
Pathophysiology of Heart Failure: A Role for Peripheral Blood Mononuclear Cells Mitochondrial Dysfunction?

 , ,
, ,  ,
,
Abstract
:1. Introduction
2. Potential Relationships between PBMC, Mitochondrial Function and HF
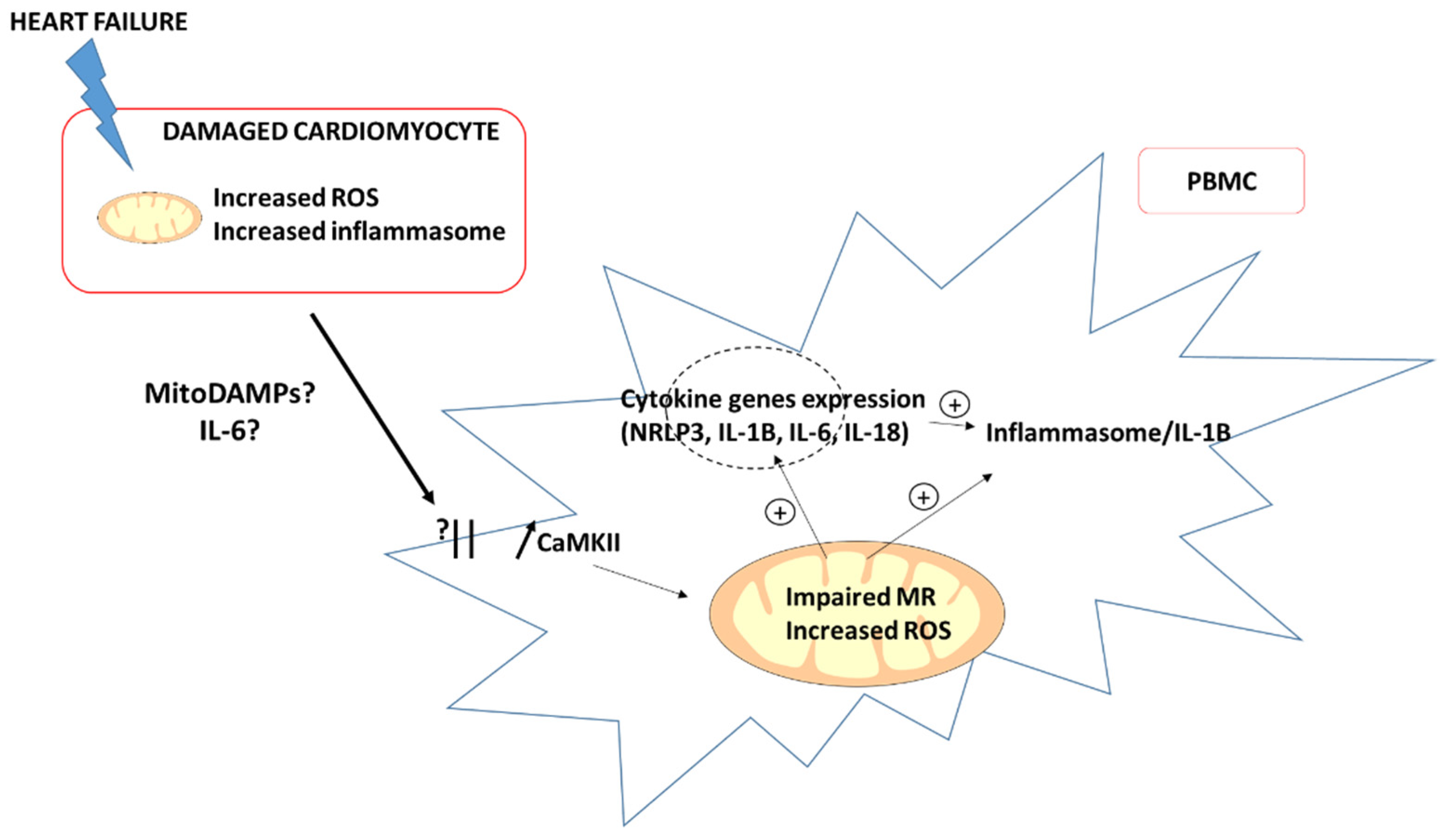
3. Potential Mechanisms Involved in Impaired Mitochondrial Respiration of PBMC in HF
4. Conclusions and Current Challenges
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Böhm, M.; Burri, H.; Butler, J.; Čelutkienė, J.; Chioncel, O.; et al. 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur. Heart J. 2021, 42, 3599–3726, Erratum in Eur. Heart J. 2021. [Google Scholar] [CrossRef]
- Maggioni, A.P.; Dahlström, U.; Filippatos, G.; Chioncel, O.; Crespo Leiro, M.; Drozdz, J.; Fruhwald, F.; Gullestad, L.; Logeart, D.; Fabbri, G.; et al. EURObservational Research Programme: Regional differences and 1-year follow-up results of the Heart Failure Pilot Survey (ESC-HF Pilot). Eur. J. Heart Fail. 2013, 15, 808–817. [Google Scholar] [CrossRef] [Green Version]
- Gabet, A.; Juillière, Y.; Lamarche-Vadel, A.; Vernay, M.; Olié, V. National trends in rate of patients hospitalized for heart failure and heart failure mortality in France, 2000–2012. Eur. J. Hear. Fail. 2015, 17, 583–590. [Google Scholar] [CrossRef]
- Lugnier, C.; Meyer, A.; Charloux, A.; Andrès, E.; Gény, B.; Talha, S. The Endocrine Function of the Heart: Physiology and Involvements of Natriuretic Peptides and Cyclic Nucleotide Phosphodiesterases in Heart Failure. J. Clin. Med. 2019, 8, 1746. [Google Scholar] [CrossRef] [Green Version]
- Anker, S.D.; Butler, J.; Filippatos, G.; Ferreira, J.P.; Bocchi, E.; Böhm, M.; Rocca, H.-P.B.; Choi, D.-J.; Chopra, V.; Chuquiure-Valenzuela, E.; et al. Empagliflozin in Heart Failure with a Preserved Ejection Fraction. N. Engl. J. Med. 2021, 385, 1451–1461. [Google Scholar] [CrossRef]
- Alfatni, A.; Riou, M.; Charles, A.-L.; Meyer, A.; Barnig, C.; Andres, E.; Lejay, A.; Talha, S.; Geny, B. Peripheral Blood Mononuclear Cells and Platelets Mitochondrial Dysfunction, Oxidative Stress, and Circulating mtDNA in Cardiovascular Diseases. J. Clin. Med. 2020, 9, 311. [Google Scholar] [CrossRef] [Green Version]
- Pottecher, J.; Guillot, M.; Belaidi, E.; Charles, A.-L.; Lejay, A.; Gharib, A.; Diemunsch, P.; Geny, B. Cyclosporine A normalizes mitochondrial coupling, reactive oxygen species production, and inflammation and partially restores skeletal muscle maximal oxidative capacity in experimental aortic cross-clamping. J. Vasc. Surg. 2013, 57, 1100–1108.e2. [Google Scholar] [CrossRef] [Green Version]
- Giordano, F.J. Oxygen, oxidative stress, hypoxia, and heart failure. J. Clin. Investig. 2005, 115, 500–508. [Google Scholar] [CrossRef]
- Nevers, T.; Salvador, A.M.; Grodecki-Pena, A.; Knapp, A.; Velázquez, F.; Aronovitz, M.; Kapur, N.K.; Karas, R.H.; Blanton, R.M.; Alcaide, P. Left Ventricular T-Cell Recruitment Contributes to the Pathogenesis of Heart Failure. Circ. Hear. Fail. 2015, 8, 776–787. [Google Scholar] [CrossRef] [Green Version]
