, 964–976. [Google Scholar] [CrossRef] [PubMed]
- Supady, A.; Weber, E.; Rieder, M.; Lother, A.; Niklaus, T.; Zahn, T.; Frech, F.; Muller, S.; Kuhl, M.; Benk, C.; et al. Cytokine adsorption in patients with severe COVID-19 pneumonia requiring extracorporeal membrane oxygenation (CYCOV): A single centre, open-label, randomised, controlled trial. Lancet Respir. Med. 2021, 9, 755–762. [Google Scholar] [CrossRef]
- Schadler, D.; Pausch, C.; Heise, D.; Meier-Hellmann, A.; Brederlau, J.; Weiler, N.; Marx, G.; Putensen, C.; Spies, C.; Jorres, A.; et al. The effect of a novel extracorporeal cytokine hemoadsorption device on IL-6 elimination in septic patients: A randomized controlled trial. PLoS ONE 2017, 12, e0187015. [Google Scholar] [CrossRef] [Green Version]
- Poli, E.C.; Alberio, L.; Bauer-Doerries, A.; Marcucci, C.; Roumy, A.; Kirsch, M.; De Stefano, E.; Liaudet, L.; Schneider, A.G. Cytokine clearance with CytoSorb(R) during cardiac surgery: A pilot randomized controlled trial. Crit. Care 2019, 23, 108. [Google Scholar] [CrossRef] [Green Version]
- Wilson, J.G.; Calfee, C.S. ARDS Subphenotypes: Understanding a Heterogeneous Syndrome. Crit. Care 2020, 24, 102. [Google Scholar] [CrossRef] [Green Version]
- Gattinoni, L.; Pelosi, P.; Suter, P.M.; Pedoto, A.; Vercesi, P.; Lissoni, A. Acute respiratory distress syndrome caused by pulmonary and extrapulmonary disease. Different syndromes? Am. J. Respir. Crit. Care Med. 1998, 158, 3–11. [Google Scholar] [CrossRef] [Green Version]
- Calfee, C.S.; Delucchi, K.; Parsons, P.E.; Thompson, B.T.; Ware, L.B.; Matthay, M.A.; Network, N.A. Subphenotypes in acute respiratory distress syndrome: Latent class analysis of data from two randomised controlled trials. Lancet Respir. Med. 2014, 2, 611–620. [Google Scholar] [CrossRef] [Green Version]
- Satturwar, S.; Fowkes, M.; Farver, C.; Wilson, A.M.; Eccher, A.; Girolami, I.; Pujadas, E.; Bryce, C.; Salem, F.; El Jamal, S.M.; et al. Postmortem Findings Associated with SARS-CoV-2: Systematic Review and Meta-analysis. Am. J. Surg. Pathol. 2021, 45, 587–603. [Google Scholar] [CrossRef] [PubMed]
- Tomashefski, J.F., Jr.; Davies, P.; Boggis, C.; Greene, R.; Zapol, W.M.; Reid, L.M. The pulmonary vascular lesions of the adult respiratory distress syndrome. Am. J. Pathol. 1983, 112, 112–126. [Google Scholar] [PubMed]
- Hariri, L.P.; North, C.M.; Shih, A.R.; Israel, R.A.; Maley, J.H.; Villalba, J.A.; Vinarsky, V.; Rubin, J.; Okin, D.A.; Sclafani, A.; et al. Lung Histopathology in Coronavirus Disease 2019 as Compared With Severe Acute Respiratory Sydrome and H1N1 Influenza: A Systematic Review. Chest 2021, 159, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Milross, L.; Majo, J.; Cooper, N.; Kaye, P.M.; Bayraktar, O.; Filby, A.; Fisher, A.J. Post-mortem lung tissue: The fossil record of the pathophysiology and immunopathology of severe COVID-19. Lancet Respir. Med. 2022, 10, 95–106. [Google Scholar] [CrossRef]
- Carsana, L.; Sonzogni, A.; Nasr, A.; Rossi, R.S.; Pellegrinelli, A.; Zerbi, P.; Rech, R.; Colombo, R.; Antinori, S.; Corbellino, M.; et al. Pulmonary post-mortem findings in a series of COVID-19 cases from northern Italy: A two-centre descriptive study. Lancet Infect. Dis. 2020, 20, 1135–1140. [Google Scholar] [CrossRef]
- Poissy, J.; Goutay, J.; Caplan, M.; Parmentier, E.; Duburcq, T.; Lassalle, F.; Jeanpierre, E.; Rauch, A.; Labreuche, J.; Susen, S.; et al. Pulmonary Embolism in Patients With COVID-19: Awareness of an Increased Prevalence. Circulation 2020, 142, 184–186. [Google Scholar] [CrossRef]
- Helms, J.; Tacquard, C.; Severac, F.; Leonard-Lorant, I.; Ohana, M.; Delabranche, X.; Merdji, H.; Clere-Jehl, R.; Schenck, M.; Fagot Gandet, F.; et al. High risk of thrombosis in patients with severe SARS-CoV-2 infection: A multicenter prospective cohort study. Intensive Care Med. 2020, 46, 1089–1098. [Google Scholar] [CrossRef]
- Villalba, J.A.; Hilburn, C.F.; Garlin, M.A.; Elliott, G.A.; Li, Y.; Kunitoki, K.; Poli, S.; Alba, G.A.; Madrigal, E.; Taso, M.; et al. Vasculopathy and Increased Vascular Congestion in Fatal COVID-19 and ARDS. Am. J. Respir. Crit. Care Med. 2022. [Google Scholar] [CrossRef]
