Abstract
Background: Elexacaftor/tezacaftor/ivacaftor (ETI) is the newest cystic fibrosis transmembrane conductance regulator (CFTR) modulator drug approved for the treatment of patients with cystic fibrosis (pwCF) aged ≥6 years with at least one copy of the F508del mutation (F) in the CFTR gene or another mutation that is responsive to treatment with ETI. This study determined the effectiveness and safety of ETI in a cohort of severely affected pwCF with an F/F genotype. Methods: Retrospective observational study in F/F pwCF treated for 48 weeks, enrolled in an ETI managed access program available to subjects with advanced lung disease (ppFEV1 < 40). Twenty-six patients from three centres were included. The main outcomes included lung function, sweat chloride concentration (SCC), nutrition, frequency of pulmonary exacerbations (PEx), CFQ-R, and safety. Results: ppFEV1 improved by 12.06 (95%CI 8.54, 15.57) from baseline after 4 weeks of treatment with ETI, 15.32 (11.3, 19.34) after 24 weeks, and 14.48 (10.64, 18.32) after 48 weeks. The increase in FEV1 was accompanied by a decrease in SCC, improvement of BMI, and noticeable reduction in PEx. An overall good safety profile was observed. Conclusions: In F/F pwCF with advanced lung disease with an F/F genotype, ETI was safe and associated with clinical improvement.
1. Introduction
The combinations of the cystic fibrosis transmembrane conductance regulator (CFTR) correctors lumacaftor and tezacaftor with the potentiator ivacaftor were the first CFTR modulator drugs approved for the treatment of patients with cystic fibrosis (CF) homozygous for the common F508del mutation. In clinical trials, lumacaftor-ivacaftor and tezacaftor-ivacaftor showed a moderate improvement in mean forced expiratory volume in one second (FEV1 % predicted), improvement in body mass index (BMI), and a reduction in PEx [1,2,3,4,5,6,7]. To further enhance the modulation of F508del-CFTR, it was hypothesised that the addition of a second corrector to a corrector–potentiator combination, acting with a complementary mechanism of action, would be necessary to more fully restore CFTR processing and trafficking. Recent randomised clinical trials showed that treatment with a triple combination of CFTR modulators, two correctors (elexacaftor-tezacaftor), and the potentiator ivacaftor, resulted in quick and sustained improvements of lung function and nutritional status and reduced the rate of PEx patients carrying at least one F508del mutation [8,9,10]. In particular, the RCT involving patients homozygous for the F508del mutation showed these encouraging results comparing the triple combination treatment (TCT) with the double treatment tezacaftor-ivacaftor and not with a placebo [8]. These results have led to the approval of elexacaftor-tezacaftor-ivacaftor (ETI) by the US Food and Drug Administration and the European Medicines Agency for patients with eligible CFTR genotypes and aged 6 years and older.
Patients with advanced pulmonary disease (e.g., percent predicted FEV1 < 40, or under evaluation for lung transplantation) are usually excluded from the Phase III clinical trials, mainly because of comorbidities [11]. No trials have been conducted in pwCF with advanced disease and long-term evaluations are also lacking in this specific subgroup. A managed access program was started in 2019 by Vertex Pharmaceuticals to give the patients homozygous for the F508del mutation and with advanced lung disease the opportunity to receive the medication free of charge whilst awaiting national reimbursed commercialisation. The aim of this study was to describe the real-world safety and effectiveness over 48 weeks of ETI treatment in a cohort of F/F pwCF.
2. Materials and Methods
2.1. Study Design and Outcome Measures
Three Italian CF centres participated in this retrospective observational study recruiting 26 subjects, enrolled in an ETI managed access program between October 2019 and July 2020.