- Blanton, R.M.; Carrillo-Salinas, F.J.; Alcaide, P. T-cell recruitment to the heart: Friendly guests or unwelcome visitors? Am. J. Physiol. Heart Circ. Physiol. 2019, 317, H124–H140. [Google Scholar] [CrossRef]
- Ismahil, M.A.; Hamid, T.; Bansal, S.S.; Patel, B.; Kingery, J.R.; Prabhu, S.D. Remodeling of the mononuclear phagocyte network underlies chronic inflammation and disease progression in heart failure: Critical importance of the cardiosplenic axis. Circ. Res. 2014, 114, 266–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bajpai, G.; Bredemeyer, A.; Li, W.; Zaitsev, K.; Koenig, A.L.; Lokshina, I.; Mohan, J.; Ivey, B.; Hsiao, H.M.; Weinheimer, C.; et al. Tissue Resident CCR2- and CCR2+ Cardiac Macrophages Differentially Orchestrate Monocyte Recruitment and Fate Specifi-cation Following Myocardial Injury. Circ. Res. 2019, 124, 263–278. [Google Scholar] [CrossRef] [PubMed]
- Abplanalp, W.T.; John, D.; Cremer, S.; Assmus, B.; Dorsheimer, L.; Hoffmann, J.; Becker-Pergola, G.; Rieger, M.A.; Zeiher, A.M.; Vasa-Nicotera, M.; et al. Single-cell RNA-sequencing reveals profound changes in circulating immune cells in patients with heart failure. Cardiovasc. Res. 2020, 117, 484–494. [Google Scholar] [CrossRef] [Green Version]
- Bonaventura, A.; Montecucco, F.; Dallegri, F.; Carbone, F.; Lüscher, T.F.; Camici, G.; Liberale, L. Novel findings in neutrophil biology and their impact on cardiovascular disease. Cardiovasc. Res. 2019, 115, 1266–1285. [Google Scholar] [CrossRef]
- Li, P.; Wang, B.; Sun, F.; Li, Y.; Li, Q.; Lang, H.; Zhao, Z.; Gao, P.; Zhao, Y.; Shang, Q.; et al. Mitochondrial respiratory dysfunctions of blood mononuclear cells link with cardiac disturbance in patients with early-stage heart failure. Sci. Rep. 2015, 5, 10229. [Google Scholar] [CrossRef] [Green Version]
- Shirakawa, R.; Yokota, T.; Nakajima, T.; Takada, S.; Yamane, M.; Furihata, T.; Maekawa, S.; Nambu, H.; Katayama, T.; Fukushima, A.; et al. Mitochondrial reactive oxygen species generation in blood cells is associated with disease severity and exercise intolerance in heart failure patients. Sci. Rep. 2019, 9, 14709. [Google Scholar] [CrossRef]
- Zhou, B.; Wang, D.D.-H.; Qiu, Y.; Airhart, S.; Liu, Y.; Stempien-Otero, A.; O’Brien, K.D.; Tian, R. Boosting NAD level suppresses inflammatory activation of PBMCs in heart failure. J. Clin. Investig. 2020, 130, 6054–6063. [Google Scholar] [CrossRef]
- Visseren, F.L.J.; Mach, F.; Smulders, Y.M.; Carballo, D.; Koskinas, K.C.; Bäck, M.; Benetos, A.; Biffi, A.; Boavida, J.M.; Capodanno, D.; et al. ESC Scientific Document Group. 2021 ESC Guidelines on cardiovascular disease prevention in clinical practice. Eur. Heart J. 2021, 42, 3227–3337. [Google Scholar] [CrossRef]
- Tomiyama, H.; Shiina, K.; Matsumoto-Nakano, C.; Ninomiya, T.; Komatsu, S.; Kimura, K.; Chikamori, T.; Yamashina, A. The Contribution of Inflammation to the Development of Hypertension Mediated by Increased Arterial Stiffness. J. Am. Hear. Assoc. 2017, 6, e005729. [Google Scholar] [CrossRef]