- Santamarina, M.G.; Boisier Riscal, D.; Beddings, I.; Contreras, R.; Baque, M.; Volpacchio, M.; Martinez Lomakin, F. COVID-19: What Iodine Maps From Perfusion CT can reveal-A Prospective Cohort Study. Crit. Care 2020, 24, 619. [Google Scholar] [CrossRef]
- Li, Q.; Huang, X.T.; Li, C.H.; Liu, D.; Lv, F.J. CT features of coronavirus disease 2019 (COVID-19) with an emphasis on the vascular enlargement pattern. Eur. J. Radiol. 2021, 134, 109442. [Google Scholar] [CrossRef] [PubMed]
- Poschenrieder, F.; Meiler, S.; Lubnow, M.; Zeman, F.; Rennert, J.; Scharf, G.; Schaible, J.; Stroszczynski, C.; Pfeifer, M.; Hamer, O.W. Severe COVID-19 pneumonia: Perfusion analysis in correlation with pulmonary embolism and vessel enlargement using dual-energy CT data. PLoS ONE 2021, 16, e0252478. [Google Scholar] [CrossRef] [PubMed]
- Patel, B.V.; Arachchillage, D.J.; Ridge, C.A.; Bianchi, P.; Doyle, J.F.; Garfield, B.; Ledot, S.; Morgan, C.; Passariello, M.; Price, S.; et al. Pulmonary Angiopathy in Severe COVID-19: Physiologic, Imaging, and Hematologic Observations. Am. J. Respir. Crit. Care Med. 2020, 202, 690–699. [Google Scholar] [CrossRef] [PubMed]
- Gattinoni, L.; Coppola, S.; Cressoni, M.; Busana, M.; Rossi, S.; Chiumello, D. COVID-19 Does Not Lead to a “Typical” Acute Respiratory Distress Syndrome. Am. J. Respir. Crit. Care Med. 2020, 201, 1299–1300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cressoni, M.; Caironi, P.; Polli, F.; Carlesso, E.; Chiumello, D.; Cadringher, P.; Quintel, M.; Ranieri, V.M.; Bugedo, G.; Gattinoni, L. Anatomical and functional intrapulmonary shunt in acute respiratory distress syndrome. Crit. Care Med. 2008, 36, 669–675. [Google Scholar] [CrossRef] [Green Version]
- Chiumello, D.; Busana, M.; Coppola, S.; Romitti, F.; Formenti, P.; Bonifazi, M.; Pozzi, T.; Palumbo, M.M.; Cressoni, M.; Herrmann, P.; et al. Physiological and quantitative CT-scan characterization of COVID-19 and typical ARDS: A matched cohort study. Intensive Care Med. 2020, 46, 2187–2196. [Google Scholar] [CrossRef]
- Barbeta, E.; Motos, A.; Torres, A.; Ceccato, A.; Ferrer, M.; Cilloniz, C.; Bueno, L.; Badia, J.R.; Castro, P.; Ferrando, C.; et al. SARS-CoV-2-induced Acute Respiratory Distress Syndrome: Pulmonary Mechanics and Gas-Exchange Abnormalities. Ann. Am. Thorac. Soc. 2020, 17, 1164–1168. [Google Scholar] [CrossRef]
- Vasques, F.; Sanderson, B.; Formenti, F.; Shankar-Hari, M.; Camporota, L. Physiological dead space ventilation, disease severity and outcome in ventilated patients with hypoxaemic respiratory failure due to coronavirus disease 2019. Intensive Care Med. 2020, 46, 2092–2093. [Google Scholar] [CrossRef]
- Camporota, L.; Sanderson, B.; Chiumello, D.; Terzi, N.; Argaud, L.; Rimmele, T.; Metuor, R.; Verstraete, A.; Cour, M.; Bohe, J.; et al. Prone Position in COVID-19 and -COVID-19 Acute Respiratory Distress Syndrome: An International Multicenter Observational Comparative Study. Crit. Care Med. 2022, 50, 633–643. [Google Scholar] [CrossRef]
- Grieco, D.L.; Bongiovanni, F.; Chen, L.; Menga, L.S.; Cutuli, S.L.; Pintaudi, G.; Carelli, S.; Michi, T.; Torrini, F.; Lombardi, G.; et al. Respiratory physiology of COVID-19-induced respiratory failure compared to ARDS of other etiologies. Crit. Care 2020, 24, 529. [Google Scholar] [CrossRef]
- Kummer, R.L.; Shapiro, R.S.; Marini, J.J.; Huelster, J.S.; Leatherman, J.W. Paradoxically Improved Respiratory Compliance With Abdominal Compression in COVID-19 ARDS. Chest 2021, 160, 1739–1742. [Google Scholar] [CrossRef] [PubMed]
- Haudebourg, A.F.; Perier, F.; Tuffet, S.; de Prost, N.; Razazi, K.; Mekontso Dessap, A.; Carteaux, G. Respiratory Mechanics of COVID-19- versus Non-COVID-19-associated Acute Respiratory Distress Syndrome. Am. J. Respir. Crit. Care Med. 2020, 202, 287–290. [Google Scholar] [CrossRef] [PubMed]
- Panwar, R.; Madotto, F.; Laffey, J.G.; van Haren, F.M.P. Compliance Phenotypes in Early Acute Respiratory Distress Syndrome before the COVID-19 Pandemic. Am. J. Respir. Crit. Care Med. 2020, 202, 1244–1252. [Google Scholar] [CrossRef] [PubMed]