A core set of clinical assessments were conducted at weeks 4, 12, 24, and 48: lung function, SCC, and BMI. The main outcome measure was absolute change from baseline in percentage of predicted FEV1 (ppFEV1). The secondary outcomes were the absolute change from baseline in SCC and in BMI (the weight in kilograms divided by the square of the height in meters) and the number of PEx through the 48 weeks of treatment. The Cystic Fibrosis Questionnaire–Revised (CFQ-R) was administered at weeks 4, 24, and 48, and the absolute change from baseline in the respiratory domain score (range, 0 to 100, with higher scores indicating a higher patient-reported quality of life with regard to respiratory symptoms; minimum clinically important difference, 4 points) as compared. Further end points included safety and side-effect issues.
2.2. Study Population
Criteria for entering the managed access program included homozygosity for the F508del mutation, together with treatment for at least 6 months with lumacaftor-ivacaftor (LI) or with tezacaftor-ivacaftor, and with severe lung disease (FEV1 < 30% predicted, or FEV1 < 40% predicted but rapidly declining and/or with ≥6 PEx in the last 12 months) or with the condition of discontinued treatment because of adverse events or drug–drug interactions. Key exclusion criteria included mechanical ventilation, severe liver disease with or without hepatic impairment, history of solid organ or haematological transplantation, history of drug or alcohol abuse in the past year, pregnancy, and sexually active patients of reproductive potential who are not willing to use appropriate contraception methods. Treatment was administered orally according to the manufacturer’s recommendations (200 mg ELX/100 mg TEZ/150 mg IVA in the morning and 150 mg IVA in the evening). Maintenance medications at baseline were continued throughout the study. The COVID-19 pandemic did not affect this study. The scheduled encounters were regularly performed, and no missing data was present.
2.3. Methods
Clinical and laboratory data (aspartate aminotransferase, alanine aminotransferase, bilirubin, and creatine-phosphokinase) were collected at baseline and at weeks 4, 12, 24, and 48 following the initiation of ETI treatment. Data of PEx and use of antibiotics were recorded for 48 weeks before initiating ETI. PEx was defined as previously described [12].
To minimise the likelihood that baseline data would be affected by inter-current clinical events, the best-recorded values during the 3 months before initiating the treatment were defined as baseline.
Vertex Pharmaceuticals played no role in the conceptualisation of this study, in the formal analysis of data, or in the writing and submission of this manuscript.
The study protocol complied with the revised Declaration of Helsinki, and all the recruited subjects provided written informed consent.
2.4. Analysis
Results are presented as means and standard deviations (SDs), means and 95% confidence interval (CI), or medians and range, as appropriate. Data were compared using the Wilcoxon signed rank test, paired t-test, or Fisher’s test as appropriate. p-values less than 0.05 were considered significant.
3. Results
Twenty-six subjects (12 females, 46.2%) were recruited. The median (range) age was 31.1 years (20.8–48.3 years). All patients included in this report had pancreatic insufficiency; CF-related diabetes (CFRD) affected 11 (42.3%) patients. The baseline demographics and characteristics of the study population are described in Table 1. All but one of the patients were on treatment with L/I. One subject had discontinued L/I because of adverse events. No patient was on the lung transplantation waiting list.

Table 1.
Baseline demographics.
3.1. Changes Following ETI Treatment
3.1.1. Lung Function
The mean (SD) value of the ppFEV1 was 29.9 (8.4) at baseline, and it quickly increased to 41.9 (11.5) after 4 weeks of treatment with ETI. The trend of improvement continued through to week 48 (Figure 1).
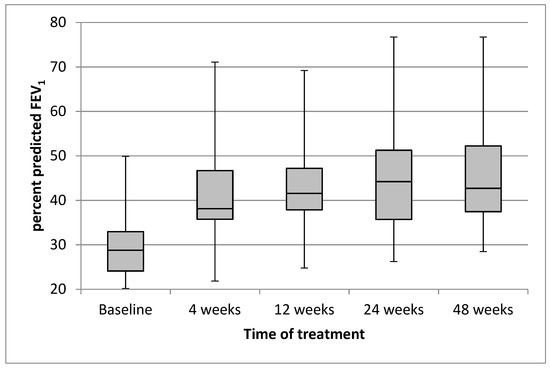
Figure 1.