- DeConne, T.M.; Muñoz, E.R.; Sanjana, F.; Hobson, J.C.; Martens, C.R. Cardiometabolic risk factors are associated with immune cell mitochondrial respiration in humans. Am. J. Physiol. Heart Circ. Physiol. 2020, 319, H481–H487. [Google Scholar] [CrossRef]
- Dai, D.-F.; Rabinovitch, P.S.; Ungvari, Z. Mitochondria and Cardiovascular Aging. Circ. Res. 2012, 110, 1109–1124. [Google Scholar] [CrossRef] [PubMed]
- Eirin, A.; Lerman, A.; Lerman, L.O. Enhancing Mitochondrial Health to Treat Hypertension. Curr. Hypertens. Rep. 2018, 20, 89. [Google Scholar] [CrossRef] [PubMed]
- Incalza, M.A.; D’Oria, R.; Natalicchio, A.; Perrini, S.; Laviola, L.; Giorgino, F. Oxidative stress and reactive oxygen species in endothelial dysfunction associated with cardiovascular and metabolic diseases. Vasc. Pharmacol. 2018, 100, 1–19. [Google Scholar] [CrossRef]
- Boralkar, K.A.; Kobayashi, Y.; Amsallem, M.; Ataam, J.A.; Moneghetti, K.J.; Cauwenberghs, N.; Horne, B.D.; Knowlton, K.U.; Maecker, H.; Kuznetsova, T.; et al. Value of Neutrophil to Lymphocyte Ratio and Its Trajectory in Patients Hospitalized With Acute Heart Failure and Preserved Ejection Fraction. Am. J. Cardiol. 2019, 125, 229–235. [Google Scholar] [CrossRef]
- Núñez, J.; Minana, G.; Bodi, V.; Nunez, E.; Sanchis, J.; Husser, O.; Llacer, A. Low Lymphocyte Count and Cardiovascular Diseases. Curr. Med. Chem. 2011, 18, 3226–3233. [Google Scholar] [CrossRef] [PubMed]
- Chacko, B.K.; Kramer, P.A.; Ravi, S.; Johnson, M.S.; Hardy, R.W.; Ballinger, S.W.; Darley-Usmar, V.M. Methods for defining distinct bioenergetic profiles in platelets, lymphocytes, monocytes, and neutrophils, and the oxidative burst from human blood. Lab. Investig. 2013, 93, 690–700. [Google Scholar] [CrossRef] [PubMed]
- Afari, M.E.; Bhat, T. Neutrophil to lymphocyte ratio (NLR) and cardiovascular diseases: An update. Expert Rev. Cardiovasc. Ther. 2016, 14, 573–577. [Google Scholar] [CrossRef]
- Curran, F.M.; Bhalraam, U.; Mohan, M.; Singh, J.S.; Anker, S.D.; Dickstein, K.; Doney, A.S.; Filippatos, G.; George, J.; Metra, M.; et al. Neutrophil-to-lymphocyte ratio and outcomes in patients with new-onset or worsening heart failure with reduced and preserved ejection fraction. ESC Heart Fail. 2021, 8, 3168–3179. [Google Scholar] [CrossRef]
- Durmus, E.; Kivrak, T.; Gerin, F.; Sunbul, M.; Sari, I.; Erdogan, O. Neutrophil-to-Lymphocyte Ratio and Platelet-to-Lymphocyte Ratio are Predictors of Heart Failure. Arq. Bras. Cardiol. 2015, 105, 606–613. [Google Scholar] [CrossRef]
- Baixauli, F.; Acin-Perez, R.; Villarroya-Beltrí, C.; Mazzeo, C.; Nuñez-Andrade, N.; Gabande-Rodriguez, E.; Ledesma, M.D.; Blázquez, A.; Martín, M.A.; Falcón-Pérez, J.M.; et al. Mitochondrial Respiration Controls Lysosomal Function during Inflammatory T Cell Responses. Cell Metab. 2015, 22, 485–498. [Google Scholar] [CrossRef] [Green Version]