- Beloncle, F.; Studer, A.; Seegers, V.; Richard, J.C.; Desprez, C.; Fage, N.; Merdji, H.; Pavlovsky, B.; Helms, J.; Cunat, S.; et al. Longitudinal changes in compliance, oxygenation and ventilatory ratio in COVID-19 versus non-COVID-19 pulmonary acute respiratory distress syndrome. Crit. Care 2021, 25, 248. [Google Scholar] [CrossRef] [PubMed]
- Gattinoni, L.; Coppola, S.; Cressoni, M.; Busana, M.; Rossi, S.; Chiumello, D. Reply by Gattinoni et al. to Hedenstierna et al., to Maley et al., to Fowler et al., to Bhatia and Mohammed, to Bos, to Koumbourlis and Motoyama, and to Haouzi et al. Am. J. Respir. Crit. Care Med. 2020, 202, 628–630. [Google Scholar] [CrossRef]
- Marini, J.J.; Gattinoni, L. Energetics and the Root Mechanical Cause for Ventilator-induced Lung Injury. Anesthesiology 2018, 128, 1062–1064. [Google Scholar] [CrossRef]
- Marini, J.J.; Rocco, P.R.M.; Gattinoni, L. Static and Dynamic Contributors to Ventilator-induced Lung Injury in Clinical Practice. Pressure, Energy, and Power. Am. J. Respir. Crit. Care Med. 2020, 201, 767–774. [Google Scholar] [CrossRef]
- Fan, E.; Del Sorbo, L.; Goligher, E.C.; Hodgson, C.L.; Munshi, L.; Walkey, A.J.; Adhikari, N.K.J.; Amato, M.B.P.; Branson, R.; Brower, R.G.; et al. An Official American Thoracic Society/European Society of Intensive Care Med.icine/Society of Critical Care Medicine Clinical Practice Guideline: Mechanical Ventilation in Adult Patients with Acute Respiratory Distress Syndrome. Am. J. Respir. Crit. Care Med. 2017, 195, 1253–1263. [Google Scholar] [CrossRef]
- Alhazzani, W.; Moller, M.H.; Arabi, Y.M.; Loeb, M.; Gong, M.N.; Fan, E.; Oczkowski, S.; Levy, M.M.; Derde, L.; Dzierba, A.; et al. Surviving Sepsis Campaign: Guidelines on the Management of Critically Ill Adults with Coronavirus Disease 2019 (COVID-19). Crit. Care Med. 2020, 48, e440–e469. [Google Scholar] [CrossRef]
- Dreyfuss, D.; Saumon, G. Ventilator-induced lung injury: Lessons from experimental studies. Am. J. Respir. Crit. Care Med. 1998, 157, 294–323. [Google Scholar] [CrossRef] [Green Version]
- Stewart, T.E.; Meade, M.O.; Cook, D.J.; Granton, J.T.; Hodder, R.V.; Lapinsky, S.E.; Mazer, C.D.; McLean, R.F.; Rogovein, T.S.; Schouten, B.D.; et al. Evaluation of a ventilation strategy to prevent barotrauma in patients at high risk for acute respiratory distress syndrome. Pressure- and Volume-Limited Ventilation Strategy Group. N. Engl. J. Med. 1998, 338, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Brochard, L.; Roudot-Thoraval, F.; Roupie, E.; Delclaux, C.; Chastre, J.; Fernandez-Mondejar, E.; Clementi, E.; Mancebo, J.; Factor, P.; Matamis, D.; et al. Tidal volume reduction for prevention of ventilator-induced lung injury in acute respiratory distress syndrome. The Multicenter Trail Group on Tidal Volume reduction in ARDS. Am. J. Respir. Crit. Care Med. 1998, 158, 1831–1838. [Google Scholar] [CrossRef] [PubMed]
- Brower, R.G.; Shanholtz, C.B.; Fessler, H.E.; Shade, D.M.; White, P., Jr.; Wiener, C.M.; Teeter, J.G.; Dodd-o, J.M.; Almog, Y.; Piantadosi, S. Prospective, randomized, controlled clinical trial comparing traditional versus reduced tidal volume ventilation in acute respiratory distress syndrome patients. Crit. Care Med. 1999, 27, 1492–1498. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Liu, X.; Xu, Y.; Xu, Z.; Huang, Y.; Chen, S.; Li, S.; Liu, D.; Lin, Z.; Li, Y. Ventilatory Ratio in Hypercapnic Mechanically Ventilated Patients with COVID-19-associated Acute Respiratory Distress Syndrome. Am. J. Respir. Crit. Care Med. 2020, 201, 1297–1299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marini, J.J.; Gattinoni, L. Management of COVID-19 Respiratory Distress. JAMA 2020, 323, 2329–2330. [Google Scholar] [CrossRef]
- Gattinoni, L.; Marini, J.J. In search of the Holy Grail: Identifying the best PEEP in ventilated patients. Intensive Care Med. 2022, 48, 728–731. [Google Scholar] [CrossRef]