Effect of treatment with elexacaftor/tezacaftor/ivacaftor on percentage of predicted FEV1 from baseline through to week 48. Each box and whisker plot shows the position of the minimum, lower quartile, median, upper quartile, and maximum of the data, at baseline and at different times of treatment.
The mean (95% CI) absolute improvement in ppFEV1 was 12.06 (8.54, 15.57; p < 0.0001) after 4 weeks of treatment with ETI, 13.22 (9.47; 16.98; p < 0.0001) after 12 weeks, 15.32 (11.3, 19.34; p < 0.0001) after 24 weeks, and 14.48 (10.64, 18.32; p < 0.0001) after 48 weeks. The changes following the treatment of ppFVC had a similar trend to those of ppFEV1 (Table 2).
Table 2.
Absolute change of lung function after treatment with ETI over the 48-week period.
All subjects showed individual positive responses with respect to ppFEV1. Seventeen (65.4%) out of twenty-six subjects showed improvements of ppFEV1 greater than 10, and six (23.1%) had increases higher than 20.
3.1.2. BMI
Mean (SD) BMI at baseline was 20.9 (2.16) kg/m2. BMI was normal (18.5–24.9 kg/m2) in 21 subjects (80.8%), and 5 subjects (19.2%) were underweight (BMI < 18.5 kg/m2). Mean (SD) BMI increased to 21.1 (3.1) after 4 weeks of ETI and improved further through to week 48, with a mean (SD) final BMI of 23.0 (2.2).
The mean (95% CI) absolute increase in BMI was 0.66 (0.37, 0.95; p < 0.0001) after 4 weeks of treatment with ETI, 1.57 (1.19, 1.94; p < 0.0001) after 12 weeks, 2.02 (1.56, 2.48; p < 0.0001) after 24 weeks, and 2.08 (1.63, 2.52; p < 0.0001) after 48 weeks of treatment (Table 3). After 24 weeks of treatment, all the underweight subjects had improved their BMI to the normal range, and three further patients gained overweight.
Table 3.
Absolute changes in sweat chloride and BMI over the 48-week treatment period.
3.1.3. Sweat Chloride
The overall mean (SD) SC concentration was 77.5 (35.3) mmol/L at baseline, and it decreased to 35.9 (20.1) mmol/L after 4 weeks of ETI (p < 0.0001) and improved further through to week 48, with a mean (SD) SC concentration decrease to 29.2 (19.5) mmol/L (Table 3).
3.1.4. Rate of Pulmonary Exacerbations
During the 48 weeks prior to starting ETI, 26 subjects suffered from 105 instances of PEx, of which 42 were treated with intravenous antibiotics (IV). Treatment with ETI for 48 weeks resulted in a 97% lower rate of PEx. Twenty-two subjects (84.6%) remained free of PEx during the period of the study, and only one patient needed IV antibiotics for a single cycle.
3.1.5. CFQ-R Respiratory Domain
The CFQ-R respiratory domain score improved significantly throughout the study period. The median (range) score was 55.5 (27.8, 88.9) at baseline, which improved significantly to 88.9 (66.7, 100) after 4 weeks (p < 0.00001), to 94.4 (61.1, 100) after 24 weeks, and was stable at 94.4 (77.8, 100) until 48 weeks of treatment (p < 0.00001). The mean (95% CI) improvements compared to baseline values were 28.4 (19.9 to 36.9), 29.1 (20.1.9 to 38.1), and 32.6 (24.6 to 40.1) points after 4, 24, and 48 weeks of treatment, respectively.
3.1.6. Treatment-Related Adverse Events
No interruption of the treatment was needed, and no patients withdrew from the medication. No ETI-related adverse events (i.e., skin rash, testicular pain, arterial hypertension, headache, anxiety, and depression) were reported. No relevant abnormal results of serum chemistry, haematology, coagulation studies, liver function tests, and urinalysis attributable to ETI treatment were reported. No deaths occurred during the study period.
