
Pathophysiology of Coagulation and Emerging Roles for Extracellular Vesicles in Coagulation Cascades and Disorders



Abstract
:1. Introduction
2. Coagulation: A Historical Overview
3. Hemostasis
3.1. Primary Hemostasis, Secondary Hemostasis, and the Cell-Based Model of Coagulation
3.2. Fibrinolysis, Anti-Coagulation Factors, and Coagulation Inhibitors
3.3. Pathophysiology of Coagulopathies
4. Novel Avenues in Thrombosis Research
4.1. Extracellular Vesicles as Novel Modulators of Coagulation

{kind=link}
{kind=link}
{kind=link}
| Disease | Alteration of the Abundance of Circulating Procoagulant Extracellular Vesicles | References |
|---|---|---|
| Thrombotic thrombocytopenic purpura (TTP) | Increased levels of circulating platelet-derived EVs | [96] |
| Idiopathic thrombocytopenic purpura (ITP) | Increased levels of circulating platelet-derived EVs | [116] |
| Heparin-induced thrombocytopenia (HIT) | Increased levels of circulating TF- expressing platelet-derived EVs | [98,117] |
| Sickle cell anemia | Elevated circulating levels of erythrocyte, platelet, monocyte, and endothelial cell-derived EVs | [93,118,119] |
| Disseminated intravascular coagulation (DIC) | Increased levels of circulating endothelial cell-derived EVs (suggested as a biomarker of DIC caused by septic shock) | [120,121,122] |
| Acute coronary syndromes (ACS) | Elevated platelet and monocyte-derived MVs Increased levels of circulating CD31+ CD42b− MVs | [99,123,124] |
| Venous thromboembolism (VTE) | Elevated levels of circulating endothelial cell and platelet-derived PSGL-1 and CD62P-expressing MVs | [125,126] |
| Acute ischemic stroke (AIS) | Elevated levels of circulating endothelial cell-derived MVs | [127] |
| Paroxysmal nocturnal hemoglobinuria (PNH) | Increased levels of circulating platelet, monocyte, and endothelial cell-derived EVs | [128,129] |
| Coronary heart disease (CHD) | Elevated levels of CD31+, CD42−, and CD144+ endothelial cell-derived EVs | [124] |
| Acute myocardial ischemia | Elevated levels of circulating CD66b+, CD62E+, and CD142+ EVs | [130] |
| ST-segment elevation myocardial infarction (STEMI) | Elevated levels of circulating leukocyte-derived CD11+, endothelial cell-derived CD105+, and TF-bearing MVs Increased levels of erythrocyte-derived but not platelet-derived MVs | [131] |
| Acute stroke (AS) | Elevated levels of circulating CD62E+ endothelial cell-derived EVs | [101] |
| Acute pulmonary embolism (APE). | Increased levels of circulating TF-expressing MVs | [132,133] |
| Atrial Fibrillation (AF) | Increased levels of circulating platelet-derived and mononuclear cell-derived EVs and reduced levels of circulating endothelial cell-derived EVs Increased levels of circulating procoagulant EVs expressing TF, PS, and P-selectin | [105,106,107,108] |
4.1.1. Microvesicles
4.1.2. Platelet-Derived Microvesicles
Exosomes
4.1.3. Platelet-Derived Exosomes
4.1.4. Apoptotic Bodies
4.2. Extracellular Vesicles as Drivers of Hypercoagulable States
4.2.1. Extracellular Vesicles in the Cancer-Associated Prothrombotic States
4.2.2. Extracellular Vesicles in the Sepsis-Associated Prothrombotic States
5. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Kahn, M.J. Hypercoagulability as a cause of stroke in adults.(Featured cme topic: Stroke). South. Med. J. 2003, 96, 350–353. [Google Scholar] [CrossRef]
- Riddel, J.P., Jr.; Aouizerat, B.E.; Miaskowski, C.; Lillicrap, D.P. Theories of blood coagulation. J. Pediatr. Oncol. Nurs. 2007, 24, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Fughelli, P.; Stella, A.; Sterpetti, A.V. Marcello malpighi (1628–1694) the revolution in medicine. Circ. Res. 2019, 124, 1430–1432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palta, S.; Saroa, R.; Palta, A. Overview of the coagulation system. Indian J. Anaesth. 2014, 58, 515–523. [Google Scholar] [CrossRef] [PubMed]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nawaz, M.; Shah, N.; Zanetti, B.R.; Maugeri, M.; Silvestre, R.N.; Fatima, F.; Neder, L.; Valadi, H. Extracellular vesicles and matrix remodeling enzymes: The emerging roles in extracellular matrix remodeling, progression of diseases and tissue repair. Cells 2018, 7, 167. [Google Scholar] [CrossRef] [Green Version]
- Van der Pol, E.; Böing, A.N.; Harrison, P.; Sturk, A.; Nieuwland, R. Classification, functions, and clinical relevance of extracellular vesicles. Pharmacol. Rev. 2012, 64, 676–705. [Google Scholar] [CrossRef] [Green Version]
- Femminò, S.; Penna, C.; Margarita, S.; Comità, S.; Brizzi, M.F.; Pagliaro, P. Extracellular vesicles and cardiovascular system: Biomarkers and cardioprotective effectors. Vasc. Pharmacol. 2020, 135, 106790. [Google Scholar] [CrossRef]
- Meletis, J.; Konstantopoulos, K. The beliefs, myths, and reality surrounding the word hema (blood) from Homer to the present. Anemia 2010, 2010, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Tsoucalas, G.; Karamanou, M.; Papaioannou, T.G.; Sgantzos, M. Theories about blood coagulation in the writings of ancient Greek medico-philosophers. Curr. Pharm. Des. 2017, 23, 1275–1278. [Google Scholar] [CrossRef]
- Babington, B.G. Some considerations with respect to the blood, founded on one or two very simple experiments on that fluid. Med. Chirurgical Trans. 1831, 16, 293. [Google Scholar] [CrossRef] [Green Version]
- Owen, C.A. A history of blood coagulation. Mayo Found. Med. 2002, 287, 1051–1052. [Google Scholar]
- Douglas, S. Coagulation history, Oxford 1951–53. Br. J. Haematol. 1999, 107, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Costa-Filho, R.; Hochleitner, G.; Wendt, M.; Teruya, A.; Spahn, D.R. Over 50 years of fibrinogen concentrate. Clin. Appl. Thromb. Hemost. 2016, 22, 109–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, A. Neue untersuchungen über die faserstoffgerinnung. Arch. Physiol. Menschen Tiere 1872, 6, 413–538. [Google Scholar] [CrossRef]
- Morawitz, P. The Chemistry of Blood Coagulation [Die Chemie der Blutgerinnung]; Hartmann, R.; Guenter, P.F., Translators; Charles C Thomas: Springfield, IL, USA, 1958. [Google Scholar]
- Willebrand, E.V. Über hereditäre Pseudohämophilie. Acta Med. Scand. 1931, 76, 521–550. [Google Scholar] [CrossRef]
- Owren, P.A. The coagulation of blood; investigations on a new clotting factor. Acta Med. Scand. 1947, 194, 1–7. [Google Scholar]
- Alexander, B.; Goldstein, R.; Landwehr, G.; Cook, C.; Addelson, E.; Wilson, C. Congenital SPCA deficiency: A hitherto unrecognized coagulation defect with hemorrhage rectified by serum and serum fractions. J. Clin. Investig. 1951, 30, 596–608. [Google Scholar] [CrossRef]
- Patek, A.J.; Stetson, R.P.; Hemophilia, I. The abnormal coagulation of the blood and its relation to the blood platelets. J. Clin. Investig. 1936, 15, 531–542. [Google Scholar] [CrossRef] [Green Version]
- Aggeler, P.M.; White, S.G.; Glendening, M.B.; Page, E.W.; Leake, T.B.; Bates, G. Plasma throinboplastin component (PTC) deficiency: A new disease resembling hemophilia. Exp. Biol. Med. 1952, 79, 692–694. [Google Scholar] [CrossRef]
- Rosenthal, R.L.; Dreskin, O.H.; Rosenthal, N. New hemophilia-like disease caused by deficiency of a third plasma thromboplastin factor. Exp. Biol. Med. 1953, 82, 171–174. [Google Scholar] [CrossRef] [PubMed]
- Neuenschwander, P. Coagulation cascade: Overview. Encyclopedia of Respiratory Medicine; Laurent, G.J., Dshapiro, S., Eds.; Elsevier Ltd.: Oxford, UK, 2006. [Google Scholar]
- Davie, E.W.; Ratnoff, O.D. Waterfall sequence for intrinsic blood clotting. Science 1964, 145, 1310–1312. [Google Scholar] [CrossRef] [PubMed]
- Macfarlane, R. An enzyme cascade in the blood clotting mechanism, and its function as a biochemical amplifier. Nature 1964, 202, 498–499. [Google Scholar] [CrossRef]
- Versteeg, H.H.; Heemskerk, J.W.; Levi, M.; Reitsma, P.H. New fundamentals in hemostasis. Physiol. Rev. 2013, 93, 327–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luchtman-Jones, L.; Broze, G.J. The current status of coagulation. Ann. Med. 1995, 27, 47–52. [Google Scholar] [CrossRef]
- Dam, H. The antihæmorrhagic vitamin of the chick.: Occurrence and chemical nature. Nature 1935, 135, 652–653. [Google Scholar] [CrossRef]
- Berkner, K.L. The vitamin K–dependent carboxylase. Annu. Rev. Nutr. 2005, 25, 127–149. [Google Scholar] [CrossRef]
- Hoffman, M.; Monroe, D.M., III. A cell-based model of hemostasis. Thromb. Haemost. 2001, 85, 958–965. [Google Scholar]
- Loscalzo, J.; Schafer, A.I. Thrombosis and Hemorrhage; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2003. [Google Scholar]
- Lippi, G.; Favaloro, E.J. Laboratory hemostasis: Milestones in clinical chemistry and laboratory medicine. Clin. Chem. Lab. Med. (CCLM) 2012, 51, 91–97. [Google Scholar] [CrossRef]
- Bonar, R.A.; Lippi, G.; Favaloro, E.J. Overview of hemostasis and thrombosis and contribution of laboratory testing to diagnosis and management of hemostasis and thrombosis disorders. In Hemostasis and Thrombosis; Springer: Berlin/Heidelberg, Germany, 2017; pp. 3–27. [Google Scholar]
- Yau, J.W.; Teoh, H.; Verma, S. Endothelial cell control of thrombosis. BMC Cardiovasc. Disord. 2015, 15, 130. [Google Scholar] [CrossRef] [Green Version]
- Gale, A.J. Current understanding of hemostasis. Toxicol. Pathol. 2010, 39, 273–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broos, K.; Feys, H.B.; de Meyer, S.F.; Vanhoorelbeke, K.; Deckmyn, H. Platelets at work in primary hemostasis. Blood Rev. 2011, 25, 155–167. [Google Scholar] [CrossRef] [PubMed]
- Ruggeri, Z.M. The role of von Willebrand factor in thrombus formation. Thromb. Res. 2007, 120, S5–S9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kehrel, B.; Wierwille, S.; Clemetson, K.J.; Anders, O.; Steiner, M.; Knight, C.G.; Farndale, R.W.; Okuma, M.; Barnes, M.J. Glycoprotein VI is a major collagen receptor for platelet activation: It recognizes the platelet-activating quaternary structure of collagen, whereas CD36, glycoprotein IIb/IIIa, and von willebrand factor do not. Blood J. Am. Soc. Hematol. 1998, 91, 491–499. [Google Scholar] [CrossRef]
- Nieswandt, B.; Brakebusch, C.; Bergmeier, W.; Schulte, V.; Bouvard, D.; Mokhtari-Nejad, R.; Lindhout, T.; Heemskerk, J.W.; Zirngibl, H.; Fässler, R. Glycoprotein VI but not α2β1 integrin is essential for platelet interaction with collagen. EMBO J. 2001, 20, 2120–2130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, B.-H.; Springer, T.A. Integrin structures and conformational signaling. Curr. Opin. Cell Biol. 2006, 18, 579–586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varga-Szabo, D.; Pleines, I.; Nieswandt, B. Cell adhesion mechanisms in platelets. Arter. Thromb. Vasc. Biol. 2008, 28, 403–412. [Google Scholar] [CrossRef]