- Barthelemy, R.; Beaucote, V.; Bordier, R.; Collet, M.; Le Gall, A.; Hong, A.; de Roquetaillade, C.; Gayat, E.; Mebazaa, A.; Chousterman, B.G. Haemodynamic impact of positive end-expiratory pressure in SARS-CoV-2 acute respiratory distress syndrome: Oxygenation versus oxygen delivery. Br. J. Anaesth. 2021, 126, e70–e72. [Google Scholar] [CrossRef]
- Suter, P.M.; Fairley, B.; Isenberg, M.D. Optimum end-expiratory airway pressure in patients with acute pulmonary failure. N. Engl. J. Med. 1975, 292, 284–289. [Google Scholar] [CrossRef]
- Dickel, S.; Grimm, C.; Popp, M.; Struwe, C.; Sachkova, A.; Golinski, M.; Seeber, C.; Fichtner, F.; Heise, D.; Kranke, P.; et al. A Nationwide Cross-Sectional Online Survey on the Treatment of COVID-19-ARDS: High Variance in Standard of Care in German ICUs. J. Clin. Med. 2021, 10, 3363. [Google Scholar] [CrossRef]
- Grasselli, G.; Zangrillo, A.; Zanella, A.; Antonelli, M.; Cabrini, L.; Castelli, A.; Cereda, D.; Coluccello, A.; Foti, G.; Fumagalli, R.; et al. Baseline Characteristics and Outcomes of 1591 Patients Infected With SARS-CoV-2 Admitted to ICUs of the Lombardy Region, Italy. JAMA 2020, 323, 1574–1581. [Google Scholar] [CrossRef] [Green Version]
- Chiumello, D.; Bonifazi, M.; Pozzi, T.; Formenti, P.; Papa, G.F.S.; Zuanetti, G.; Coppola, S. Positive end-expiratory pressure in COVID-19 acute respiratory distress syndrome: The heterogeneous effects. Crit. Care 2021, 25, 431. [Google Scholar] [CrossRef] [PubMed]
- Sinha, P.; Calfee, C.S.; Beitler, J.R.; Soni, N.; Ho, K.; Matthay, M.A.; Kallet, R.H. Physiologic Analysis and Clinical Performance of the Ventilatory Ratio in Acute Respiratory Distress Syndrome. Am. J. Respir. Crit. Care Med. 2019, 199, 333–341. [Google Scholar] [CrossRef] [PubMed]
- Beitler, J.R.; Sarge, T.; Banner-Goodspeed, V.M.; Gong, M.N.; Cook, D.; Novack, V.; Loring, S.H.; Talmor, D.; Group, E.P.-S. Effect of Titrating Positive End-Expiratory Pressure (PEEP) With an Esophageal Pressure-Guided Strategy vs an Empirical High PEEP-Fio2 Strategy on Death and Days Free from Mechanical Ventilation Among Patients With Acute Respiratory Distress Syndrome: A Randomized Clinical Trial. JAMA 2019, 321, 846–857. [Google Scholar] [CrossRef] [Green Version]
- Pan, C.; Chen, L.; Lu, C.; Zhang, W.; Xia, J.A.; Sklar, M.C.; Du, B.; Brochard, L.; Qiu, H. Lung Recruitability in COVID-19-associated Acute Respiratory Distress Syndrome: A Single-Center Observational Study. Am. J. Respir. Crit. Care Med. 2020, 201, 1294–1297. [Google Scholar] [CrossRef] [Green Version]
- Ball, L.; Robba, C.; Maiello, L.; Herrmann, J.; Gerard, S.E.; Xin, Y.; Battaglini, D.; Brunetti, I.; Minetti, G.; Seitun, S.; et al. Computed tomography assessment of PEEP-induced alveolar recruitment in patients with severe COVID-19 pneumonia. Crit. Care 2021, 25, 81. [Google Scholar] [CrossRef] [PubMed]
- Protti, A.; Santini, A.; Pennati, F.; Chiurazzi, C.; Cressoni, M.; Ferrari, M.; Iapichino, G.E.; Carenzo, L.; Lanza, E.; Picardo, G.; et al. Lung Response to a Higher Positive End-Expiratory Pressure in Mechanically Ventilated Patients With COVID-19. Chest 2022, 161, 979–988. [Google Scholar] [CrossRef] [PubMed]
- Roesthuis, L.; van den Berg, M.; van der Hoeven, H. Advanced respiratory monitoring in COVID-19 patients: Use less PEEP! Crit. Care 2020, 24, 230. [Google Scholar] [CrossRef]
- Perier, F.; Tuffet, S.; Maraffi, T.; Alcala, G.; Victor, M.; Haudebourg, A.F.; De Prost, N.; Amato, M.; Carteaux, G.; Mekontso Dessap, A. Effect of Positive End-Expiratory Pressure and Proning on Ventilation and Perfusion in COVID-19 Acute Respiratory Distress Syndrome. Am. J. Respir. Crit. Care Med. 2020, 202, 1713–1717. [Google Scholar] [CrossRef]
- Mauri, T.; Spinelli, E.; Scotti, E.; Colussi, G.; Basile, M.C.; Crotti, S.; Tubiolo, D.; Tagliabue, P.; Zanella, A.; Grasselli, G.; et al. Potential for Lung Recruitment and Ventilation-Perfusion Mismatch in Patients With the Acute Respiratory Distress Syndrome From Coronavirus Disease 2019. Crit. Care Med. 2020, 48, 1129–1134. [Google Scholar] [CrossRef]