4. Discussion
TCT with ETI significantly improved pulmonary function and CFQ-R respiratory domain over 48 weeks in 26 pwCF homozygous for the F508del CFTR mutation and advanced lung disease. Moreover, the TCT resulted in a significant reduction in the annual rate of PEx and in the need for antibiotic therapy. Finally, ETI treatment also resulted in improvements of extra-pulmonary outcomes (BMI and SCC).
The pivotal phase III RCT included F/F pwCF, and moderate lung disease showed the clinical efficacy of a 4-week treatment of ETI, resulting in an improved ppFEV1 (10.0%) and CFQ-R respiratory domain (17.4 points) and an SCC concentration that was 45.1 mmol/L lower compared with an active comparator (tezacaftor plus ivacaftor alone) [8]. Although the RCT excluded patients with more advanced (ppFEV1 < 40%) pulmonary disease at the time of screening, a subgroup of 10 enrolled patients had ppFEV1 < 40% on day 1 of the study, but no result is reported about this little subgroup. A further phase IIIB RCT including F/F pwCF was conducted for 24 weeks, with a fast improvement of ppFEV1 (11.2) after 4 weeks of treatment with ETI that sustained through to 24 weeks [13]. Our patients, all but one treated with L/I before beginning TCT, showed a similar increase in pulmonary function in the first 4 weeks of ETI treatment (mean improvement of ppFEV1 12.1) that was sustained through to 24 and 48 weeks (mean improvements of ppFEV1 15.3 and 14.5, respectively).
Several reports from different countries have shown the early effects of ETI in F/F pwCF and severe pulmonary disease. In a single-centre open-label cohort study conducted in Ireland, ppFEV1, BMI, and PEx frequency improved among 14 patients (8 were homozygous for F508del and were previously on CFTR modulator therapy) with ppFEV1 < 40 who were treated with ETI, from a mean ppFEV1 of 27 to 36 [14]. A large study reported the experience of all French CF centres, with a cohort of 245 patients, with 100 F/F. A subgroup of these F/F subjects, previously treated with a CFTR modulator, demonstrated a mean improvement in ppFEV1 of 11 and 13 after TCT for one and three months, respectively [15]. A short-term retrospective study performed across three large academic CF centres in the United States (US) showed a mean increase in ppFEV1 of 7.1 in a group of 32 F/F subjects after a mean treatment of 39 days [16]. Another retrospective observational study from the US over one year of ETI administration in a mixed population (F/MF, F/F, F/G, F/RF) of 22 adult patients with ppFEV1 < 40, showed a mean improvement of ppFEV1 5.5 after 1 month and of 7.6 after 12 months [17].
Finally, a recent prospective, large observational study in 487 pwCF aged ≥ 12 years with ≥1 F508del allele from the US showed that ETI treatment resulted in large improvements in lung function, respiratory symptoms, and BMI. In particular, 209 F/F subjects were present in the study cohort who were previously treated with L/I or T/I. This F/F subgroup had a mean (SD) ppFEV1 of 81.1 (22.5) when starting the treatment with ETI, and showed a mean increase in ppFEV1 of 7.96 after 1 month, 8.63 after 3 months, and 9.21 after 6 months [18].
Our results agree with all these reports with the follow-up period extended over 48 weeks, much longer than previously reported, confirming that the improvements are as clinically significant as sustained (Table 4).
Table 4.
Comparison of the main results of this study with previous reports.
Another relevant aspect is the distribution of individual positive responses of ppFEV1 following the ETI treatment. About 81% of patients had an increase in ppFEV1 of ≥ 5 (and 65.4% ≥ 10) and only 19% of patients had an increase in ppFEV1 of ≤ 5. This result is comparable to that observed in the French cohort, where an increase in ppFEV1 of ≥ 5 was present in 91% of patients and ≥ 10 in 69% [19], and this is better than in the US academic study, where at least 5% improvements of FEV1 occurred in 64% of subjects [16].