- Clere-Jehl, R.; Helms, J.; Kassem, M.; Le Borgne, P.; Delabranche, X.; Charles, A.-L.; Geny, B.; Meziani, F.; Bilbault, P. Septic Shock Alters Mitochondrial Respiration of Lymphoid Cell-Lines and Human Peripheral Blood Mononuclear Cells: The Role of Plasma. Shock 2019, 51, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Amitrano, A.M.; Berry, B.J.; Lim, K.; Kim, K.-D.; Waugh, R.E.; Wojtovich, A.P.; Kim, M. Optical Control of CD8+ T Cell Metabolism and Effector Functions. Front. Immunol. 2021, 12, 2309. [Google Scholar] [CrossRef] [PubMed]
- Faas, M.M.; De Vos, P. Mitochondrial function in immune cells in health and disease. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165845. [Google Scholar] [CrossRef] [PubMed]
- Suetomi, T.; Miyamoto, S.; Brown, J.H. Inflammation in nonischemic heart disease: Initiation by cardiomyocyte CaMKII and NLRP3 inflammasome signaling. Am. J. Physiol. Circ. Physiol. 2019, 317, H877–H890. [Google Scholar] [CrossRef] [PubMed]
- West, A.P. Mitochondrial dysfunction as a trigger of innate immune responses and inflammation. Toxicology 2017, 391, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Chow, S.L.; Maisel, A.S.; Anand, I.; Bozkurt, B.; de Boer, R.A.; Felker, G.M.; Fonarow, G.C.; Greenberg, B.; Januzzi, J.L., Jr.; Kiernan, M.S.; et al. Role of biomarkers for the prevention, assessment, and management of heart failure: A scientific statement from the American heart association. Circulation 2017, 135, e1054–e1091. [Google Scholar] [CrossRef]
- Fontes, J.A.; Rose, N.R.; Cihakova, D. The varying faces of IL-6: From cardiac protection to cardiac failure. Cytokine 2015, 74, 62–68. [Google Scholar] [CrossRef] [Green Version]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef] [Green Version]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225, Erratum in Nature 2011, 475, 122. [Google Scholar] [CrossRef]
- Gulick, T.; Chung, M.K.; Pieper, S.J.; Lange, L.G.; Schreiner, G.F. Interleukin 1 and tumor necrosis factor inhibit car-diac myocyte beta-adrenergic responsiveness. Proc. Natl. Acad. Sci. USA 1989, 86, 6753–6757. [Google Scholar] [CrossRef] [Green Version]
- Tatsumi, T.; Matoba, S.; Kawahara, A.; Keira, N.; Shiraishi, J.; Akashi, K.; Kobara, M.; Tanaka, T.; Katamura, M.; Nakagawa, C.; et al. Cytokine-induced nitric oxide production inhibits mitochondrial energy production and impairs con-tractile function in rat cardiac myocytes. J. Am. Coll. Cardiol. 2000, 35, 1338–1346. [Google Scholar] [CrossRef] [Green Version]
- Everett, B.M.; Cornel, J.H.; Lainscak, M.; Anker, S.D.; Abbate, A.; Thuren, T.; Libby, P.; Glynn, R.J.; Ridker, P.M. Anti-Inflammatory Therapy With Canakinumab for the Prevention of Hospitalization for Heart Failure. Circulation 2019, 139, 1289–1299. [Google Scholar] [CrossRef] [PubMed]