- Brass, L.; Furie, B.; Lijnen, H.; Esmon, C. Hematology: Basic Principles and Practice; Churchill-Livingston, Inc.: New York, NY, USA, 2000. [Google Scholar]
- Kirchhofer, D.; Nemerson, Y. Initiation of blood coagulation: The tissue factor/factor VIIa complex. Curr. Opin. Biotechnol. 1996, 7, 386–391. [Google Scholar] [CrossRef]
- Dahlbäck, B. Blood coagulation. Lancet 2000, 355, 1627–1632. [Google Scholar] [CrossRef]
- Boron, W.; Boulpaep, E. Medical Physiology: Updated Edition; Schmitt, W.R., Dudlick, M., Eds.; Elsevier Saunders Inc.: Philadelphia, PA, USA, 2005. [Google Scholar]
- Smith, S.A.; Travers, R.J.; Morrissey, J.H. How it all starts: Initiation of the clotting cascade. Crit. Rev. Biochem. Mol. Biol. 2015, 50, 326–336. [Google Scholar] [CrossRef] [Green Version]
- Lane, D.A.; Philippou, H.; Huntington, J.A. Directing thrombin. Blood 2005, 106, 2605–2612. [Google Scholar] [CrossRef]
- Kahn, M.L.; Zheng, Y.-W.; Huang, W.; Bigornia, V.; Zeng, D.; Moff, S.; Farese, R.V.; Tam, C.; Coughlin, S.R. A dual thrombin receptor system for platelet activation. Nature 1998, 394, 690–694. [Google Scholar] [CrossRef]
- Monroe, D.; Hoffman, M.; Roberts, H. Transmission of a procoagulant signal from tissue factor-bearing cells to platelets. Blood Coagul. Fibrinolysis 1996, 7, 459–464. [Google Scholar] [CrossRef]
- Díaz-Ricart, M.; Estebanell, E.; Lozano, M.; Aznar-Salatti, J.; White, J.G.; Ordinas, A.; Escolar, G. Thrombin facilitates primary platelet adhesion onto vascular surfaces in the absence of plasma adhesive proteins: Studies under flow conditions. Haematologica 2000, 85, 280–288. [Google Scholar]
- Hung, D.; Vu, T.-K.; Wheaton, V.; Ishii, K.; Coughlin, S. Cloned platelet thrombin receptor is necessary for thrombin-induced platelet activation. J. Clin. Investig. 1992, 89, 1350–1353. [Google Scholar] [CrossRef]
- Hultin, M.B. Modulation of thrombin-mediated activation of factor VIII: C by calcium ions, phospholipid, and platelets. Blood 1985, 66, 53–58. [Google Scholar] [CrossRef] [Green Version]
- Monkovic, D.D.; Tracy, P.B. Activation of human factor V by factor Xa and thrombin. Biochemistry 1990, 29, 1118–1128. [Google Scholar] [CrossRef]
- Oliver, J.A.; Monroe, D.M.; Roberts, H.R.; Hoffman, M. Thrombin activates factor XI on activated platelets in the absence of factor XII. Arter. Thromb. Vasc. Biol. 1999, 19, 170–177. [Google Scholar] [CrossRef] [Green Version]
- Baglia, F.A.; Walsh, P.N. Prothrombin is a cofactor for the binding of factor XI to the platelet surface and for platelet-mediated factor XI activation by thrombin. Biochemistry 1998, 37, 2271–2281. [Google Scholar] [CrossRef] [PubMed]
- Baglia, F.A.; Walsh, P.N. Thrombin-mediated feedback activation of factor XI on the activated platelet surface is preferred over contact activation by factor XIIa or factor XIa. J. Biol. Chem. 2000, 275, 20514–20519. [Google Scholar] [CrossRef] [Green Version]
- Khatib, R.; Wilson, F. Pharmacology of Medications Used in the Treatment of Atherosclerotic Cardiovascular Disease. In Refrence Module in Biomedical Sciences; Elsevier Ltd.: Oxford, UK, 2018; pp. 68–88. [Google Scholar]
- Wood, J.P.; Ellery, P.E.; Maroney, S.A.; Mast, A.E. Biology of tissue factor pathway inhibitor. Blood J. Am. Soc. Hematol. 2014, 123, 2934–2943. [Google Scholar] [CrossRef] [Green Version]
- Weisel, J.W.; Litvinov, R.I. Mechanisms of fibrin polymerization and clinical implications. Blood J. Am. Soc. Hematol. 2013, 121, 1712–1719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muszbek, L.; Katona, É.; Kerényi, A. Assessment of factor XIII. In Hemostasis and Thrombosis; Springer: Berlin/Heidelberg, Germany, 2017; pp. 277–293. [Google Scholar]
- Szymanski, L.M.; Pate, R.R.; Durstine, J.L. Effects of maximal exercise and venous occlusion on fibrinolytic activity in physically active and inactive men. J. Appl. Physiol. 1994, 77, 2305–2310. [Google Scholar] [CrossRef]
- Cesarman-Maus, G.; Hajjar, K.A. Molecular mechanisms of fibrinolysis. Br. J. Haematol. 2005, 129, 307–321. [Google Scholar] [CrossRef]
- Hoylaerts, M.; Rijken, D.; Lijnen, H.; Collen, D. Kinetics of the activation of plasminogen by human tissue plasminogen activator. Role of fibrin. J. Biol. Chem. 1982, 257, 2912–2919. [Google Scholar] [CrossRef]
- Bajzar, L.; Manuel, R.; Nesheim, M.E. Purification and characterization of TAFI, a thrombin-activable fibrinolysis inhibitor. J. Biol. Chem. 1995, 270, 14477–14484. [Google Scholar] [CrossRef] [Green Version]
- Bouma, B.N.; Mosnier, L.O. Thrombin activatable fibrinolysis inhibitor (TAFI)—How does thrombin regulate fibrinolysis? Ann. Med. 2006, 38, 378–388. [Google Scholar] [CrossRef] [PubMed]
- Esmon, C.T. The protein C pathway. Chest 2003, 124, 26S–32S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mosnier, L.O.; Meijers, J.C.; Bouma, B.N. Regulation of fibrinolysis in plasma by TAFI and protein C is dependent on the concentration of thrombomodulin. Thromb. Haemost. 2001, 85, 5–11. [Google Scholar] [PubMed]
- Taylor, F.B., Jr.; Peer, G.T.; Lockhart, M.S.; Ferrell, G.; Esmon, C.T. Endothelial cell protein C receptor plays an important role in protein C activation in vivo. Blood J. Am. Soc. Hematol. 2001, 97, 1685–1688. [Google Scholar] [CrossRef] [Green Version]
- Bajzar, L.; Nesheim, M.; Tracy, P. The profibrinolytic effect of activated protein C in clots formed from plasma is TAFI-dependent. Blood 1996, 88, 2093–2100. [Google Scholar] [CrossRef] [PubMed]
- Edlund, M. Menorrhagia—A symptom not sufficiently surveyed. The path to diagnosis and treatment lined with ambiguity and misunderstandings. Lakartidningen 2001, 98, 5505–5506, 5509–5510. [Google Scholar] [PubMed]
- Martlew, V. Peri-operative management of patients with coagulation disorders. Br. J. Anaesth. 2000, 85, 446–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurth, A.; Ludwig, G.; Scharrer, I. Prevalence, pathophysiology, diagnosis, and treatment of Willebrand syndrome in orthopedic-trauma patients. Der Orthop. 1999, 28, 366–374. [Google Scholar] [CrossRef]
- Van, M.W.; De, R.B.; Van, S.D.V.; Van, S.B. Vitamin K, an update for the paediatrician. Eur. J. Pediatr. 2009, 168, 127–134. [Google Scholar]
- Pinjala, R.; Reddy, L.; Nihar, R.; Praveen, G.; Sandeep, M. Thrombophilia–How Far and How Much to Investigate? Indian J. Surg. 2012, 74, 157–162. [Google Scholar] [CrossRef] [Green Version]
- Kyrle, P.A.; Rosendaal, F.R.; Eichinger, S. Risk assessment for recurrent venous thrombosis. Lancet 2010, 376, 2032–2039. [Google Scholar] [CrossRef]
- Ambruso, D.R.; Leonard, B.D.; Bies, R.D.; Jacobson, L.; Hathaway, W.E.; Reeve, E.B. Antithrombin III deficiency: Decreased synthesis of a biochemically normal molecule. Blood 1982, 60, 78–83. [Google Scholar] [CrossRef] [Green Version]
- Segers, K.; Dahlbäck, B.; Nicolaes, G.A. Coagulation factor V and thrombophilia: Background and mechanisms. Thromb. Haemost. 2007, 98, 530–542. [Google Scholar] [CrossRef] [Green Version]
- Koster, T.; Vandenbroucke, J.; Rosendaal, F.; de Ronde, H.; Briët, E.; Bertina, R.M. Venous thrombosis due to poor anticoagulant response to activated protein C: Leiden thrombophilia study. Lancet 1993, 342, 1503–1506. [Google Scholar] [CrossRef]
- Tait, R.; Walker, I.D.; Perry, D.; Islam, S.; Daly, M.; McCall, F.; Conkie, J.; Carrell, R. Prevalence of antithrombin deficiency in the healthy population. Br. J. Haematol. 1994, 87, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Foster, D.C.; Yoshitake, S.; Davie, E.W. The nucleotide sequence of the gene for human protein C. Proc. Natl. Acad. Sci. USA 1985, 82, 4673–4677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broekmans, A.W.; Veltkamp, J.J.; Bertina, R.M. Congenital protein C deficiency and venous thromboembolism: A study of three Dutch families. N. Engl. J. Med. 1983, 309, 340–344. [Google Scholar] [CrossRef] [PubMed]
- Reitsma, P.; Bernardi, F.; Doig, R.; Gandrille, S.; Greengard, J.; Ireland, H.; Krawczak, M.; Lind, B.; Long, G.; Poort, S. Protein C deficiency: A database of mutations, 1995 update. Thromb. Haemost. 1995, 74, 876–889. [Google Scholar] [CrossRef]
- Puhm, F.; Boilard, E.; Machlus, K.R. Platelet extracellular vesicles: Beyond the blood. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 87–96. [Google Scholar] [CrossRef]
- Yuana, Y.; Sturk, A.; Nieuwland, R. Extracellular vesicles in physiological and pathological conditions. Blood Rev. 2013, 27, 31–39. [Google Scholar] [CrossRef] [Green Version]
- Doyle, L.M.; Wang, M.Z. Overview of extracellular vesicles, their origin, composition, purpose, and methods for exosome isolation and analysis. Cells 2019, 8, 727. [Google Scholar] [CrossRef] [Green Version]
- Berckmans, R.J.; Nieuwland, R.; Böing, A.N.; Romijn, F.P.; Hack, C.E.; Sturk, A. Cell-derived microparticles circulate in healthy humans and support low grade thrombin generation. Thromb. Haemost. 2001, 85, 639–649. [Google Scholar]
- Berckmans, R.J.; Lacroix, R.; Hau, C.M.; Sturk, A.; Nieuwland, R. Extracellular vesicles and coagulation in blood from healthy humans revisited. J. Extracell. Vesicles 2019, 8, 1688936. [Google Scholar] [CrossRef]
- Das, K.; Keshava, S.; Ansari, S.A.; Kondreddy, V.; Esmon, C.T.; Griffin, J.H.; Pendurthi, U.R.; Rao, L.V.M. Factor VIIa induces extracellular vesicles from the endothelium: A potential mechanism for its hemostatic effect. Blood 2021, 137, 3428–3442. [Google Scholar] [CrossRef]
- Das, K.; Keshava, S.; Pendurthi, U.R.; Rao, L.V.M. Factor VIIa suppresses inflammation and barrier disruption through the release of EEVs and transfer of microRNA 10a. Blood 2022, 139, 118–133. [Google Scholar] [CrossRef] [PubMed]
- Nakatani, E.; Naito, Y.; Ishibashi, K.; Ohkura, N.; Atsumi, G.I. Extracellular vesicles derived from 3T3-L1 adipocytes enhance procoagulant activity. Biol. Pharm. Bull. 2022, 45, 178–183. [Google Scholar] [CrossRef] [PubMed]
- Marchini, J.F.; Manica, A.; Crestani, P.; Dutzmann, J.; Folco, E.J.; Weber, H.; Libby, P.; Croce, K. Oxidized low-density lipoprotein induces macrophage production of prothrombotic microparticles. J. Am. Heart Assoc. 2020, 9, e015878. [Google Scholar] [CrossRef] [PubMed]
- Dragovic, R.A.; Gardiner, C.; Brooks, A.S.; Tannetta, D.S.; Ferguson, D.J.; Hole, P.; Carr, B.; Redman, C.W.; Harris, A.L.; Dobson, P.J. Sizing and phenotyping of cellular vesicles using nanoparticle tracking analysis. Nanomed. Nanotechnol. Biol. Med. 2011, 7, 780–788. [Google Scholar] [CrossRef] [Green Version]