The objective increase in lung function of our patients treated with ETI was also reflected by the subjective evaluation obtained by the CFQ-R respiratory domain score. The mean increase observed after 4 weeks of treatment was 28.4 points, greater than that in the pivotal RCT (16.0 points) [8]. On the other hand, the mean score at baseline (60.4) of subjects in this study was lower than that in the pivotal trial (70.6 to 72.6), showing more severe pulmonary disease. The improvement of the score was sustained during the 48 weeks of follow-up, showing the stabilisation of the lung disease.
The extra pulmonary outcomes (decrease in SCC and BMI gain) in this study were also relevant. The mean SCC decrease observed after 4 weeks of treatment was −32.6 mmol/L, confirming the recovery of CFTR activity. The difference is lower than that observed in the pivotal trial (−43.4), but the mean (SD) baseline SCC in our study was lower (77.5 mmol/L (35.3) vs. 90.0 (12.3) and 91.4 (11.0)). The lower baseline SCC in our patients could be due to the prolonged treatment with L/I. Indeed, it is more similar to the mean (SD) baseline SCC (85.6 (15.2)) of the F/F subjects recruited in the US observational study, treated with two-drug modulators before the switch to the TCT [18]. Changes in SCC are considered to provide a direct indicator of systemic CFTR function, and this data has been confirmed in parallel measurements of CFTR function in airway and intestinal epithelia in patients with CF and one or two F508del alleles [20].
An important endpoint in this study was the effect of the ETI treatment on healthcare utilisation. The number of PEx dramatically decreased by 97% during the 48 weeks of treatment compared with the year prior to the commencement of ETI. About 85% of the patients were free of PEx after initiating ETI, and only one patient needed a single course of IV antibiotic therapy. These results are comparable to those of the open label extension study of the pivotal trial [10] but need to be cautiously interpreted. Indeed, the study period happened at the same time as limitation of contacts during the COVID-19 lockdown measures were imposed. Real-world data from multiple case records with long follow-up periods are mandatory to confirm this trend. These data, combined with clinical trial results, will help to understand the changing healthcare needs of people with CF [21].
The improved use of body energy is proved by the BMI gain. At week 4, treatment with ETI resulted in a mean increase in BMI of 0.66 kg/m2 (95% CI 0.37 to 0.95; p < 0.0001), similar to that in the phase III trial. The BMI gain showed gradual increase during the follow up, with a mean increase of 2.08 (95% CI 1.63 to 2.52) after 48 weeks of treatment. At this time, no subject was malnourished (they were 19.2% at the starting point), and 15% of the patients were overweight. A similar increase in BMI after using CFTR modulators in severely affected pwCF was described in patients with the F/MF genotype [22] and in patients with mutations determining CFTR residual function [23]. Although over the last decades an emphasis has been on promoting BMI gain to optimise pulmonary outcomes, the risk of overweightness/obesity in an adult population should be monitored in the long-term [24,25,26].
About the safety of ETI treatment, over the 48 weeks of treatment, no interruption of the treatment was needed, and no patients withdrew from the medication. The overall safety data of this study was consistent with clinical trials and real-world findings. In our small sample of pwCF over a 48-week period, ETI treatment was not associated with new safety concerns. In any case, the collection of safety data over longer periods of treatment is essential to strengthen these observations.
Real-world evidence data about the effectiveness and safety of new drugs in pwCF with advanced lung disease, who are at high risk of comorbidities and of rapid worsening in pulmonary function and quality of life, are very important because these subgroups of patients are usually not eligible for clinical trials [11,27].