- Chen, L.; Del Sorbo, L.; Grieco, D.L.; Junhasavasdikul, D.; Rittayamai, N.; Soliman, I.; Sklar, M.C.; Rauseo, M.; Ferguson, N.D.; Fan, E.; et al. Potential for Lung Recruitment Estimated by the Recruitment-to-Inflation Ratio in Acute Respiratory Distress Syndrome. A Clinical Trial. Am. J. Respir. Crit. Care Med. 2020, 201, 178–187. [Google Scholar] [CrossRef]
- Gattinoni, L.; Camporota, L.; Marini, J.J. Prone Position and COVID-19: Mechanisms and Effects. Crit. Care Med. 2022, 50, 873–875. [Google Scholar] [CrossRef] [PubMed]
- Rouby, J.J.; Puybasset, L.; Nieszkowska, A.; Lu, Q. Acute respiratory distress syndrome: Lessons from computed tomography of the whole lung. Crit. Care Med. 2003, 31, S285–S295. [Google Scholar] [CrossRef] [PubMed]
- Albert, R.K.; Hubmayr, R.D. The prone position eliminates compression of the lungs by the heart. Am. J. Respir. Crit. Care Med. 2000, 161, 1660–1665. [Google Scholar] [CrossRef]
- Pelosi, P.; D’Andrea, L.; Vitale, G.; Pesenti, A.; Gattinoni, L. Vertical gradient of regional lung inflation in adult respiratory distress syndrome. Am. J. Respir. Crit. Care Med. 1994, 149, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Gattinoni, L.; Taccone, P.; Carlesso, E.; Marini, J.J. Prone position in acute respiratory distress syndrome. Rationale, indications, and limits. Am. J. Respir. Crit. Care Med. 2013, 188, 1286–1293. [Google Scholar] [CrossRef] [PubMed]
- Guerin, C.; Albert, R.K.; Beitler, J.; Gattinoni, L.; Jaber, S.; Marini, J.J.; Munshi, L.; Papazian, L.; Pesenti, A.; Vieillard-Baron, A.; et al. Prone position in ARDS patients: Why, when, how and for whom. Intensive Care Med. 2020, 46, 2385–2396. [Google Scholar] [CrossRef]
- Mentzelopoulos, S.D.; Roussos, C.; Zakynthinos, S.G. Prone position reduces lung stress and strain in severe acute respiratory distress syndrome. Eur. Respir. J. 2005, 25, 534–544. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Zhang, Y.; Li, Y.; Song, C.; Lin, F.; Pan, P. The Application of Awake-Prone Positioning Among Non-intubated Patients With COVID-19-Related ARDS: A Narrative Review. Front. Med. (Lausanne) 2022, 9, 817689. [Google Scholar] [CrossRef]
- Langer, T.; Brioni, M.; Guzzardella, A.; Carlesso, E.; Cabrini, L.; Castelli, G.; Dalla Corte, F.; De Robertis, E.; Favarato, M.; Forastieri, A.; et al. Prone position in intubated, mechanically ventilated patients with COVID-19: A multi-centric study of more than 1000 patients. Crit. Care 2021, 25, 128. [Google Scholar] [CrossRef]
- Bellani, G.; Laffey, J.G.; Pham, T.; Fan, E. The LUNG SAFE study: A presentation of the prevalence of ARDS according to the Berlin Definition! Crit. Care 2016, 20, 268. [Google Scholar] [CrossRef] [Green Version]
- Guerin, C.; Reignier, J.; Richard, J.C.; Beuret, P.; Gacouin, A.; Boulain, T.; Mercier, E.; Badet, M.; Mercat, A.; Baudin, O.; et al. Prone positioning in severe acute respiratory distress syndrome. N. Engl. J. Med. 2013, 368, 2159–2168. [Google Scholar] [CrossRef] [PubMed]
- Mathews, K.S.; Soh, H.; Shaefi, S.; Wang, W.; Bose, S.; Coca, S.; Gupta, S.; Hayek, S.S.; Srivastava, A.; Brenner, S.K.; et al. Prone Positioning and Survival in Mechanically Ventilated Patients With Coronavirus Disease 2019-Related Respiratory Failure. Crit. Care Med. 2021, 49, 1026–1037. [Google Scholar] [CrossRef] [PubMed]
- Fossali, T.; Pavlovsky, B.; Ottolina, D.; Colombo, R.; Basile, M.C.; Castelli, A.; Rech, R.; Borghi, B.; Ianniello, A.; Flor, N.; et al. Effects of Prone Position on Lung Recruitment and Ventilation-Perfusion Matching in Patients With COVID-19 Acute Respiratory Distress Syndrome: A Combined CT Scan/Electrical Impedance Tomography Study. Crit. Care Med. 2022, 50, 723–732. [Google Scholar] [CrossRef] [PubMed]
- Zarantonello, F.; Sella, N.; Pettenuzzo, T.; Andreatta, G.; Calore, A.; Dotto, D.; De Cassai, A.; Calabrese, F.; Boscolo, A.; Navalesi, P. Early physiological effects of prone positioning in COVID-19 Acute Respiratory Distress Syndrome. Anesthesiology 2022. Online ahead of print. [Google Scholar] [CrossRef]
- Rossi, S.; Palumbo, M.M.; Sverzellati, N.; Busana, M.; Malchiodi, L.; Bresciani, P.; Ceccarelli, P.; Sani, E.; Romitti, F.; Bonifazi, M.; et al. Mechanisms of oxygenation responses to proning and recruitment in COVID-19 pneumonia. Intensive Care Med. 2022, 48, 56–66. [Google Scholar] [CrossRef]