- Shet, A.S.; Aras, O.; Gupta, K.; Hass, M.J.; Rausch, D.J.; Saba, N.; Koopmeiners, L.; Key, N.S.; Hebbel, R.P. Sickle blood contains tissue factor–positive microparticles derived from endothelial cells and monocytes. Blood 2003, 102, 2678–2683. [Google Scholar] [CrossRef] [Green Version]
- Eitan, E.; Green, J.; Bodogai, M.; Mode, N.A.; Bæk, R.; Jørgensen, M.M.; Freeman, D.W.; Witwer, K.W.; Zonderman, A.B.; Biragyn, A. Age-related changes in plasma extracellular vesicle characteristics and internalization by leukocytes. Sci. Rep. 2017, 7, 1342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forest, A.; Pautas, E.; Ray, P.; Bonnet, D.; Verny, M.; Amabile, N.; Boulanger, C.; Riou, B.; Tedgui, A.; Mallat, Z. Circulating microparticles and procoagulant activity in elderly patients. J. Gerontol. Ser. A Biomed. Sci. Med. Sci. 2010, 65, 414–420. [Google Scholar] [CrossRef]
- Galli, M.; Grassi, A.; Barbui, T. Platelet-derived microvesicles in thrombotic thrombocytopenic purpura and hemolytic uremic syndrome. Thromb. Haemost. 1996, 75, 427–431. [Google Scholar] [CrossRef]
- Tomer, A.; Harker, L.A.; Kasey, S.; Eckman, J.R. Thrombogenesis in sickle cell disease. J. Lab. Clin. Med. 2001, 137, 398–407. [Google Scholar] [CrossRef]
- Hughes, M.; Hayward, C.P.; Warkentin, T.E.; Horsewood, P.; Chorneyko, K.A.; Kelton, J.G. Morphological analysis of microparticle generation in heparin-induced thrombocytopenia. Blood J. Am. Soc. Hematol. 2000, 96, 188–194. [Google Scholar]
- Mallat, Z.; Benamer, H.; Hugel, B.; Benessiano, J.; Steg, P.G.; Freyssinet, J.-M.; Tedgui, A. Elevated levels of shed membrane microparticles with procoagulant potential in the peripheral circulating blood of patients with acute coronary syndromes. Circulation 2000, 101, 841–843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.J.; Jy, W.; Horstman, L.L.; Janania, J.; Reyes, Y.; Kelley, R.E.; Ahn, Y.S. Elevated platelet microparticles in transient ischemic attacks, lacunar infarcts, and multiinfarct dementias. Thromb. Res. 1993, 72, 295–304. [Google Scholar] [CrossRef]
- Jung, K.H.; Chu, K.; Lee, S.T.; Park, H.K.; Bahn, J.J.; Kim, D.H.; Kim, J.H.; Kim, M.; Lee, S.K.; Roh, J.K. Circulating endothelial microparticles as a marker of cerebrovascular disease. Ann. Neurol. Off. J. Am. Neurol. Assoc. Child Neurol. Soc. 2009, 66, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Deng, F.; Wang, S.; Zhang, L. Endothelial microparticles act as novel diagnostic and therapeutic biomarkers of diabetes and its complications: A literature review. BioMed Res. Int. 2016, 2016, 9802026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez, M.C.; Andriantsitohaina, R. Extracellular vesicles in metabolic syndrome. Circ. Res. 2017, 120, 1674–1686. [Google Scholar] [CrossRef]
- Alaaeddine, R.A.; AlZaim, I.; Hammoud, S.H.; Arakji, A.; Eid, A.H.; Abd-Elrahman, K.S.; El-Yazbi, A.F. The pleiotropic effects of antithrombotic drugs in the metabolic–cardiovascular–neurodegenerative disease continuum: Impact beyond reduced clotting. Clin. Sci. 2021, 135, 1015–1051. [Google Scholar] [CrossRef]
- Berezin, A.E.; Berezin, A.A. Extracellular vesicles and thrombogenicity in atrial fibrillation. Int. J. Mol. Sci. 2022, 23, 1774. [Google Scholar] [CrossRef]
- Mørk, M.; Andreasen, J.J.; Rasmussen, L.H.; Lip, G.Y.H.; Pedersen, S.; Bæk, R.; Jørgensen, M.M.; Kristensen, S.R. Elevated blood plasma levels of tissue factor-bearing extracellular vesicles in patients with atrial fibrillation. Thromb. Res. 2019, 173, 141–150. [Google Scholar] [CrossRef]
- Wang, H.; Yan, H.M.; Tang, M.X.; Wang, Z.H.; Zhong, M.; Zhang, Y.; Deng, J.T.; Zhang, W. Increased serum levels of microvesicles in nonvalvular atrial fibrillation determinated by ELISA using a specific monoclonal antibody AD-1. Clin. Chim. Acta 2010, 411, 1700–1704. [Google Scholar] [CrossRef]
- Wang, L.; Bi, Y.; Yu, M.; Li, T.; Tong, D.; Yang, X.; Zhang, C.; Guo, L.; Wang, C.; Kou, Y.; et al. Phosphatidylserine-exposing blood cells and microparticles induce procoagulant activity in non-valvular atrial fibrillation. Int. J. Cardiol. 2018, 258, 138–143. [Google Scholar] [CrossRef]
- Almeida, V.H.; Rondon, A.M.; Gomes, T.; Monteiro, R.Q. Novel aspects of extracellular vesicles as mediators of cancer-associated thrombosis. Cells 2019, 8, 716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Ni, Y.Q.; Xu, H.; Xiang, Q.Y.; Zhao, Y.; Zhan, J.K.; He, J.Y.; Li, S.; Liu, Y.S. Roles and mechanisms of exosomal non-coding RNAs in human health and diseases. Signal Transduct. Target. Ther. 2021, 6, 383. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wen, Q.; Chen, H.; Wu, X.; Liu, B.; Li, H.; Su, L.; Tong, H. Exosomes derived from heat stroke cases carry miRNAs associated with inflammation and coagulation cascade. Front. Immunol. 2021, 12, 624753. [Google Scholar] [CrossRef] [PubMed]
- Jansen, F.; Yang, X.; Hoelscher, M.; Cattelan, A.; Schmitz, T.; Proebsting, S.; Wenzel, D.; Vosen, S.; Franklin, B.S.; Fleischmann, B.K.; et al. Endothelial microparticle-mediated transfer of microRNA-126 promotes vascular endothelial cell repair via spred1 and is abrogated in glucose-damaged endothelial microparticles. Circulation 2013, 128, 2026–2038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, S.; Zhang, Y.; Li, Y.; Luo, L.; Zhao, Y.; Yao, Y. Extracellular vesicles in cardiovascular diseases. Cell Death Discov. 2020, 6, 68. [Google Scholar] [CrossRef]
- Arroyo, A.B.; de los Reyes-García, A.M.; Teruel-Montoya, R.; Vicente, V.; González-Conejero, R.; Martínez, C. microRNAs in the haemostatic system: More than witnesses of thromboembolic diseases? Thromb. Res. 2018, 166, 1–9. [Google Scholar] [CrossRef]
- Tay, J.; Tiao, J.; Hughes, Q.; Jorritsma, J.; Gilmore, G.; Baker, R. Circulating microRNA as thrombosis sentinels: Caveats and considerations. Semin. Thromb. Hemost. 2018, 44, 206–215. [Google Scholar] [CrossRef]
- Wenche, J.; Horstman, L.L.; Arce, M.; Ahn, Y.S. Clinical significance of platelet microparticles in autoimmune thrombocytopenias. J. Lab. Clin. Med. 1992, 119, 334–345. [Google Scholar]
- Kasthuri, R.S.; Glover, S.L.; Jonas, W.; McEachron, T.; Pawlinski, R.; Arepally, G.M.; Key, N.S.; Mackman, N. PF4/heparin-antibody complex induces monocyte tissue factor expression and release of tissue factor positive microparticles by activation of FcγRI. Blood J. Am. Soc. Hematol. 2012, 119, 5285–5293. [Google Scholar] [CrossRef]
- Lapping-Carr, G.; Gemel, J.; Mao, Y.; Beyer, E.C. Circulating extracellular vesicles and endothelial damage in sickle cell disease. Front. Physiol. 2020, 11, 1063. [Google Scholar] [CrossRef]
- Nader, E.; Garnier, Y.; Connes, P.; Romana, M. Extracellular vesicles in sickle cell disease: Plasma concentration, blood cell types origin distribution and biological properties. Front. Med. 2021, 8, 728693. [Google Scholar] [CrossRef] [PubMed]
- Biasucci, L.M.; Porto, I.; di Vito, L.; de Maria, G.L.; Leone, A.M.; Tinelli, G.; Tritarelli, A.; di Rocco, G.; Snider, F.; Capogrossi, M.C. Differences in microparticle release in patients with acute coronary syndrome and stable angina. Circ. J. 2012, 76, 2174–2182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, C.; Zhao, W.-B.; Chen, Y.; Hu, H.-Y. Higher plasma concentrations of platelet microparticles in patients with acute coronary syndrome: A systematic review and meta-analysis. Can. J. Cardiol. 2016, 32, 1325.e1–1325.e10. [Google Scholar] [CrossRef] [PubMed]
- Delabranche, X.; Boisramé-Helms, J.; Asfar, P.; Berger, A.; Mootien, Y.; Lavigne, T.; Grunebaum, L.; Lanza, F.; Gachet, C.; Freyssinet, J.-M. Microparticles are new biomarkers of septic shock-induced disseminated intravascular coagulopathy. Intensiv. Care Med. 2013, 39, 1695–1703. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, N.; Nomura, S.; Kamihata, H.; Kimura, Y.; Iwasaka, T. Increased level of oxidized LDL-dependent monocytederived microparticles in acute coronary syndrome. Thromb. Haemost. 2004, 91, 146–154. [Google Scholar] [CrossRef]
- Wang, B.; Li, T.; Han, X.; Li, Y.; Cheng, W.; Wang, L.; Lu, Z.; Yang, J.; Zhao, M. The level of circulating microparticles in patients with coronary heart disease: A systematic review and meta-analysis. J. Cardiovasc. Transl. Res. 2019, 13, 702–712. [Google Scholar] [CrossRef]
- Chirinos, J.A.; Heresi, G.A.; Velasquez, H.; Jy, W.; Jimenez, J.J.; Ahn, E.; Horstman, L.L.; Soriano, A.O.; Zambrano, J.P.; Ahn, Y.S. Elevation of endothelial microparticles, platelets, and leukocyte activation in patients with venous thromboembolism. J. Am. Coll. Cardiol. 2005, 45, 1467–1471. [Google Scholar] [CrossRef]
- Jamaly, S.; Basavaraj, M.G.; Starikova, I.; Olsen, R.; Brækkan, S.K.; Hansen, J.B. Elevated plasma levels of P-selectin glycoprotein ligand-1-positive microvesicles in patients with unprovoked venous thromboembolism. J. Thromb. Haemost. 2018, 16, 1546–1554. [Google Scholar] [CrossRef]
- Simak, J.; Gelderman, M.; Yu, H.; Wright, V.; Baird, A. Circulating endothelial microparticles in acute ischemic stroke: A link to severity, lesion volume and outcome. J. Thromb. Haemost. 2006, 4, 1296–1302. [Google Scholar] [CrossRef]
- Wiedmer, T.; Hall, S.; Ortel, T.; Kane, W.; Rosse, W.; Sims, P. Complement-induced vesiculation and exposure of membrane prothrombinase sites in platelets of paroxysmal nocturnal hemoglobinuria. Blood 1993, 82, 1192–1196. [Google Scholar] [CrossRef] [Green Version]
- Kozuma, Y.; Sawahata, Y.; Takei, Y.; Chiba, S.; Ninomiya, H. Procoagulant properties of microparticles released from red blood cells in paroxysmal nocturnal haemoglobinuria. Br. J. Haematol. 2011, 152, 631–639. [Google Scholar] [CrossRef]
- Suades, R.; Padró, T.; Crespo, J.; Ramaiola, I.; Martin-Yuste, V.; Sabaté, M.; Sans-Roselló, J.; Sionis, A.; Badimon, L. Circulating microparticle signature in coronary and peripheral blood of ST elevation myocardial infarction patients in relation to pain-to-PCI elapsed time. Int. J. Cardiol. 2016, 202, 378–387. [Google Scholar] [CrossRef] [PubMed]
- Morel, O.; Pereira, B.; Averous, G.; Faure, A.; Jesel, L.; Germain, P.; Grunebaum, L.; Ohlmann, P.; Freyssinet, J.-M.; Bareiss, P. Increased levels of procoagulant tissue factor-bearing microparticles within the occluded coronary artery of patients with ST-segment elevation myocardial infarction: Role of endothelial damage and leukocyte activation. Atherosclerosis 2009, 204, 636–641. [Google Scholar] [CrossRef] [PubMed]
- Bal, L.; Ederhy, S.; di Angelantonio, E.; Toti, F.; Zobairi, F.; Dufaitre, G.; Meuleman, C.; Mallat, Z.; Boccara, F.; Tedgui, A. Factors influencing the level of circulating procoagulant microparticles in acute pulmonary embolism. Arch. Cardiovasc. Dis. 2010, 103, 394–403. [Google Scholar] [CrossRef] [Green Version]