This study had the limitations of a retrospective design, including a small number of patients and the lack of a control group. We strove to work out these problems by performing within-group comparisons. Indeed, this was a descriptive, rather than analytical, study. Moreover, quantitative microbiological data and imaging are lacking in this study. There is evidence that pwCF with CFTR gating mutations were less likely to eradicate bacteria colonising their airways, even when treated with the CFTR potentiator ivacaftor [28,29]. L/I decreased the total bacterial load and increased the diversity of the airway microbiome in F508del homozygous patients [30]. ETI treatment has showed to confer additional benefits relative to previous CFTR modulators in patients with F508del homozygosity [8], F508del-gating or F508del-residual function genotypes [31]. To evaluate the microbiological effect of more effective CFTR rescue, longer observation times are needed.
Longer follow-up periods are required to validate the observed improvements with respect to clinical stability, slowing down or stopping the decline in pulmonary function, reducing the incidence of complications, and improving the survival [32].
5. Conclusions
Treatment with ETI was shown to be effective and safe in homozygous F/F pwCF with advanced lung disease. Further studies are needed to define whether the slowing down or reversion of severe pulmonary disease could be obtained in pwCF, and whether more people with severe CF will gain life-changing benefits from CFTR modulation.
Author Contributions
Conceptualisation, V.C., P.I., V.T. and D.S.; data curation, P.I., C.C., A.P. and M.F.; formal analysis, V.C., V.T. and D.S.; investigation, C.C., L.F., S.B., A.C., L.S. and G.M.; methodology, A.C. and D.S.; project administration, M.D.; resources, N.F.; supervision, N.F.; validation, V.T., A.P. and M.F.; visualisation, L.F. and G.T.; writing—original draft, V.C. and D.S.; writing—review and editing, V.C., P.I., G.T. and D.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
The study was conducted according to the guidelines of the Declaration of Helsinki, and the collection of data for each patient was an integral part of the approval by each local Ethics Committee of the compassionate use program.
Informed Consent Statement
Written informed consent was obtained from all subjects involved in the study.
Conflicts of Interest
D.S. has been principal investigator in several clinical trials conducted by Vertex Pharmaceuticals and for which his institution has received funding; he has received fees for participation in advisory boards, satellite symposia, or specific projects. V.C. and PI have received fees for participation in advisory boards and consultancy from Vertex Pharmaceuticals. C.C., G.M. and M.D. report supported attendance at meetings and travel from Vertex Pharmaceuticals. V.T., L.F., M.F., A.P., S.B., A.C., L.S. and N.F. have nothing to disclose.
References
- Mall, M.A.; Mayer-Hamblett, N.; Rowe, S.M. Cystic Fibrosis: Emergence of Highly Effective Targeted Therapeutics and Potential Clinical Implications. Am. J. Respir. Crit. Care Med. 2020, 201, 1193–1208. [Google Scholar] [CrossRef] [PubMed]
- Konstan, M.W.; McKone, E.F.; Moss, R.B.; Marigowda, G.; Tian, S.; Waltz, D.; Huang, X.; Lubarsky, B.; Rubin, J.; Millar, S.J.; et al. Assessment of safety and efficacy of long-term treatment with combination lumacaftor and ivacaftor therapy in patients with cystic fibrosis homozygous for the F508del-CFTR mutation (PROGRESS): A phase 3, extension study. Lancet Respir. Med. 2017, 5, 107–118. [Google Scholar] [CrossRef]