- Protti, A.; Santini, A.; Pennati, F.; Chiurazzi, C.; Ferrari, M.; Iapichino, G.E.; Carenzo, L.; Dalla Corte, F.; Lanza, E.; Martinetti, N.; et al. Lung response to prone positioning in mechanically-ventilated patients with COVID-19. Crit. Care 2022, 26, 127. [Google Scholar] [CrossRef]
- Brodie, D.; Slutsky, A.S.; Combes, A. Extracorporeal Life Support for Adults with Respiratory Failure and Related Indications: A Review. JAMA 2019, 322, 557–568. [Google Scholar] [CrossRef]
- Fernando, S.M.; Qureshi, D.; Tanuseputro, P.; Fan, E.; Munshi, L.; Rochwerg, B.; Talarico, R.; Scales, D.C.; Brodie, D.; Dhanani, S.; et al. Mortality and costs following extracorporeal membrane oxygenation in critically ill adults: A population-based cohort study. Intensive Care Med. 2019, 45, 1580–1589. [Google Scholar] [CrossRef]
- Nesseler, N.; Fadel, G.; Mansour, A.; Para, M.; Falcoz, P.E.; Mongardon, N.; Porto, A.; Bertier, A.; Levy, B.; Cadoz, C.; et al. Extracorporeal Membrane Oxygenation for Respiratory Failure Related to COVID-19: A Nationwide Cohort Study. Anesthesiology 2022, 136, 732–748. [Google Scholar] [CrossRef]
- Combes, A.; Hajage, D.; Capellier, G.; Demoule, A.; Lavoue, S.; Guervilly, C.; Da Silva, D.; Zafrani, L.; Tirot, P.; Veber, B.; et al. Extracorporeal Membrane Oxygenation for Severe Acute Respiratory Distress Syndrome. N. Engl. J. Med. 2018, 378, 1965–1975. [Google Scholar] [CrossRef]
- Badulak, J.; Antonini, M.V.; Stead, C.M.; Shekerdemian, L.; Raman, L.; Paden, M.L.; Agerstrand, C.; Bartlett, R.H.; Barrett, N.; Combes, A.; et al. Extracorporeal Membrane Oxygenation for COVID-19: Updated 2021 Guidelines from the Extracorporeal Life Support Organization. ASAIO J. 2021, 67, 485–495. [Google Scholar] [CrossRef]
- Gattinoni, L.; Gattarello, S.; Steinberg, I.; Busana, M.; Palermo, P.; Lazzari, S.; Romitti, F.; Quintel, M.; Meissner, K.; Marini, J.J.; et al. COVID-19 pneumonia: Pathophysiology and management. Eur. Respir. Rev. 2021, 30, 210138. [Google Scholar] [CrossRef] [PubMed]
- Bain, W.; Yang, H.; Shah, F.A.; Suber, T.; Drohan, C.; Al-Yousif, N.; DeSensi, R.S.; Bensen, N.; Schaefer, C.; Rosborough, B.R.; et al. COVID-19 versus Non-COVID-19 Acute Respiratory Distress Syndrome: Comparison of Demographics, Physiologic Parameters, Inflammatory Biomarkers, and Clinical Outcomes. Ann. Am. Thorac. Soc. 2021, 18, 1202–1210. [Google Scholar] [CrossRef] [PubMed]
- Dalia, A.A.; Convissar, D.; Crowley, J.; Raz, Y.; Funamoto, M.; Wiener-Kronish, J.; Shelton, K. The role of extracorporeal membrane oxygenation in COVID-19. J. Cardiothorac. Vasc. Anesth. 2022, 36, 3668–3675. [Google Scholar] [CrossRef] [PubMed]
- McNamee, J.J.; Gillies, M.A.; Barrett, N.A.; Perkins, G.D.; Tunnicliffe, W.; Young, D.; Bentley, A.; Harrison, D.A.; Brodie, D.; Boyle, A.J.; et al. Effect of Lower Tidal Volume Ventilation Facilitated by Extracorporeal Carbon Dioxide Removal vs Standard Care Ventilation on 90-Day Mortality in Patients with Acute Hypoxemic Respiratory Failure: The REST Randomized Clinical Trial. JAMA 2021, 326, 1013–1023. [Google Scholar] [CrossRef]
- Ramanathan, K.; Shekar, K.; Ling, R.R.; Barbaro, R.P.; Wong, S.N.; Tan, C.S.; Rochwerg, B.; Fernando, S.M.; Takeda, S.; MacLaren, G.; et al. Extracorporeal membrane oxygenation for COVID-19: A systematic review and meta-analysis. Crit. Care 2021, 25, 211. [Google Scholar] [CrossRef]
- Ling, R.R.; Ramanathan, K.; Sim, J.J.L.; Wong, S.N.; Chen, Y.; Amin, F.; Fernando, S.M.; Rochwerg, B.; Fan, E.; Barbaro, R.P.; et al. Evolving outcomes of extracorporeal membrane oxygenation during the first 2 years of the COVID-19 pandemic: A systematic review and meta-analysis. Crit. Care 2022, 26, 147. [Google Scholar] [CrossRef]
- Barbaro, R.P.; MacLaren, G.; Boonstra, P.S.; Combes, A.; Agerstrand, C.; Annich, G.; Diaz, R.; Fan, E.; Hryniewicz, K.; Lorusso, R.; et al. Extracorporeal membrane oxygenation for COVID-19: Evolving outcomes from the international Extracorporeal Life Support Organization Registry. Lancet 2021, 398, 1230–1238. [Google Scholar] [CrossRef]