- Bal, L.; Ederhy, S.; di Angelantonio, E.; Toti, F.; Zobairi, F.; Dufaitre, G.; Meuleman, C.; Mallat, Z.; Boccara, F.; Tedgui, A. Circulating procoagulant microparticles in acute pulmonary embolism: A case-control study. Int. J. Cardiol. 2010, 145, 321–322. [Google Scholar] [CrossRef]
- Wolf, P. The nature and significance of platelet products in human plasma. Br. J. Haematol. 1967, 13, 269–288. [Google Scholar] [CrossRef]
- Davidson, S.M.; Andreadou, I.; Barile, L.; Birnbaum, Y.; Cabrera-Fuentes, H.A.; Cohen, M.V.; Downey, J.M.; Girao, H.; Pagliaro, P.; Penna, C. Circulating blood cells and extracellular vesicles in acute cardioprotection. Cardiovasc. Res. 2019, 115, 1156–1166. [Google Scholar] [CrossRef]
- Sims, P.J.; Faioni, E.; Wiedmer, T.; Shattil, S. Complement proteins C5b-9 cause release of membrane vesicles from the platelet surface that are enriched in the membrane receptor for coagulation factor Va and express prothrombinase activity. J. Biol. Chem. 1988, 263, 18205–18212. [Google Scholar] [CrossRef]
- Giesen, P.L.; Rauch, U.; Bohrmann, B.; Kling, D.; Roqué, M.; Fallon, J.T.; Badimon, J.J.; Himber, J.; Riederer, M.A.; Nemerson, Y. Blood-borne tissue factor: Another view of thrombosis. Proc. Natl. Acad. Sci. USA 1999, 96, 2311–2315. [Google Scholar] [CrossRef] [Green Version]
- VanWijk, M.J.; VanBavel, E.; Sturk, A.; Nieuwland, R. Microparticles in cardiovascular diseases. Cardiovasc. Res. 2003, 59, 277–287. [Google Scholar] [CrossRef]
- Zifkos, K.; Dubois, C.; Schäfer, K. Extracellular vesicles and thrombosis: Update on the clinical and experimental evidence. Int. J. Mol. Sci. 2021, 22, 9317. [Google Scholar] [CrossRef]
- Ferreira, P.M.; Bozbas, E.; Tannetta, S.D.; Alroqaiba, N.; Zhou, R.; Crawley, J.T.B.; Gibbins, J.M.; Jones, C.I.; Ahnström, J.; Yaqoob, P. Mode of induction of platelet-derived extracellular vesicles is a critical determinant of their phenotype and function. Sci. Rep. 2020, 10, 18061. [Google Scholar] [CrossRef] [PubMed]
- Tucher, C.; Bode, K.; Schiller, P.; Claßen, L.; Birr, C.; Souto-Carneiro, M.M.; Blank, N.; Lorenz, H.-M.; Schiller, M. Extracellular vesicle subtypes released from activated or apoptotic T-lymphocytes carry a specific and stimulus-dependent protein cargo. Front. Immunol. 2018, 9, 534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dasgupta, S.K.; Abdel-Monem, H.; Niravath, P.; Le, A.; Bellera, R.V.; Langlois, K.; Nagata, S.; Rumbaut, R.E.; Thiagarajan, P. Lactadherin and clearance of platelet-derived microvesicles. Blood J. Am. Soc. Hematol. 2009, 113, 1332–1339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mulcahy, L.A.; Pink, R.C.; Carter, D.R.F. Routes and mechanisms of extracellular vesicle uptake. J. Extracell. Vesicles 2014, 3, 24641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tushuizen, M.E.; Diamant, M.; Sturk, A.; Nieuwland, R. Cell-derived microparticles in the pathogenesis of cardiovascular disease: Friend or foe? Arterioscler. Thromb. Vasc. Biol. 2011, 31, 4–9. [Google Scholar] [CrossRef] [Green Version]
- Flaumenhaft, R.; Mairuhu, A.T.; Italiano, J.E. Platelet-and megakaryocyte-derived microparticles. In Seminars in Thrombosis and Hemostasis; Thieme Medical Publishers: New York, NY, USA, 2010. [Google Scholar]
- Suades, R.; Padró, T.; Vilahur, G.; Badimon, L. Circulating and platelet-derived microparticles in human blood enhance thrombosis on atherosclerotic plaques. Thromb. Haemost. 2012, 108, 1208–1219. [Google Scholar] [CrossRef]
- Kay, J.G.; Grinstein, S. Phosphatidylserine-mediated cellular signaling. Lipid-Mediat. Protein Signal. 2013, 991, 177–193. [Google Scholar]
- Reddy, E.C.; Rand, M.L. Procoagulant phosphatidylserine-exposing platelets in vitro and in vivo. Front. Cardiovasc. Med. 2020, 7, 15. [Google Scholar] [CrossRef] [Green Version]
- Yang, A.; Chen, F.; He, C.; Zhou, J.; Lu, Y.; Dai, J.; Birge, R.B.; Wu, Y. The procoagulant activity of apoptotic cells is mediated by interaction with factor XII. Front. Immunol. 2017, 8, 1188. [Google Scholar] [CrossRef] [Green Version]
- Boulanger, C.M.; Loyer, X.; Rautou, P.-E.; Amabile, N. Extracellular vesicles in coronary artery disease. Nat. Rev. Cardiol. 2017, 14, 259–272. [Google Scholar] [CrossRef] [PubMed]
- Sluijter, J.P.G.; Davidson, S.M.; Boulanger, C.M.; Buzas, E.I.; de Kleijn, D.P.V.; Engel, F.B.; Giricz, Z.; Hausenloy, D.J.; Kishore, R.; Lecour, S. Extracellular vesicles in diagnostics and therapy of the ischaemic heart: Position paper from the working group on cellular biology of the heart of the European society of cardiology. Cardiovasc. Res. 2018, 114, 19–34. [Google Scholar] [CrossRef] [PubMed]
- Preston, R.A.; Jy, W.; Jimenez, J.J.; Mauro, L.M.; Horstman, L.L.; Valle, M.; Aime, G.; Ahn, Y.S. Effects of severe hypertension on endothelial and platelet microparticles. Hypertension 2003, 41, 211–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biro, E.; Sturk-Maquelin, K.; Vogel, G.; Meuleman, D.; Smit, M.; Hack, C.; Sturk, A.; Nieuwland, R. Human cell-derived microparticles promote thrombus formation in vivo in a tissue factor-dependent manner. J. Thromb. Haemost. 2003, 1, 2561–2568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falati, S.; Liu, Q.; Gross, P.; Merrill-Skoloff, G.; Chou, J.; Vandendries, E.; Celi, A.; Croce, K.; Furie, B.C.; Furie, B. Accumulation of tissue factor into developing thrombi in vivo is dependent upon microparticle P-selectin glycoprotein ligand 1 and platelet P-selectin. J. Exp. Med. 2003, 197, 1585–1598. [Google Scholar] [CrossRef]
- Kou, Y.; Zou, L.; Liu, R.; Zhao, X.; Wang, Y.; Zhang, C.; Dong, Z.; Kou, J.; Bi, Y.; Fu, L. Intravascular cells and circulating microparticles induce procoagulant activity via phosphatidylserine exposure in heart failure. J. Thromb. Thrombolysis 2019, 48, 187–194. [Google Scholar] [CrossRef]
- Chiva-Blanch, G.; Laake, K.; Myhre, P.; Bratseth, V.; Arnesen, H.; Solheim, S.; Badimon, L.; Seljeflot, I. Platelet-, monocyte-derived and tissue factor-carrying circulating microparticles are related to acute myocardial infarction severity. PLoS ONE 2017, 12, e0172558. [Google Scholar] [CrossRef]
- Wang, L.; Bi, Y.; Cao, M.; Ma, R.; Wu, X.; Zhang, Y.; Ding, W.; Liu, Y.; Yu, Q.; Zhang, Y. Microparticles and blood cells induce procoagulant activity via phosphatidylserine exposure in NSTEMI patients following stent implantation. Int. J. Cardiol. 2016, 223, 121–128. [Google Scholar] [CrossRef]
- Ueba, T.; Nomura, S.; Inami, N.; Nishikawa, T.; Kajiwara, M.; Iwata, R.; Yamashita, K. Plasma level of platelet-derived microparticles is associated with coronary heart disease risk score in healthy men. J. Atheroscler. Thromb. 2010, 17, 342–349. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; Wu, X.; Si, Y.; Yao, Z.; Dong, Z.; Novakovic, V.A.; Guo, L.; Tong, D.; Chen, H.; Bi, Y. Increased blood cell phosphatidylserine exposure and circulating microparticles contribute to procoagulant activity after carotid artery stenting. J. Neurosurg. 2017, 127, 1041–1054. [Google Scholar] [CrossRef] [Green Version]
- Nylander, S.; Femia, E.; Scavone, M.; Berntsson, P.; Asztély, A.K.; Nelander, K.; Löfgren, L.; Nilsson, R.; Cattaneo, M. Ticagrelor inhibits human platelet aggregation via adenosine in addition to P2Y12 antagonism. J. Thromb. Haemost. 2013, 11, 1867–1876. [Google Scholar] [CrossRef]
- Bernal-Mizrachi, L.; Jy, W.; Jimenez, J.J.; Pastor, J.; Mauro, L.M.; Horstman, L.L.; de Marchena, E.; Ahn, Y.S. High levels of circulating endothelial microparticles in patients with acute coronary syndromes. Am. Heart J. 2003, 145, 962–970. [Google Scholar] [CrossRef]
- Lipets, E.; Antonova, O.; Shustova, O.; Losenkova, K.; Mazurov, A.; Ataullakhanov, F. Use of Thrombodynamics for revealing the participation of platelet, erythrocyte, endothelial, and monocyte microparticles in coagulation activation and propagation. PLoS ONE 2020, 15, e0227932. [Google Scholar] [CrossRef] [PubMed]
- Andrews, A.M.; Rizzo, V. Microparticle-induced activation of the vascular endothelium requires caveolin-1/caveolae. PLoS ONE 2016, 11, e0149272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shustova, O.N.; Antonova, O.A.; Golubeva, N.V.; Khaspekova, S.G.; Yakushkin, V.V.; Aksuk, S.A.; Alchinova, I.B.; Karganov, M.Y.; Mazurov, A.V. Differential procoagulant activity of microparticles derived from monocytes, granulocytes, platelets and endothelial cells: Impact of active tissue factor. Blood Coagul. Fibrinolysis 2017, 28, 373–382. [Google Scholar] [CrossRef]
- Van Der Meijden, P.; van Schilfgaarde, M.; van Oerle, R.; Renne, T.; Cate, H.T.; Spronk, H. Platelet-and erythrocyte-derived microparticles trigger thrombin generation via factor XIIa. J. Thromb. Haemost. 2012, 10, 1355–1362. [Google Scholar] [CrossRef] [PubMed]
- Aleman, M.M.; Gardiner, C.; Harrison, P.; Wolberg, A.S. Differential contributions of monocyte-and platelet-derived microparticles towards thrombin generation and fibrin formation and stability. J. Thromb. Haemost. 2011, 9, 2251–2261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hohensinner, P.J.; Mayer, J.; Kichbacher, J.; Kral-Pointner, J.; Thaler, B.; Kaun, C.; Hell, L.; Haider, P.; Mussbacher, M.; Schmid, J.A.; et al. Alternative activation of human macrophages enhances tissue factor expression and production of extracellular vesicles. Haematologica 2021, 106, 454–463. [Google Scholar] [CrossRef] [Green Version]
- George, J.N.; Thoi, L.L.; McManus, L.M.; Reimann, T.A. Isolation of human platelet membrane microparticles from plasma and serum. Blood 1982, 60, 834–840. [Google Scholar] [CrossRef] [Green Version]
- George, J.N.; Pickett, E.B.; Saucerman, S.; McEver, R.P.; Kunicki, T.J.; Kieffer, N.; Newman, P.J. Platelet surface glycoproteins. Studies on resting and activated platelets and platelet membrane microparticles in normal subjects, and observations in patients during adult respiratory distress syndrome and cardiac surgery. J. Clin. Investig. 1986, 78, 340–348. [Google Scholar] [CrossRef]
- Italiano, J.E., Jr.; Mairuhu, A.T.; Flaumenhaft, R. Clinical relevance of microparticles from platelets and megakaryocytes. Curr. Opin. Hematol. 2010, 17, 578–584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flaumenhaft, R. Formation and fate of platelet microparticles. Blood Cells Mol. Dis. 2006, 36, 182–187. [Google Scholar] [CrossRef] [PubMed]