- Van Tassell, B.W.; Canada, J.; Carbone, S.; Trankle, C.; Buckley, L.; Erdle, C.O.; Abouzaki, N.A.; Dixon, D.; Kadariya, D.; Christopher, S.; et al. Interleukin-1 Blockade in Recently Decompensated Systolic Heart Failure: Results From REDHART (Recently Decompensated Heart Failure Anakinra Response Trial). Circ. Hear. Fail. 2017, 10, e004373. [Google Scholar] [CrossRef] [PubMed]
- Bilchick, K.; Kothari, H.; Narayan, A.; Garmey, J.; Omar, A.; Capaldo, B.; McNamara, C. Cardiac resynchronization therapy reduces expression of inflammation-promoting genes related to interleukin-1β in heart failure. Cardiovasc. Res. 2019, 116, 1311–1322. [Google Scholar] [CrossRef] [Green Version]
- Lopaschuk, G.D.; Verma, S. Mechanisms of Cardiovascular Benefits of Sodium Glucose Co-Transporter 2 (SGLT2) Inhibitors. JACC Basic Transl. Sci. 2020, 5, 632–644. [Google Scholar] [CrossRef]
- Shao, Q.; Meng, L.; Lee, S.; Tse, G.; Gong, M.; Zhang, Z.; Zhao, J.; Zhao, Y.; Li, G.; Liu, T. Empagliflozin, a sodium glucose co-transporter-2 inhibitor, alleviates atrial remodeling and improves mitochondrial function in high-fat diet/streptozotocin-induced diabetic rats. Cardiovasc. Diabetol. 2019, 18, 165. [Google Scholar] [CrossRef] [Green Version]
- Ferrannini, E.; Muscelli, E.; Frascerra, S.; Baldi, S.; Mari, A.; Heise, T.; Broedl, U.C.; Woerle, H.-J. Metabolic response to sodium-glucose cotransporter 2 inhibition in type 2 diabetic patients. J. Clin. Investig. 2014, 124, 499–508, Erratum in J. Clin. Investig. 2014, 124, 1868. [Google Scholar] [CrossRef] [Green Version]
- Packer, M. Critical examination of mechanisms underlying the reduction in heart failure events with SGLT2 inhibitors: Identification of a molecular link between their actions to stimulate erythrocytosis and to alleviate cellular stress. Cardiovasc. Res. 2020, 117, 74–84. [Google Scholar] [CrossRef]
- Schönenberger, M.J.; Kovacs, W.J. Hypoxia signaling pathways: Modulators of oxygen-related organelles. Front. Cell. Dev. Biol. 2015, 3, 42. [Google Scholar] [CrossRef]
- Wang, T.; Liu, H.; Lian, G.; Zhang, S.Y.; Wang, X.; Jiang, C. HIF1α-induced glycolysis metabolism is essential to the acti-vation of inflammatory macrophages. Mediat. Inflamm. 2017, 2017, 9029327. [Google Scholar] [CrossRef] [Green Version]
- Murphy, S.P.; Kakkar, R.; McCarthy, C.P.; Januzzi, J.L., Jr. Inflammation in Heart Failure. J. Am. Coll. Cardiol. 2020, 75, 1324–1340. [Google Scholar] [CrossRef] [PubMed]
- Harouki, N.; Nicol, L.; Remy-Jouet, I.; Henry, J.-P.; Dumesnil, A.; Lejeune, A.; Renet, S.; Golding, F.; Djerada, Z.; Wecker, D.; et al. The IL-1β Antibody Gevokizumab Limits Cardiac Remodeling and Coronary Dysfunction in Rats With Heart Failure. JACC Basic Transl. Sci. 2017, 2, 418–430. [Google Scholar] [CrossRef] [PubMed]
- Kobara, M.; Noda, K.; Kitamura, M.; Okamoto, A.; Shiraishi, T.; Toba, H.; Matsubara, H.; Nakata, T. Antibody against interleukin-6 receptor attenuates left ventricular remodelling after myocardial infarction in mice. Cardiovasc. Res. 2010, 87, 424–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maayah, Z.H.; Takahara, S.; Dyck, J.R.B. The beneficial effects of reducing NLRP3 inflammasome activation in the cardiotoxicity and the anti-cancer effects of doxorubicin. Arch. Toxicol. 2020, 95, 1–9. [Google Scholar] [CrossRef]