- Broman, L.M.; Eksborg, S.; Lo Coco, V.; De Piero, M.E.; Belohlavek, J.; Lorusso, R.; Euro, E.C.-W.G.; Euro, E.S.C. Extracorporeal membrane oxygenation for COVID-19 during first and second waves. Lancet Respir. Med. 2021, 9, e80–e81. [Google Scholar] [CrossRef]
- Chong, W.H.; Saha, B.K.; Medarov, B.I. Clinical Characteristics Between Survivors and Nonsurvivors of COVID-19 Patients Requiring Extracorporeal Membrane Oxygenation (ECMO) Support: A Systematic Review and Meta-Analysis. J. Intensive Care Med. 2022, 37, 304–318. [Google Scholar] [CrossRef]
- Kannapadi, N.V.; Jami, M.; Premraj, L.; Etchill, E.W.; Giuliano, K.; Bush, E.L.; Kim, B.S.; Seal, S.; Whitman, G.; Cho, S.M. Neurological Complications in COVID-19 Patients With ECMO Support: A Systematic Review and Meta-Analysis. Heart Lung Circ. 2022, 31, 292–298. [Google Scholar] [CrossRef] [PubMed]
- Paternoster, G.; Bertini, P.; Innelli, P.; Trambaiolo, P.; Landoni, G.; Franchi, F.; Scolletta, S.; Guarracino, F. Right Ventricular Dysfunction in Patients With COVID-19: A Systematic Review and Meta-analysis. J. Cardiothorac. Vasc. Anesth. 2021, 35, 3319–3324. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Guo, Z.; Li, B.; Zhang, X.; Tian, R.; Wu, W.; Zhang, Z.; Lu, Y.; Chen, N.; Clifford, S.P.; et al. Extracorporeal Membrane Oxygenation for Coronavirus Disease 2019 in Shanghai, China. ASAIO J. 2020, 66, 475–481. [Google Scholar] [CrossRef] [PubMed]
- Dreier, E.; Malfertheiner, M.V.; Dienemann, T.; Fisser, C.; Foltan, M.; Geismann, F.; Graf, B.; Lunz, D.; Maier, L.S.; Muller, T.; et al. ECMO in COVID-19-prolonged therapy needed? A retrospective analysis of outcome and prognostic factors. Perfusion 2021, 36, 582–591. [Google Scholar] [CrossRef]
- Cypel, M.; Keshavjee, S. When to consider lung transplantation for COVID-19. Lancet Respir. Med. 2020, 8, 944–946. [Google Scholar] [CrossRef]
- Giani, M.; Rezoagli, E.; Guervilly, C.; Rilinger, J.; Duburcq, T.; Petit, M.; Textoris, L.; Garcia, B.; Wengenmayer, T.; Grasselli, G.; et al. Prone positioning during venovenous extracorporeal membrane oxygenation for acute respiratory distress syndrome: A pooled individual patient data analysis. Crit. Care 2022, 26, 8. [Google Scholar] [CrossRef] [PubMed]
- Poon, W.H.; Ramanathan, K.; Ling, R.R.; Yang, I.X.; Tan, C.S.; Schmidt, M.; Shekar, K. Prone positioning during venovenous extracorporeal membrane oxygenation for acute respiratory distress syndrome: A systematic review and meta-analysis. Crit. Care 2021, 25, 292. [Google Scholar] [CrossRef]
- Laghlam, D.; Charpentier, J.; Hamou, Z.A.; Nguyen, L.S.; Pene, F.; Cariou, A.; Mira, J.P.; Jozwiak, M. Effects of Prone Positioning on Respiratory Mechanics and Oxygenation in Critically Ill Patients With COVID-19 Requiring Venovenous Extracorporeal Membrane Oxygenation. Front. Med. 2021, 8, 810393. [Google Scholar] [CrossRef]
- Giani, M.; Martucci, G.; Madotto, F.; Belliato, M.; Fanelli, V.; Garofalo, E.; Forlini, C.; Lucchini, A.; Panarello, G.; Bottino, N.; et al. Prone Positioning during Venovenous Extracorporeal Membrane Oxygenation in Acute Respiratory Distress Syndrome. A Multicenter Cohort Study and Propensity-matched Analysis. Ann. Am. Thorac. Soc. 2021, 18, 495–501. [Google Scholar] [CrossRef]
- Iwashyna, T.J.; Burke, J.F.; Sussman, J.B.; Prescott, H.C.; Hayward, R.A.; Angus, D.C. Implications of Heterogeneity of Treatment Effect for Reporting and Analysis of Randomized Trials in Critical Care. Am. J. Respir. Crit. Care Med. 2015, 192, 1045–1051. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
| Timing | Within 1 week of known clinical insult or new or worsening respiratory symptoms | |
| Chest imaging | Bilateral opacities on CXR or CT not fully explained by effusions, lobar/lung collapse, or nodules | |
| Origin of edema | Respiratory failure not fully explained by cardiac failure or fluid overload | |
| Oxygenation | Mild | 200 mm Hg < PaO2/FiO2 ≤ 300 mm Hg with PEEP or CPAP ≥ 5 cm H2O |