- Flaumenhaft, R.; Dilks, J.R.; Richardson, J.; Alden, E.; Patel-Hett, S.R.; Battinelli, E.; Klement, G.L.; Sola-Visner, M.; Italiano, J.E., Jr. Megakaryocyte-derived microparticles: Direct visualization and distinction from platelet-derived microparticles. Blood J. Am. Soc. Hematol. 2009, 113, 1112–1121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Zee, P.M.; Biró, E.; Ko, Y.; de Winter, R.J.; Hack, C.E.; Sturk, A.; Nieuwland, R. P-selectin- and CD63-exposing platelet microparticles reflect platelet activation in peripheral arterial disease and myocardial infarction. Clin. Chem. 2006, 52, 657–664. [Google Scholar] [CrossRef] [PubMed]
- Fackler, O.T.; Grosse, R. Cell motility through plasma membrane blebbing. J. Cell Biol. 2008, 181, 879–884. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.K.; Song, K.S.; Chung, J.H.; Lee, K.R.; Lee, S.N. Platelet microparticles induce angiogenesis in vitro. Br. J. Haematol. 2004, 124, 376–384. [Google Scholar] [CrossRef]
- Stępień, E.; Stankiewicz, E.; Zalewski, J.; Godlewski, J.; Zmudka, K.; Wybrańska, I. Number of microparticles generated during acute myocardial infarction and stable angina correlates with platelet activation. Arch. Med. Res. 2012, 43, 31–35. [Google Scholar] [CrossRef]
- Gąsecka, A.; Rogula, S.; Eyileten, C.; Postuła, M.; Jaguszewski, M.J.; Kochman, J.; Mazurek, T.; Nieuwland, R.; Filipiak, K.J. Role of P2Y receptors in platelet extracellular vesicle release. Int. J. Mol. Sci. 2020, 21, 6065. [Google Scholar] [CrossRef]
- Ponomareva, A.; Nevzorova, T.; Mordakhanova, E.; Andrianova, I.; Rauova, L.; Litvinov, R.; Weisel, J. Intracellular origin and ultrastructure of platelet-derived microparticles. J. Thromb. Haemost. 2017, 15, 1655–1667. [Google Scholar] [CrossRef] [Green Version]
- Aatonen, M.T.; Öhman, T.; Nyman, T.A.; Laitinen, S.; Grönholm, M.; Siljander, P.R.-M. Isolation and characterization of platelet-derived extracellular vesicles. J. Extracell. Vesicles 2014, 3, 24692. [Google Scholar] [CrossRef]
- Siljander, P.; Carpen, O.; Lassila, R. Platelet-derived microparticles associate with fibrin during thrombosis. Blood 1996, 87, 4651–4663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brisson, A.R.; Tan, S.; Linares, R.; Gounou, C.; Arraud, N. Extracellular vesicles from activated platelets: A semiquantitative cryo-electron microscopy and immuno-gold labeling study. Platelets 2017, 28, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Goetzl, E.J.; Goetzl, L.; Karliner, J.S.; Tang, N.; Pulliam, L. Human plasma platelet-derived exosomes: Effects of aspirin. FASEB J. 2016, 30, 2058–2063. [Google Scholar] [CrossRef] [Green Version]
- Connor, D.E.; Ly, K.; Aslam, A.; Boland, J.; Low, J.; Jarvis, S.; Muller, D.W.; Joseph, J.E. Effects of antiplatelet therapy on platelet extracellular vesicle release and procoagulant activity in health and in cardiovascular disease. Platelets 2016, 27, 805–811. [Google Scholar] [CrossRef] [PubMed]
- Gasecka, A.; Nieuwland, R.; van der Pol, E.; Hajji, N.; Ćwiek, A.; Pluta, K.; Konwerski, M.; Filipiak, K.J. P2Y12 antagonist ticagrelor inhibits the release of procoagulant extracellular vesicles from activated platelets. Cardiol. J. 2020, 26, 782–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christersson, C.; Johnell, M.; Siegbahn, A. The influence of direct thrombin inhibitors on the formation of platelet-leukocyte aggregates and tissue factor expression. Thromb. Res. 2010, 126, e327–e333. [Google Scholar] [CrossRef] [PubMed]
- Pontiggia, L.; Steiner, B.; Ulrichts, H.; Deckmyn, H.; Forestier, M.; Beer, J.H. Platelet microparticle formation and thrombin generation under high shear are effectively suppressed by a monoclonal antibody against GPIbα. Thromb. Haemost. 2006, 96, 774–780. [Google Scholar] [CrossRef] [PubMed]
- Shantsila, E.; Kamphuisen, P.W.; Lip, G.Y. Circulating microparticles in cardiovascular disease: Implications for atherogenesis and atherothrombosis. J. Thromb. Haemost. 2010, 8, 2358–2368. [Google Scholar] [CrossRef]
- Brill, A.; Dashevsky, O.; Rivo, J.; Gozal, Y.; Varon, D. Platelet-derived microparticles induce angiogenesis and stimulate post-ischemic revascularization. Cardiovasc. Res. 2005, 67, 30–38. [Google Scholar] [CrossRef]
- Burnier, L.; Fontana, P.; Kwak, B.R.; Angelillo-Scherrer, A. Cell-derived microparticles in haemostasis and vascular medicine. Thromb. Haemost. 2009, 101, 439–451. [Google Scholar] [CrossRef]
- Castaman, G.; Yu-Feng, L.; Battistin, E.; Rodeghiero, F. Characterization of a novel bleeding disorder with isolated prolonged bleeding time and deficiency of platelet microvesicle generation. Br. J. Haematol. 1997, 96, 458–463. [Google Scholar] [CrossRef] [PubMed]
- Gemmell, C.; Sefton, M.; Yeo, E. Platelet-derived microparticle formation involves glycoprotein IIb-IIIa. Inhibition by RGDS and a Glanzmann’s thrombasthenia defect. J. Biol. Chem. 1993, 268, 14586–14589. [Google Scholar] [CrossRef]
- Fujii, T.; Sakata, A.; Nishimura, S.; Eto, K.; Nagata, S. TMEM16F is required for phosphatidylserine exposure and microparticle release in activated mouse platelets. Proc. Natl. Acad. Sci. USA 2015, 112, 12800–12805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diehl, P.; Fricke, A.; Sander, L.; Stamm, J.; Bassler, N.; Htun, N.; Ziemann, M.; Helbing, T.; El-Osta, A.; Jowett, J.B.; et al. Microparticles: Major transport vehicles for distinct microRNAs in circulation. Cardiovasc. Res. 2012, 93, 633–644. [Google Scholar] [CrossRef] [PubMed]
- Hunter, M.P.; Ismail, N.; Zhang, X.; Aguda, B.D.; Lee, E.J.; Yu, L.; Xiao, T.; Schafer, J.; Lee, M.L.; Schmittgen, T.D.; et al. Detection of microRNA expression in human peripheral blood microvesicles. PLoS ONE 2008, 3, e3694. [Google Scholar] [CrossRef] [Green Version]
- De Rosa, S.; Fichtlscherer, S.; Lehmann, R.; Assmus, B.; Dimmeler, S.; Zeiher, A.M. Transcoronary concentration gradients of circulating microRNAs. Circulation 2011, 124, 1936–1944. [Google Scholar] [CrossRef] [Green Version]
- Kuwabara, Y.; Ono, K.; Horie, T.; Nishi, H.; Nagao, K.; Kinoshita, M.; Watanabe, S.; Baba, O.; Kojima, Y.; Shizuta, S. Increased microRNA-1 and microRNA-133a levels in serum of patients with cardiovascular disease indicate myocardial damage. Circ. Cardiovasc. Genet. 2011, 4, 446–454. [Google Scholar] [CrossRef]
- Qu, M.; Zou, X.; Fang, F.; Wang, S.; Xu, L.; Zeng, Q.; Fan, Z.; Chen, L.; Yue, W.; Xie, X.; et al. Platelet-derived microparticles enhance megakaryocyte differentiation and platelet generation via miR-1915-3p. Nat. Commun. 2020, 11, 4964. [Google Scholar] [CrossRef]
- Harding, C.; Heuser, J.; Stahl, P. Receptor-mediated endocytosis of transferrin and recycling of the transferrin receptor in rat reticulocytes. J. Cell Biol. 1983, 97, 329–339. [Google Scholar] [CrossRef]
- Johnstone, R.; Mathew, A.; Mason, A.; Teng, K. Exosome formation during maturation of mammalian and avian reticulocytes: Evidence that exosome release is a major route for externalization of obsolete membrane proteins. J. Cell. Physiol. 1991, 147, 27–36. [Google Scholar] [CrossRef]
- Lima, L.G.; Oliveira, A.S.; Campos, L.C.; Bonamino, M.; Chammas, R.; Werneck, C.C.; Vicente, C.P.; Barcinski, M.A.; Petersen, L.C.; Monteiro, R.Q. Malignant transformation in melanocytes is associated with increased production of procoagulant microvesicles. Thromb. Haemost. 2011, 106, 712–723. [Google Scholar] [CrossRef] [PubMed]
- Rak, J. Microparticles in cancer. In Seminars in Thrombosis and Hemostasis; Thieme Medical Publishers: New York, NY, USA, 2010. [Google Scholar]
- Feigelson, S.W.; Grabovsky, V.; Shamri, R.; Levy, S.; Alon, R. The CD81 tetraspanin facilitates instantaneous leukocyte VLA-4 adhesion strengthening to vascular cell adhesion molecule 1 (VCAM-1) under shear flow. J. Biol. Chem. 2003, 278, 51203–51212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reyes, R.; Monjas, A.; Yánez-Mó, M.; Cardeñes, B.; Morlino, G.; Gilsanz, A.; Machado-Pineda, Y.; Lafuente, E.; Monk, P.; Sánchez-Madrid, F. Different states of integrin LFA-1 aggregation are controlled through its association with tetraspanin CD9. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2015, 1853, 2464–2480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dasgupta, S.K.; Le, A.; Chavakis, T.; Rumbaut, R.E.; Thiagarajan, P. Developmental endothelial locus-1 (Del-1) mediates clearance of platelet microparticles by the endothelium. Circulation 2012, 125, 1664–1672. [Google Scholar] [CrossRef] [Green Version]
- Pegtel, D.M.; Gould, S.J. Exosomes. Ann. Rev. Biochem. 2019, 88, 487–514. [Google Scholar] [CrossRef] [PubMed]
- Théry, C.; Zitvogel, L.; Amigorena, S. Exosomes: Composition, biogenesis and function. Nat. Rev. Immunol. 2002, 2, 569–579. [Google Scholar] [CrossRef]
- Takahashi, A.; Okada, R.; Nagao, K.; Kawamata, Y.; Hanyu, A.; Yoshimoto, S.; Takasugi, M.; Watanabe, S.; Kanemaki, M.T.; Obuse, C. Exosomes maintain cellular homeostasis by excreting harmful DNA from cells. Nat. Commun. 2017, 8, 15287. [Google Scholar] [CrossRef] [Green Version]
- Ostrowski, M.; Carmo, N.B.; Krumeich, S.; Fanget, I.; Raposo, G.; Savina, A.; Moita, C.F.; Schauer, K.; Hume, A.N.; Freitas, R.P. Rab27a and Rab27b control different steps of the exosome secretion pathway. Nat. Cell Biol. 2010, 12, 19–30. [Google Scholar] [CrossRef] [Green Version]
- Hyenne, V.; Apaydin, A.; Rodriguez, D.; Spiegelhalter, C.; Hoff-Yoessle, S.; Diem, M.; Tak, S.; Lefebvre, O.; Schwab, Y.; Goetz, J.G. RAL-1 controls multivesicular body biogenesis and exosome secretion. J. Cell Biol. 2015, 211, 27–37. [Google Scholar] [CrossRef] [Green Version]
- Patel, B.; Patel, J.; Cho, J.; Manne, S.; Bonala, S.; Henske, E.; Roegiers, F.; Markiewski, M.; Karbowniczek, M. Exosomes mediate the acquisition of the disease phenotypes by cells with normal genome in tuberous sclerosis complex. Oncogene 2016, 35, 3027–3036. [Google Scholar] [CrossRef]
- Akbar, N.; Digby, J.E.; Cahill, T.J.; Tavare, A.N.; Corbin, A.L.; Saluja, S.; Dawkins, S.; Edgar, L.; Rawlings, N.; Ziberna, K. Endothelium-derived extracellular vesicles promote splenic monocyte mobilization in myocardial infarction. JCI Insight 2017, 2, e93344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.M.; Choi, E.-J.; Kim, J.H.; Kim, T.D.; Kim, Y.-K.; Kang, C.; Gho, Y.S. A membranous form of ICAM-1 on exosomes efficiently blocks leukocyte adhesion to activated endothelial cells. Biochem. Biophys. Res. Commun. 2010, 397, 251–256. [Google Scholar] [CrossRef] [PubMed]
- Van Niel, G.; d’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef] [PubMed]
- Toth, B.; Lok, C.A.; Böing, A.; Diamant, M.; van der Post, J.A.; Friese, K.; Nieuwland, R. Microparticles and exosomes: Impact on normal and complicated pregnancy. Am. J. Reprod. Immunol. 2007, 58, 389–402. [Google Scholar] [CrossRef] [PubMed]