- Hartman, M.-L.; Shirihai, O.S.; Holbrook, M.; Xu, G.; Kocherla, M.; Shah, A.; Fetterman, J.L.; A Kluge, M.; A Frame, A.; Hamburg, N.M.; et al. Relation of mitochondrial oxygen consumption in peripheral blood mononuclear cells to vascular function in type 2 diabetes mellitus. Vasc. Med. 2014, 19, 67–74. [Google Scholar] [CrossRef] [Green Version]
- Ederlé, C.; Charles, A.L.; Khayath, N.; Poirot, A.; Meyer, A.; Clere-Jehl, R.; Andres, E.; De Blay, F.; Geny, B. Mitochondrial Function in Peripheral Blood Mononuclear Cells (PBMC) Is Enhanced, Together with Increased Reactive Oxygen Species, in Severe Asthmatic Patients in Exacerbation. J. Clin. Med. 2019, 8, 1613. [Google Scholar] [CrossRef] [Green Version]
- Riou, M.; Alfatni, A.; Charles, A.L.; Andrès, E.; Pistea, C.; Charloux, A.; Geny, B. New Insights into the Implication of Mitochondrial Dysfunction in Tissue, Peripheral Blood Mononuclear Cells, and Platelets during Lung Diseases. J. Clin. Med. 2020, 9, 1253. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Study 1 [15]. (Li et al., 2015) | Study 2 [16]. (Shirakawa et al., 2019) | Study 3 [17]. (Zhou B et al., 2020) | |
|---|---|---|---|
| Type | Comparative (vs. controls) | Comparative according to NYHA | Comparative (vs. controls) |
| Population | 25 chronic HFpEF (>50%) asymptomatic patients | 31 chronic HFrEF (<35%) patients | 19 stage-D HFrEF (<35%) patients |
| Mitochondrial respiration | Impaired | Impaired according to NYHA | Impaired |
| Mitochondrial ROSs in PBMC | Decreased antioxidant capacity | Increased | Increased |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sauer, F.; Riou, M.; Charles, A.-L.; Meyer, A.; Andres, E.; Geny, B.; Talha, S. Pathophysiology of Heart Failure: A Role for Peripheral Blood Mononuclear Cells Mitochondrial Dysfunction? J. Clin. Med. 2022, 11, 741. https://doi.org/10.3390/jcm11030741
Sauer F, Riou M, Charles A-L, Meyer A, Andres E, Geny B, Talha S. Pathophysiology of Heart Failure: A Role for Peripheral Blood Mononuclear Cells Mitochondrial Dysfunction? Journal of Clinical Medicine. 2022; 11(3):741. https://doi.org/10.3390/jcm11030741
Chicago/Turabian StyleSauer, François, Marianne Riou, Anne-Laure Charles, Alain Meyer, Emmanuel Andres, Bernard Geny, and Samy Talha. 2022. "Pathophysiology of Heart Failure: A Role for Peripheral Blood Mononuclear Cells Mitochondrial Dysfunction?" Journal of Clinical Medicine 11, no. 3: 741. https://doi.org/10.3390/jcm11030741
APA StyleSauer, F., Riou, M., Charles, A.-L., Meyer, A., Andres, E., Geny, B., & Talha, S. (2022). Pathophysiology of Heart Failure: A Role for Peripheral Blood Mononuclear Cells Mitochondrial Dysfunction? Journal of Clinical Medicine, 11(3), 741. https://doi.org/10.3390/jcm11030741