| Moderate | 100 mm Hg < PaO2/FiO2 ≤ 200 mm Hg with PEEP ≤ 5 cm H2O | |
| Severe | PaO2/FiO2 ≤ 100 mm Hg with PEEP ≥ 5 cm H2O | |
| Typical ARDS | C-ARDS | |
|---|---|---|
| Etiology | Diverse, pulmonary or extrapulmonary (e.g., bacterial or viral pneumonia, severe trauma, aspiration, sepsis, etc.) | SARS-CoV-2 infection of alveolar type 2 cells (primarily) |
| Hypoxemia (PaO2/FiO2 ≤ 300 mmHg at a PEEP level of ≥ 5 cmH2O) | Acute onset (e.g., within <48 h after the clinical insult), or progressive onset (i.e., within 7 days after the clinical insult) | Progressive onset (i.e., within 7 or more days after the onset of COVID-19 symptoms) * |
| Lung compliance at hypoxemia onset | Usually low (e.g., <40 cmH2O/L) | Usually high (e.g., >40 cmH2O/L) |
| Recruitment potential | Low or high, depending on the extent/nature of lung unit involvement and associated atelectasis | Initially low—may increase with disease progression and development of edema and atelectasis |
| Functional-to-anatomical shunt ratio/hyperperfusion of gasless tissue * | Usually 0.5–2.0/no | Usually > 2.0/yes |
| Alveolar capillary microthrombosis/new vessel growth | Present/present | Diffuse (~9 times more prevalent)/marked (2.7 times higher) |
| Clinical benefit from lung-protective ventilation | Proven | Highly likely |
| Clinical benefit from prone positioning | Proven | Highly likely |
| Clinical benefit from corticosteroids | Likely; more high-quality evidence needed | Proven |
| Clinical benefit from targeted anti-inflammatory interventions | Uncertain; lack of intervention-specific evidence | Proven |
| Clinical benefit from ECMO | Likely | Possible; high-quality evidence still needed |
| Intervention | Mechanism of Action | Evidence for Efficacy |
|---|---|---|
| Remdesivir day 1: 200 mg IV days 2–10: 100 mg IV | Inhibition of the viral RNA-dependent, RNA polymerase | Shortens the time to recovery in hospitalized COVID-19 patients |
| Dexamethasone days 1–10 *: 6 mg IV | Anti-inflammator linked to the activation of the glucocorticoid receptor | Reduces the probability of in-hospital death in critically ill COVID-19 patients |
| Tocilizumab single dose: 8 mg/kg IV (max. 800 mg) | Interleukin 6 antagonism | Reduces the probability of in-hospital death in critically ill COVID-19 patients |
| Baracitinib days 1–14 *: 4 mg † oral or enteral | Janus kinase inhibition | Reduces the probability of in-hospital death in critically ill COVID-19 patients |
| Anakinra days 1–10 *: 100 mg subcutaneously | Interleukin 1 alpha/beta antagonism | Reduces the probability of in-hospital death in critically ill COVID-19 patients |
| Prone positioning for at least 16 h per day until PaO2/FiO2 ≥150 mmHg at PEEP ≤10 cmH2O and FiO2 ≤ 0.6 | Attenuation of lung stress and strain Reversal of compression atelectasis Increased homogeneity of ventilation Improved ventilation/perfusion matching | Reduces the probability of in-hospital death in moderate to severe ARDS |
| Extracorporeal membrane oxygenation | Minimization of lung stress and strain (“lung rest”) with very low tidal volumes and ventilation pressures | Possible mortality benefit in severe ARDS |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Selickman, J.; Vrettou, C.S.; Mentzelopoulos, S.D.; Marini, J.J. COVID-19-Related ARDS: Key Mechanistic Features and Treatments. J. Clin. Med. 2022, 11, 4896. https://doi.org/10.3390/jcm11164896
Selickman J, Vrettou CS, Mentzelopoulos SD, Marini JJ. COVID-19-Related ARDS: Key Mechanistic Features and Treatments. Journal of Clinical Medicine. 2022; 11(16):4896. https://doi.org/10.3390/jcm11164896
Chicago/Turabian StyleSelickman, John, Charikleia S. Vrettou, Spyros D. Mentzelopoulos, and John J. Marini. 2022. "COVID-19-Related ARDS: Key Mechanistic Features and Treatments" Journal of Clinical Medicine 11, no. 16: 4896. https://doi.org/10.3390/jcm11164896
APA StyleSelickman, J., Vrettou, C. S., Mentzelopoulos, S. D., & Marini, J. J. (2022). COVID-19-Related ARDS: Key Mechanistic Features and Treatments. Journal of Clinical Medicine, 11(16), 4896. https://doi.org/10.3390/jcm11164896