- Azevedo, L.C.P.; Janiszewski, M.; Pontieri, V.; Pedro, M.d.; Bassi, E.; Tucci, P.J.F.; Laurindo, F.R.M. Platelet-derived exosomes from septic shock patients induce myocardial dysfunction. Crit. Care 2007, 11, R120. [Google Scholar] [CrossRef] [Green Version]
- Srikanthan, S.; Li, W.; Silverstein, R.L.; McIntyre, T.M. Exosome poly-ubiquitin inhibits platelet activation, downregulates CD 36 and inhibits pro-atherothombotic cellular functions. J. Thromb. Haemost. 2014, 12, 1906–1917. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Tan, M.; Xiang, Q.; Zhou, Z.; Yan, H. Thrombin-activated platelet-derived exosomes regulate endothelial cell expression of ICAM-1 via microRNA-223 during the thrombosis-inflammation response. Thromb. Res. 2017, 154, 96–105. [Google Scholar] [CrossRef]
- Tan, M.; Yan, H.-B.; Li, J.-N.; Li, W.-K.; Fu, Y.-Y.; Chen, W.; Zhou, Z. Thrombin stimulated platelet-derived exosomes inhibit platelet-derived growth factor receptor-beta expression in vascular smooth muscle cells. Cell. Physiol. Biochem. 2016, 38, 2348–2365. [Google Scholar] [CrossRef]
- Wang, H.; Wang, Q.; Kleiman, K.; Guo, C.; Eitzman, D.T. Hematopoietic deficiency of miR-223 attenuates thrombosis in response to photochemical injury in mice. Sci. Rep. 2017, 7, 1606. [Google Scholar] [CrossRef]
- Marketou, M.; Kontaraki, J.; Papadakis, J.; Kochiadakis, G.; Vrentzos, G.; Maragkoudakis, S.; Fragkiadakis, K.; Katsouli, E.; Plataki, M.; Patrianakos, A.; et al. Platelet microRNAs in hypertensive patients with and without cardiovascular disease. J. Hum. Hypertens. 2018, 33, 149–156. [Google Scholar] [CrossRef]
- Alexandru, N.; Constantin, A.; Nemecz, M.; Comarita, I.K.; Vilcu, A.; Procopciuc, A.; Tanko, G.; Georgescu, A. Hypertension associated with hyperlipidemia induced different microRNA expression profiles in plasma, platelets, and platelet-derived microvesicles; effects of endothelial progenitor cell therapy. Front. Med. 2019, 6, 280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazar, S.; Wurtzel, J.G.T.; Chen, X.; Ma, P.; Goldfinger, L.E. High-efficiency unassisted transfection of platelets with naked double-stranded miRNAs modulates signal-activated translation and platelet function. Platelets 2021, 32, 794–806. [Google Scholar] [CrossRef] [PubMed]
- Pienimaeki-Roemer, A.; Konovalova, T.; Musri, M.M.; Sigruener, A.; Boettcher, A.; Meister, G.; Schmitz, G. Transcriptomic profiling of platelet senescence and platelet extracellular vesicles. Transfusion 2017, 57, 144–156. [Google Scholar] [CrossRef]
- Konkoth, A.; Saraswat, R.; Dubrou, C.; Sabatier, F.; Leroyer, A.S.; Lacroix, R.; Duchez, A.C.; Dignat-George, F. Multifaceted role of extracellular vesicles in atherosclerosis. Atherosclerosis 2021, 319, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.O.; Whelan, C.; Nash, J.; James, P.E. Extracellular vesicles in atherosclerosis research. Methods Mol. Biol. 2022, 2419, 349–359. [Google Scholar] [PubMed]
- Pfeffer, S.R. Unsolved mysteries in membrane traffic. Annu. Rev. Biochem. 2007, 76, 629–645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azevedo, L.C. Microparticles and exosomes: Are they part of important pathways in sepsis pathophysiology? In Severe Sepsis and Septic Shock-Understanding a Serious Killer; Intechopen: London, UK, 2012. [Google Scholar]
- van Dommelen, S.M.; Vader, P.; Lakhal, S.; Kooijmans, S.; van Solinge, W.W.; Wood, M.J.; Schiffelers, R.M. Microvesicles and exosomes: Opportunities for cell-derived membrane vesicles in drug delivery. J. Control. Release 2012, 161, 635–644. [Google Scholar] [CrossRef]
- Hristov, M.; Erl, W.; Linder, S.; Weber, P.C. Apoptotic bodies from endothelial cells enhance the number and initiate the differentiation of human endothelial progenitor cells in vitro. Blood 2004, 104, 2761–2766. [Google Scholar] [CrossRef]
- Zhu, Z.; Zhang, D.; Lee, H.; Menon, A.A.; Wu, J.; Hu, K.; Jin, Y. Macrophage-derived apoptotic bodies promote the proliferation of the recipient cells via shuttling microRNA-221/222. J. Leukoc. Biol. 2017, 101, 1349–1359. [Google Scholar] [CrossRef] [Green Version]
- Holmgren, L.; Szeles, A.; Rajnavölgyi, E.; Folkman, J.; Klein, G.; Ernberg, I.; Falk, K.I. Horizontal transfer of DNA by the uptake of apoptotic bodies. Blood J. Am. Soc. Hematol. 1999, 93, 3956–3963. [Google Scholar]
- Bergsmedh, A.; Szeles, A.; Henriksson, M.; Bratt, A.; Folkman, M.J.; Spetz, A.-L.; Holmgren, L. Horizontal transfer of oncogenes by uptake of apoptotic bodies. Proc. Natl. Acad. Sci. USA 2001, 98, 6407–6411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jansen, F.; Yang, X.; Hoyer, F.F.; Paul, K.; Heiermann, N.; Becher, M.U.; Hussein, N.A.; Kebschull, M.; Bedorf, J.; Franklin, B.S. Endothelial microparticle uptake in target cells is annexin I/phosphatidylserine receptor dependent and prevents apoptosis. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1925–1935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janiszewski, M.; Carmo, A.O.D.; Pedro, M.A.; Silva, E.; Knobel, E.; Laurindo, F.R. Platelet-derived exosomes of septic individuals possess proapoptotic NAD(P)H oxidase activity: A novel vascular redox pathway. Crit. Care Med. 2004, 32, 818–825. [Google Scholar] [CrossRef] [PubMed]
- Khorana, A.A.; Francis, C.W.; Culakova, E.; Kuderer, N.M.; Lyman, G.H. Frequency, risk factors, and trends for venous thromboembolism among hospitalized cancer patients. Cancer Interdiscip. Int. J. Am. Cancer Soc. 2007, 110, 2339–2346. [Google Scholar] [CrossRef] [PubMed]
- Abdol Razak, N.B.; Jones, G.; Bhandari, M.; Berndt, M.C.; Metharom, P. Cancer-associated thrombosis: An overview of mechanisms, risk factors, and treatment. Cancers 2018, 10, 380. [Google Scholar] [CrossRef] [Green Version]
- Al Saleh, H.A.; Haas-Neill, S.; Al-Hashimi, A.; Kapoor, A.; Shayegan, B.; Austin, R.C.; Al-Nedawi, K. Thrombotic characteristics of extracellular vesicles derived from prostate cancer cells. Prostate 2018, 78, 953–961. [Google Scholar] [CrossRef]
- Lima, L.G.; Leal, A.C.; Vargas, G.; Porto-Carreiro, I.; Monteiro, R.Q. Intercellular transfer of tissue factor via the uptake of tumor-derived microvesicles. Thromb. Res. 2013, 132, 450–456. [Google Scholar] [CrossRef]
- Zarfati, M.; Katz, T.; Avivi, I.; Brenner, B.; Aharon, A. PO-45—The role of microvesicles in multiple myeloma progression. Thromb. Res. 2016, 140 (Suppl. 1), S193. [Google Scholar] [CrossRef]
- Gomes, F.G.; Sandim, V.; Almeida, V.H.; Rondon, A.M.; Succar, B.B.; Hottz, E.D.; Leal, A.C.; Verçoza, B.R.F.; Rodrigues, J.C.F.; Bozza, P.T. Breast-cancer extracellular vesicles induce platelet activation and aggregation by tissue factor-independent and-dependent mechanisms. Thromb. Res. 2017, 159, 24–32. [Google Scholar] [CrossRef]
- Muhsin-Sharafaldine, M.-R.; Saunderson, S.C.; Dunn, A.C.; Faed, J.M.; Kleffmann, T.; McLellan, A.D. Procoagulant and immunogenic properties of melanoma exosomes, microvesicles and apoptotic vesicles. Oncotarget 2016, 7, 56279. [Google Scholar] [CrossRef]
- Zhang, C.; Yang, Z.; Zhou, P.; Yu, M.; Li, B.; Liu, Y.; Jin, J.; Liu, W.; Jing, H.; Du, J.; et al. Phosphatidylserine-exposing tumor-derived microparticles exacerbate coagulation and cancer cell transendothelial migration in triple-negative breast cancer. Theranostics 2021, 11, 6445–6460. [Google Scholar] [CrossRef] [PubMed]
- Hron, G.; Kollars, M.; Weber, H.; Sagaster, V.; Quehenberger, P.; Eichinger, S.; Kyrle, P.A.; Weltermann, A. Tissue factor-positive microparticles: Cellular origin and association with coagulation activation in patients with colorectal cancer. Thromb. Haemost. 2007, 97, 119–123. [Google Scholar] [CrossRef] [PubMed]
- Zwicker, J.I.; Liebman, H.A.; Neuberg, D.; Lacroix, R.; Bauer, K.A.; Furie, B.C.; Furie, B. Tumor-derived tissue factor–bearing microparticles are associated with venous thromboembolic events in malignancy. Clin. Cancer Res. 2009, 15, 6830–6840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manly, D.A.; Wang, J.; Glover, S.L.; Kasthuri, R.; Liebman, H.A.; Key, N.S.; Mackman, N. Increased microparticle tissue factor activity in cancer patients with venous thromboembolism. Thromb. Res. 2010, 125, 511–512. [Google Scholar] [CrossRef] [Green Version]
- Campello, E.; Spiezia, L.; Radu, C.M.; Bulato, C.; Castelli, M.; Gavasso, S.; Simioni, P. Endothelial, platelet, and tissue factor-bearing microparticles in cancer patients with and without venous thromboembolism. Thromb. Res. 2011, 127, 473–477. [Google Scholar] [CrossRef]
- Hell, L.; Däullary, T.; Burghart, V.; Mauracher, L.M.; Grilz, E.; Moser, B.; Kramer, G.; Schmid, J.A.; Ay, C.; Pabinger, I.; et al. Extracellular vesicle-associated tissue factor activity in prostate cancer patients with disseminated intravascular coagulation. Cancers 2021, 13, 1487. [Google Scholar] [CrossRef]
- Stark, K.; Schubert, I.; Joshi, U.; Kilani, B.; Hoseinpour, P.; Thakur, M.; Grünauer, P.; Pfeiler, S.; Schmidergall, T.; Stockhausen, S. Distinct pathogenesis of pancreatic cancer microvesicle—Associated venous thrombosis identifies new antithrombotic targets in vivo. Arter. Thromb. Vasc. Biol. 2018, 38, 772–786. [Google Scholar] [CrossRef] [Green Version]
- Lacroix, R.; Vallier, L.; Bonifay, A.; Simoncini, S.; Mege, D.; Aubert, M.; Panicot-Dubois, L.; Dubois, C.; Dignat-George, F. Microvesicles and cancer associated thrombosis. In Seminars in Thrombosis and Hemostasis; Thieme Medical Publishers: New York, NY, USA, 2019. [Google Scholar]
- Hisada, Y.; Mackman, N. Cancer cell-derived tissue factor positive extracellular vesicles: Biomarkers of thrombosis and survival. Curr. Opin. Hematol. 2019, 26, 349. [Google Scholar] [CrossRef]
- van Es, N.; Hisada, Y.; di Nisio, M.; Cesarman, G.; Kleinjan, A.; Mahé, I.; Otten, H.-M.; Kamphuisen, P.W.; Berckmans, R.J.; Büller, H.R. Extracellular vesicles exposing tissue factor for the prediction of venous thromboembolism in patients with cancer: A prospective cohort study. Thromb. Res. 2018, 166, 54–59. [Google Scholar] [CrossRef]
- Lazar, S.; Goldfinger, L.E. Platelets and extracellular vesicles and their cross talk with cancer. Blood 2021, 137, 3192–3200. [Google Scholar] [CrossRef]
- Żmigrodzka, M.; Witkowska-Piłaszewicz, O.; Winnicka, A. Platelets extracellular vesicles as regulators of cancer progression—An updated perspective. Int. J. Mol. Sci. 2020, 21, 5195. [Google Scholar] [CrossRef] [PubMed]
- Gkolfinopoulos, S.; Jones, R.L.; Constantinidou, A. The emerging role of platelets in the formation of the micrometastatic niche: Current evidence and future perspectives. Front. Oncol. 2020, 10, 374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hisada, Y.; Mackman, N. Tissue factor and extracellular vesicles: Activation of coagulation and impact on survival in cancer. Cancers 2021, 13, 3839. [Google Scholar] [CrossRef] [PubMed]
- Khorana, A.; Francis, C.; Menzies, K.; Wang, J.-G.; Hyrien, O.; Hathcock, J.; Mackman, N.; Taubman, M. Plasma tissue factor may be predictive of venous thromboembolism in pancreatic cancer. J. Thromb. Haemost. 2008, 6, 1983–1985. [Google Scholar] [CrossRef]
- Faille, D.; Bourrienne, M.-C.; de Raucourt, E.; de Chaisemartin, L.; Granger, V.; Lacroix, R.; Panicot-Dubois, L.; Hammel, P.; Lévy, P.; Ruszniewski, P. Biomarkers for the risk of thrombosis in pancreatic adenocarcinoma are related to cancer process. Oncotarget 2018, 9, 26453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tesselaar, M.; Romijn, F.; van der Linden, I.; Prins, F.; Bertina, R.; Osanto, S. Microparticle-associated tissue factor activity: A link between cancer and thrombosis? J. Thromb. Haemost. 2007, 5, 520–527. [Google Scholar] [CrossRef]
- Toth, B.; Liebhardt, S.; Steinig, K.; Ditsch, N.; Rank, A.; Bauerfeind, I.; Spannagl, M.; Friese, K.; Reininger, A.J. Platelet-derived microparticles and coagulation activation in breast cancer patients. Thromb. Haemost. 2008, 100, 663–669. [Google Scholar] [CrossRef] [Green Version]
- Hernández, C.; Orbe, J.; Roncal, C.; Alvarez-Hernandez, M.; de Lizarrondo, S.M.; Alves, M.T.; Mata, J.G.; Páramo, J.A. Tissue factor expressed by microparticles is associated with mortality but not with thrombosis in cancer patients. Thromb. Haemost. 2013, 110, 598–608. [Google Scholar] [CrossRef]
- Thaler, J.; Ay, C.; Mackman, N.; Bertina, R.; Kaider, A.; Marosi, C.; Key, N.; Barcel, D.; Scheithauer, W.; Kornek, G. Microparticle-associated tissue factor activity, venous thromboembolism and mortality in pancreatic, gastric, colorectal and brain cancer patients. J. Thromb. Haemost. 2012, 10, 1363–1370. [Google Scholar] [CrossRef]
- Durrieu, L.; Bharadwaj, A.; Waisman, D.M. Analysis of the thrombotic and fibrinolytic activities of tumor cell-derived extracellular vesicles. Blood Adv. 2018, 2, 1054–1065. [Google Scholar] [CrossRef] [Green Version]
- Garnier, D.; Magnus, N.; Lee, T.H.; Bentley, V.; Meehan, B.; Milsom, C.; Montermini, L.; Kislinger, T.; Rak, J. Cancer cells induced to express mesenchymal phenotype release exosome-like extracellular vesicles carrying tissue factor. J. Biol. Chem. 2012, 287, 43565–43572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, G.M.; Panicot-Dubois, L.; Lacroix, R.; Dignat-George, F.; Lombardo, D.; Dubois, C. Cancer cell—Derived microparticles bearing P-selectin glycoprotein ligand 1 accelerate thrombus formation in vivo. J. Exp. Med. 2009, 206, 1913–1927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hisada, Y.; Ay, C.; Auriemma, A.; Cooley, B.; Mackman, N. Human pancreatic tumors grown in mice release tissue factor-positive microvesicles that increase venous clot size. J. Thromb. Haemost. 2017, 15, 2208–2217. [Google Scholar] [CrossRef] [Green Version]
- Thomas, G.; Brill, A.; Mezouar, S.; Crescence, L.; Gallant, M.; Dubois, C.; Wagner, D. Tissue factor expressed by circulating cancer cell-derived microparticles drastically increases the incidence of deep vein thrombosis in mice. J. Thromb. Haemost. 2015, 13, 1310–1319. [Google Scholar] [CrossRef]
- Davila, M.; Amirkhosravi, A.; Coll, E.; Desai, H.; Robles, L.; Colon, J.; Baker, C.; Francis, J. Tissue factor-bearing microparticles derived from tumor cells: Impact on coagulation activation. J. Thromb. Haemost. 2008, 6, 1517–1524. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.-G.; Geddings, J.E.; Aleman, M.M.; Cardenas, J.C.; Chantrathammachart, P.; Williams, J.C.; Kirchhofer, D.; Bogdanov, V.Y.; Bach, R.R.; Rak, J. Tumor-derived tissue factor activates coagulation and enhances thrombosis in a mouse xenograft model of human pancreatic cancer. Blood J. Am. Soc. Hematol. 2012, 119, 5543–5552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasano, T.; Cho, M.S.; Rodriguez-Aguayo, C.; Bayraktar, E.; Taki, M.; Afshar-Kharghan, V.; Sood, A.K. Role of tissue-factor bearing extracellular vesicles released from ovarian cancer cells in platelet aggregation in vitro and venous thrombosis in mice. Thromb. Update 2021, 2, 100020. [Google Scholar] [CrossRef]
- Mezouar, S.; Darbousset, R.; Dignat-George, F.; Panicot-Dubois, L.; Dubois, C. Inhibition of platelet activation prevents the P-selectin and integrin-dependent accumulation of cancer cell microparticles and reduces tumor growth and metastasis in vivo. Int. J. Cancer 2014, 136, 462–475. [Google Scholar] [CrossRef] [Green Version]
- Tawil, N.; Bassawon, R.; Meehan, B.; Nehme, A.; Montermini, L.; Gayden, T.; de Jay, N.; Spinelli, C.; Chennakrishnaiah, S.; Choi, D.; et al. Glioblastoma cell populations with distinct oncogenic programs release podoplanin as procoagulant extracellular vesicles. Blood Adv. 2021, 5, 1682–1694. [Google Scholar] [CrossRef]
- Nickel, K.F.; Ronquist, G.; Langer, F.; Labberton, L.; Fuchs, T.A.; Bokemeyer, C.; Sauter, G.; Graefen, M.; Mackman, N.; Stavrou, E.X. The polyphosphate–factor XII pathway drives coagulation in prostate cancer-associated thrombosis. Blood J. Am. Soc. Hematol. 2015, 126, 1379–1389. [Google Scholar] [CrossRef] [Green Version]
- Shim, Y.J.; Chatterjee, V.; Swaidani, S.; Alluri, R.K.; Kundu, S.; Merkulova, A.; Angelini, D.; You, D.; Whitney, S.A.; Feener, E.P.; et al. Polyphosphate expression by cancer cell extracellular vesicles mediates binding of factor XII and contact activation. Blood Adv. 2021, 5, 4741–4751. [Google Scholar] [CrossRef] [PubMed]
- Larsson, M.; Rayzman, V.; Nolte, M.W.; Nickel, K.F.; Björkqvist, J.; Jämsä, A.; Hardy, M.P.; Fries, M.; Schmidbauer, S.; Hedenqvist, P. A factor XIIa inhibitory antibody provides thromboprotection in extracorporeal circulation without increasing bleeding risk. Sci. Transl. Med. 2014, 6, 222ra17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nickel, K.F.; Labberton, L.; Long, A.T.; Langer, F.; Fuchs, T.A.; Stavrou, E.X.; Butler, L.M.; Renné, T. The polyphosphate/factor XII pathway in cancer-associated thrombosis: Novel perspectives for safe anticoagulation in patients with malignancies. Thromb. Res. 2016, 141, S4–S7. [Google Scholar] [CrossRef]
- Demers, M.; Krause, D.S.; Schatzberg, D.; Martinod, K.; Voorhees, J.R.; Fuchs, T.A.; Scadden, D.T.; Wagner, D.D. Cancers predispose neutrophils to release extracellular DNA traps that contribute to cancer-associated thrombosis. Proc. Natl. Acad. Sci. USA 2012, 109, 13076–13081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demers, M.; Wagner, D.D. NETosis: A new factor in tumor progression and cancer-associated thrombosis. Semin. Thromb. Hemost. 2014, 40, 277–283. [Google Scholar] [CrossRef] [Green Version]
- Cedervall, J.; Zhang, Y.; Olsson, A.K. Tumor-induced NETosis as a risk factor for metastasis and organ failure. Cancer Res. 2016, 76, 4311–4315. [Google Scholar] [CrossRef] [Green Version]
- Noubouossie, D.F.; Reeves, B.N.; Strahl, B.D.; Key, N.S. Neutrophils: Back in the thrombosis spotlight. Blood 2019, 133, 2186–2197. [Google Scholar] [CrossRef]
- Chapman, E.A.; Lyon, M.; Simpson, D.; Mason, D.; Beynon, R.J.; Moots, R.J.; Wright, H.L. Caught in a trap? Proteomic analysis of neutrophil extracellular traps in rheumatoid arthritis and systemic lupus erythematosus. Front. Immunol. 2019, 10, 423. [Google Scholar] [CrossRef]
- Noubouossie, D.F.; Whelihan, M.F.; Yu, Y.B.; Sparkenbaugh, E.; Pawlinski, R.; Monroe, D.M.; Key, N.S. In vitro activation of coagulation by human neutrophil DNA and histone proteins but not neutrophil extracellular traps. Blood 2017, 129, 1021–1029. [Google Scholar] [CrossRef]
- Martinod, K.; Wagner, D.D. Thrombosis: Tangled up in NETs. Blood 2014, 123, 2768–2776. [Google Scholar] [CrossRef]
- Martinod, K.; Demers, M.; Fuchs, T.A.; Wong, S.L.; Brill, A.; Gallant, M.; Hu, J.; Wang, Y.; Wagner, D.D. Neutrophil histone modification by peptidylarginine deiminase 4 is critical for deep vein thrombosis in mice. Proc. Natl. Acad. Sci. USA 2013, 110, 8674–8679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDonald, B.; Davis, R.P.; Kim, S.J.; Tse, M.; Esmon, C.T.; Kolaczkowska, E.; Jenne, C.N. Platelets and neutrophil extracellular traps collaborate to promote intravascular coagulation during sepsis in mice. Blood 2017, 129, 1357–1367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dou, H.; Kotini, A.; Liu, W.; Fidler, T.; Endo-Umeda, K.; Sun, X.; Olszewska, M.; Xiao, T.; Abramowicz, S.; Yalcinkaya, M.; et al. Oxidized phospholipids promote NETosis and arterial thrombosis in LNK(SH2B3) deficiency. Circulation 2021, 144, 1940–1954. [Google Scholar] [CrossRef] [PubMed]
- Etulain, J.; Martinod, K.; Wong, S.L.; Cifuni, S.M.; Schattner, M.; Wagner, D.D. P-selectin promotes neutrophil extracellular trap formation in mice. Blood 2015, 126, 242–246. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Diamond, S.L. Fibrin modulates shear-induced NETosis in sterile occlusive thrombi formed under haemodynamic flow. Thromb. Haemost. 2019, 119, 586–593. [Google Scholar] [CrossRef] [PubMed]
- Healy, L.D.; Puy, C.; Fernández, J.A.; Mitrugno, A.; Keshari, R.S.; Taku, N.A.; Chu, T.T.; Xu, X.; Gruber, A.; Lupu, F.; et al. Activated protein C inhibits neutrophil extracellular trap formation in vitro and activation in vivo. J. Biol. Chem. 2017, 292, 8616–8629. [Google Scholar] [CrossRef] [Green Version]
- Von Brühl, M.-L.; Stark, K.; Steinhart, A.; Chandraratne, S.; Konrad, I.; Lorenz, M.; Khandoga, A.; Tirniceriu, A.; Coletti, R.; Köllnberger, M. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J. Exp. Med. 2012, 209, 819–835. [Google Scholar] [CrossRef]
- Clark, S.R.; Ma, A.C.; Tavener, S.A.; McDonald, B.; Goodarzi, Z.; Kelly, M.M.; Patel, K.D.; Chakrabarti, S.; McAvoy, E.; Sinclair, G.D. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat. Med. 2007, 13, 463–469. [Google Scholar] [CrossRef]
- Mizurini, D.M.; Aslan, J.S.; Gomes, T.; Ma, D.; Francischetti, I.M.; Monteiro, R.Q. Salivary thromboxane A2-binding proteins from triatomine vectors of Chagas disease inhibit platelet-mediated neutrophil extracellular traps (NETs) formation and arterial thrombosis. PLoS Negl. Trop. Dis. 2015, 9, e0003869. [Google Scholar] [CrossRef]
- Laridan, E.; Martinod, K.; de Meyer, S.F. Neutrophil extracellular traps in arterial and venous thrombosis. In Seminars in Thrombosis and Hemostasis; Thieme Publishing: New York, NY, USA, 2019. [Google Scholar]
- Fuchs, T.A.; Bhandari, A.A.; Wagner, D.D. Histones induce rapid and profound thrombocytopenia in mice. Blood 2011, 118, 3708–3714. [Google Scholar] [CrossRef] [Green Version]
- Elaskalani, O.; Razak, N.B.A.; Metharom, P. Neutrophil extracellular traps induce aggregation of washed human platelets independently of extracellular DNA and histones. Cell Commun. Signal. 2018, 16, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leal, A.C.; Mizurini, D.M.; Gomes, T.; Rochael, N.C.; Saraiva, E.M.; Dias, M.S.; Werneck, C.C.; Sielski, M.S.; Vicente, C.P.; Monteiro, R.Q. Tumor-derived exosomes induce the formation of neutrophil extracellular traps: Implications for the establishment of cancer-associated thrombosis. Sci. Rep. 2017, 7, 6438. [Google Scholar] [CrossRef] [PubMed]
- Boone, B.A.; Murthy, P.; Miller-Ocuin, J.; Doerfler, W.R.; Ellis, J.T.; Liang, X.; Ross, M.A.; Wallace, C.T.; Sperry, J.L.; Lotze, M.T. Chloroquine reduces hypercoagulability in pancreatic cancer through inhibition of neutrophil extracellular traps. BMC Cancer 2018, 18, 678. [Google Scholar] [CrossRef] [Green Version]
- Mauracher, L.M.; Posch, F.; Martinod, K.; Grilz, E.; Däullary, T.; Hell, L.; Brostjan, C.; Zielinski, C.; Ay, C.; Wagner, D. Citrullinated histone H3, a biomarker of neutrophil extracellular trap formation, predicts the risk of venous thromboembolism in cancer patients. J. Thromb. Haemost. 2018, 16, 508–518. [Google Scholar] [CrossRef]
- Seo, J.D.; Gu, J.-Y.; Jung, H.S.; Kim, Y.J.; Kim, H.K. Contact system activation and neutrophil extracellular trap markers: Risk factors for portal vein thrombosis in patients with hepatocellular carcinoma. Clin. Appl. Thromb. Hemost. 2019, 25, 1076029618825310. [Google Scholar] [CrossRef] [Green Version]
- Hisada, Y.; Grover, S.P.; Maqsood, A.; Houston, R.; Ay, C.; Noubouossie, D.F.; Cooley, B.C.; Wallén, H.; Key, N.S.; Thålin, C. Neutrophils and neutrophil extracellular traps enhance venous thrombosis in mice bearing human pancreatic tumors. Haematologica 2020, 105, 218–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nieuwland, R.; Berckmans, R.J.; McGregor, S.; Böing, A.N.; Romijn, F.P.T.M.; Westendorp, R.G.; Hack, C.E.; Sturk, A. Cellular origin and procoagulant properties of microparticles in meningococcal sepsis. Blood J. Am. Soc. Hematol. 2000, 95, 930–935. [Google Scholar] [CrossRef]
- Wang, J.G.; Manly, D.; Kirchhofer, D.; Pawlinski, R.; Mackman, N. Levels of microparticle tissue factor activity correlate with coagulation activation in endotoxemic mice. J. Thromb. Haemost. 2009, 7, 1092–1098. [Google Scholar] [CrossRef]
- Zafrani, L.; Gerotziafas, G.; Byrnes, C.; Hu, X.; Perez, J.; Lévi, C.; Placier, S.; Letavernier, E.; Leelahavanichkul, A.; Haymann, J.-P. Calpastatin controls polymicrobial sepsis by limiting procoagulant microparticle release. Am. J. Respir. Crit. Care Med. 2012, 185, 744–755. [Google Scholar] [CrossRef] [Green Version]
- Raeven, P.; Zipperle, J.; Drechsler, S. Extracellular vesicles as markers and mediators in sepsis. Theranostics 2018, 8, 3348. [Google Scholar] [CrossRef]
- Lehner, G.F.; Harler, U.; Haller, V.M.; Feistritzer, C.; Hasslacher, J.; Dunzendorfer, S.; Bellmann, R.; Joannidis, M. Characterization of microvesicles in septic shock using high-sensitivity flow cytometry. Shock Inj. Inflamm. Sepsis Lab. Clin. Approaches 2016, 46, 373–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohuchi, M.; Fujino, K.; Kishimoto, T.; Yamane, T.; Hamamoto, T.; Tabata, T.; Tsujita, Y.; Matsushita, M.; Takahashi, K.; Matsumura, K. Association of the plasma platelet-derived microparticles to platelet count ratio with hospital mortality and disseminated intravascular coagulopathy in critically lll patients. J. Atheroscler. Thromb. 2015, 22, 29439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burgelman, M.; Vandendriessche, C.; Vandenbroucke, R.E. Extracellular vesicles: A double-edged sword in sepsis. Pharmaceuticals 2021, 14, 829. [Google Scholar] [CrossRef] [PubMed]
- Andersson, U.; Tracey, K.J. HMGB1 is a therapeutic target for sterile inflammation and infection. Ann. Rev. Immunol. 2011, 29, 139–162. [Google Scholar] [CrossRef] [Green Version]
- Colombo, M.; Raposo, G.; Théry, C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu. Rev. Cell Dev. Biol. 2014, 30, 255–289. [Google Scholar] [CrossRef]
- Zwicker, J.I. Tissue factor–bearing microparticles and cancer. In Seminars in Thrombosis and Hemostasis; Thieme Medical Publishers: New York, NY, USA, 2008. [Google Scholar]
- Freyssinet, J.-M.; Toti, F. Formation of procoagulant microparticles and properties. Thromb. Res. 2010, 125, S46–S48. [Google Scholar] [CrossRef]
- de Lizarrondo, S.M.; Roncal, C.; Calvayrac, O.; Rodríguez, C.; Varo, N.; Purroy, A.; Lorente, L.; Rodríguez, J.A.; Doeuvre, L.; Hervás-Stubbs, S. Synergistic effect of thrombin and CD40 ligand on endothelial matrix metalloproteinase-10 expression and microparticle generation in vitro and in vivo. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1477–1487. [Google Scholar] [CrossRef] [Green Version]
- Iba, T.; Ogura, H. Role of extracellular vesicles in the development of sepsis-induced coagulopathy. J. Intensiv. Care 2018, 6, 68. [Google Scholar] [CrossRef]
- Horstman, L.L.; Ahn, Y.S. Platelet microparticles: A wide-angle perspective. Crit. Rev. Oncol. Hematol. 1999, 30, 111–142. [Google Scholar] [CrossRef]
- Matsumoto, H.; Yamakawa, K.; Ogura, H.; Koh, T.; Matsumoto, N.; Shimazu, T. Clinical significance of tissue factor and CD13 double-positive microparticles in SIRS patients with trauma and severe sepsis. Shock Inj. Inflamm. Sepsis Lab. Clin. Approaches 2017, 47, 409–415. [Google Scholar] [CrossRef]
- Woei-A-Jin, F.S.H.; van der Starre, W.E.; Tesselaar, M.E.; Rodriguez, P.G.; van Nieuwkoop, C.; Bertina, R.M.; van Dissel, J.T.; Osanto, S. Procoagulant tissue factor activity on microparticles is associated with disease severity and bacteremia in febrile urinary tract infections. Thromb. Res. 2014, 133, 799–803. [Google Scholar] [CrossRef] [PubMed]
- Kleinjan, A.; Böing, A.N.; Sturk, A.; Nieuwland, R. Microparticles in vascular disorders: How tissue factor-exposing vesicles contribute to pathology and physiology. Thromb. Res. 2012, 130, S71–S73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hellum, M.; Øvstebø, R.; Brusletto, B.S.; Berg, J.P.; Brandtzaeg, P.; Henriksson, C.E. Microparticle-associated tissue factor activity correlates with plasma levels of bacterial lipopolysaccharides in meningococcal septic shock. Thromb. Res. 2014, 133, 507–514. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Meng, H.; Ma, R.; He, Z.; Wu, X.; Cao, M.; Yao, Z.; Zhao, L.; Li, T.; Deng, R. Circulating microparticles, blood cells, and endothelium induce procoagulant activity in sepsis through phosphatidylserine exposure. Shock 2016, 45, 299–307. [Google Scholar] [CrossRef]
- Tripisciano, C.; Weiss, R.; Eichhorn, T.; Spittler, A.; Heuser, T.; Fischer, M.B.; Weber, V. Different potential of extracellular vesicles to support thrombin generation: Contributions of phosphatidylserine, tissue factor, and cellular origin. Sci. Rep. 2017, 7, 6522. [Google Scholar] [CrossRef]
- Radic, M.; Marion, T.; Monestier, M. Nucleosomes are exposed at the cell surface in apoptosis. J. Immunol. 2004, 172, 6692–6700. [Google Scholar] [CrossRef] [Green Version]
- Liaw, P.C.; Ito, T.; Iba, T.; Thachil, J.; Zeerleder, S. DAMP and DIC: The role of extracellular DNA and DNA-binding proteins in the pathogenesis of DIC. Blood Rev. 2016, 30, 257–261. [Google Scholar] [CrossRef]
- Weiss, R.; Gröger, M.; Rauscher, S.; Fendl, B.; Eichhorn, T.; Fischer, M.B.; Spittler, A.; Weber, V. Differential interaction of platelet-derived extracellular vesicles with leukocyte subsets in human whole blood. Sci. Rep. 2018, 8, 6598. [Google Scholar] [CrossRef]
- Im, Y.; Yoo, H.; Lee, J.Y.; Park, J.; Suh, G.Y.; Jeon, K. Association of plasma exosomes with severity of organ failure and mortality in patients with sepsis. J. Cell. Mol. Med. 2020, 24, 9439–9445. [Google Scholar] [CrossRef]
- Reithmair, M.; Buschmann, D.; Märte, M.; Kirchner, B.; Hagl, D.; Kaufmann, I.; Pfob, M.; Chouker, A.; Steinlein, O.K.; Pfaffl, M.W.; et al. Cellular and extracellular miRNAs are blood-compartment-specific diagnostic targets in sepsis. J. Cell. Mol. Med. 2017, 21, 2403–2411. [Google Scholar] [CrossRef]
- Hirschberger, S.; Hinske, L.C.; Kreth, S. MiRNAs: Dynamic regulators of immune cell functions in inflammation and cancer. Cancer Lett. 2018, 431, 11–21. [Google Scholar] [CrossRef]
- Jiao, Y.; Li, W.; Wang, W.; Tong, X.; Xia, R.; Fan, J.; Du, J.; Zhang, C.; Shi, X. Platelet-derived exosomes promote neutrophil extracellular trap formation during septic shock. Crit. Care 2020, 24, 380. [Google Scholar] [CrossRef] [PubMed]
- Usman, W.M.; Pham, T.C.; Kwok, Y.Y.; Vu, L.T.; Ma, V.; Peng, B.; Chan, Y.S.; Wei, L.; Chin, S.M.; Azad, A. Efficient RNA drug delivery using red blood cell extracellular vesicles. Nat. Commun. 2018, 9, 2359. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; el Andaloussi, S.; Wood, M.J. Exosomes and microvesicles: Extracellular vesicles for genetic information transfer and gene therapy. Hum. Mol. Genet. 2012, 21, R125–R134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pawlowski, C.L.; Li, W.; Sun, M.; Ravichandran, K.; Hickman, D.; Kos, C.; Kaur, G.; Gupta, A.S. Platelet microparticle-inspired clot-responsive nanomedicine for targeted fibrinolysis. Biomaterials 2017, 128, 94–108. [Google Scholar] [CrossRef] [PubMed]
- Disharoon, D.; Marr, D.W.; Neeves, K.B. Engineered microparticles and nanoparticles for fibrinolysis. J. Thromb. Haemost. 2019, 17, 2004–2015. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Greening, D.W.; Zhu, H.-J.; Takahashi, N.; Simpson, R.J. Extracellular vesicle isolation and characterization: Toward clinical application. J. Clin. Investig. 2016, 126, 1152–1162. [Google Scholar] [CrossRef] [Green Version]

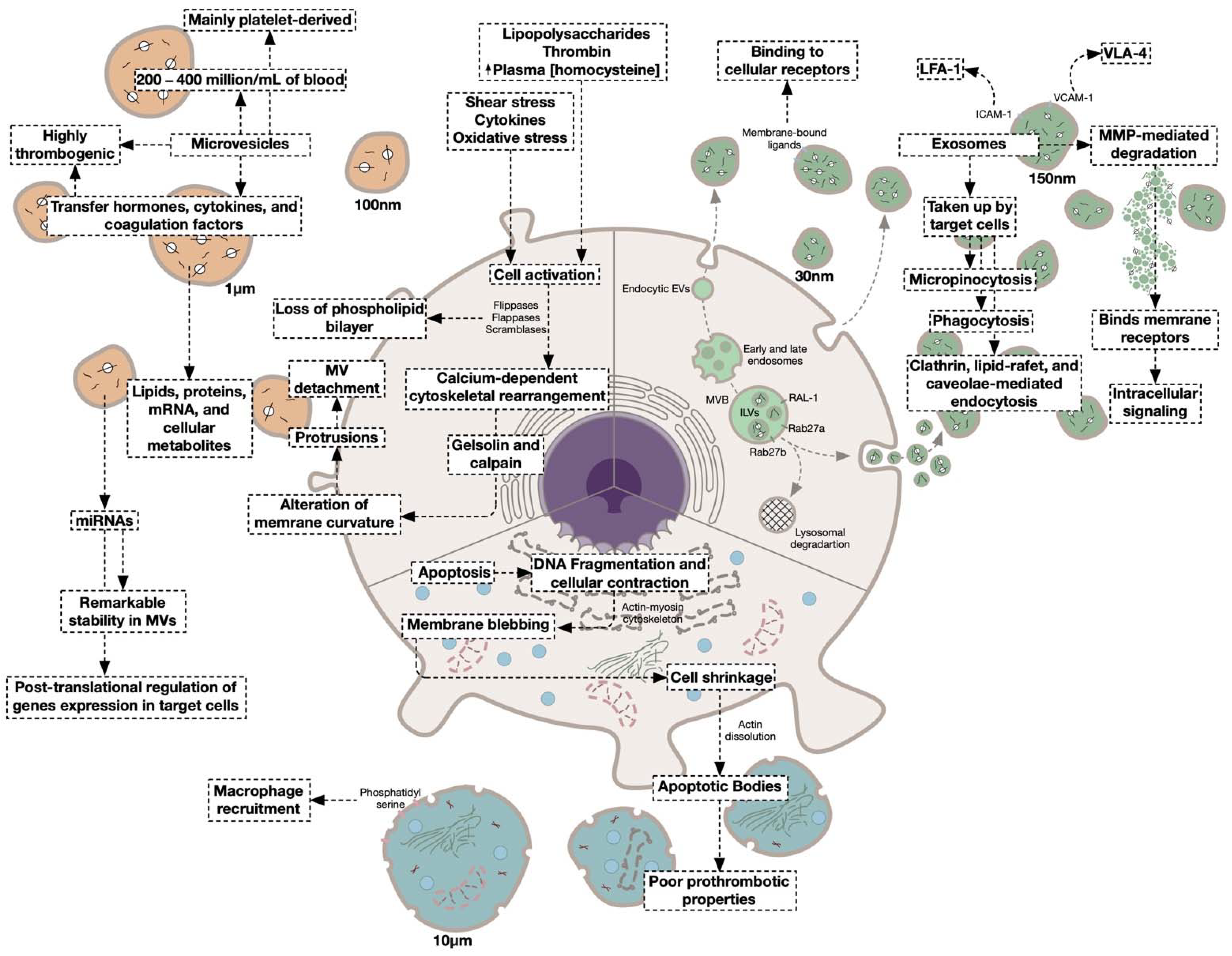

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al-Koussa, H.; AlZaim, I.; El-Sabban, M.E. Pathophysiology of Coagulation and Emerging Roles for Extracellular Vesicles in Coagulation Cascades and Disorders. J. Clin. Med. 2022, 11, 4932. https://doi.org/10.3390/jcm11164932
Al-Koussa H, AlZaim I, El-Sabban ME. Pathophysiology of Coagulation and Emerging Roles for Extracellular Vesicles in Coagulation Cascades and Disorders. Journal of Clinical Medicine. 2022; 11(16):4932. https://doi.org/10.3390/jcm11164932
Chicago/Turabian StyleAl-Koussa, Houssam, Ibrahim AlZaim, and Marwan E. El-Sabban. 2022. "Pathophysiology of Coagulation and Emerging Roles for Extracellular Vesicles in Coagulation Cascades and Disorders" Journal of Clinical Medicine 11, no. 16: 4932. https://doi.org/10.3390/jcm11164932