- Wainwright, C.E.; Elborn, J.S.; Ramsey, B.W.; Marigowda, G.; Huang, X.; Cipolli, M.; Colombo, C.; Davies, J.C.; de Boeck, K.; Flume, P.A.; et al. Lumacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for F508del CFTR. N. Engl. J. Med. 2015, 373, 220–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor-Cousar, J.L.; Jain, M.; Barto, T.L.; Haddad, T.; Atkinson, J.; Tian, S.; Tang, R.; Marigowda, G.; Waltz, D.; Pilewski, J.; et al. Lumacaftor/ivacaftor in patients with cystic fibrosis and advanced lung disease homozygous for F508del-CFTR. J. Cyst. Fibros. 2018, 17, 228–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McColley, S.A.; Konstan, M.W.; Ramsey, B.W.; Stuart Elborn, J.; Boyle, M.P.; Wainwright, C.E.; Waltz, D.; Vera Llonch, M.; Marigowda, G.; Jiang, J.G.; et al. Lumacaftor/Ivacaftor reduces pulmonary exacerbations in patients irrespective of initial changes in FEV1. J. Cyst. Fibros. 2019, 18, 94–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flume, P.A.; Biner, R.F.; Downey, D.G.; Brown, C.; Jain, M.; Fischer, R.; De Boeck, K.; Sawicki, G.S.; Chang, P.; Paz-Diaz, H.; et al. Long-term safety and efficacy of tezacaftor-ivacaftor in individuals with cystic fibrosis aged 12 years or older who are homozygous or heterozygous for F508del CFTR (EXTEND): An open-label extension study. Lancet Respir. Med. 2021, 9, 733–746. [Google Scholar] [CrossRef]
- Taylor-Cousar, J.L.; Munck, A.; McKone, E.F.; van der Ent, C.K.; Moeller, A.; Simard, C.; Wang, L.T.; Ingenito, E.P.; McKee, C.; Lu, Y.; et al. Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for F508del. N. Engl. J. Med. 2017, 377, 2013–2023. [Google Scholar] [CrossRef]
- Heijerman, H.G.M.; McKone, E.F.; Downey, D.G.; Van Braeckel, E.; Rowe, S.M.; Tullis, E.; Mall, M.A.; Welter, J.J.; Ramsey, B.W.; McKee, C.M.; et al. Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: A double-blind, randomised, phase 3 trial. Lancet 2019, 394, 1940–1948, Erratum in Lancet 2020, 395, 1694. [Google Scholar] [CrossRef]
- Middleton, P.G.; Mall, M.A.; Drevinek, P.; Lands, L.C.; McKone, E.F.; Polineni, D.; Ramsey, B.W.; Taylor-Cousar, J.L.; Tullis, E.; Vermeulen, F.; et al. Elexacaftor-Tezacaftor-Ivacaftor for Cystic Fibrosis with a Single F508del Allele. N. Engl. J. Med. 2019, 381, 1809–1819. [Google Scholar] [CrossRef]
- Griese, M.; Costa, S.; Linnemann, R.W.; Mall, M.A.; McKone, E.F.; Polineni, D.; Quon, B.S.; Ringshausen, F.C.; Taylor-Cousar, J.L.; Withers, N.J.; et al. Safety and Efficacy of Elexacaftor/Tezacaftor/Ivacaftor for 24 Weeks or Longer in People with Cystic Fibrosis and One or More F508del Alleles: Interim Results of an Open-Label Phase 3 Clinical Trial. Am. J. Respir. Crit. Care Med. 2021, 203, 381–385. [Google Scholar] [CrossRef]
- Ramos, K.J.; Pilewski, J.M.; Taylor-Cousar, J.L. Challenges in the use of highly effective modulator treatment for cystic fibrosis. J. Cyst. Fibros. 2021, 20, 381–387. [Google Scholar] [CrossRef] [PubMed]
- Davies, J.C.; Wainwright, C.E.; Canny, G.J.; Chilvers, M.A.; Howenstine, M.S.; Munck, A.; Mainz, J.G.; Rodriguez, S.; Li, H.; Yen, K.; et al. Efficacy and safety of ivacaftor in patients aged 6 to 11 years with cystic fibrosis with a G551D mutation. Am. J. Respir. Crit. Care Med. 2013, 187, 1219–1225. [Google Scholar] [CrossRef] [Green Version]
- Sutharsan, S.; McKone, E.F.; Downey, D.G.; Duckers, J.; MacGregor, G.; Tullis, E.; Van Braeckel, E.; Wainwright, C.E.; Watson, D.; Ahluwalia, N.; et al. Efficacy and safety of elexacaftor plus tezacaftor plus ivacaftor versus tezacaftor plus ivacaftor in people with cystic fibrosis homozygous for F508del-CFTR: A 24-week, multicentre, randomised, double-blind, active-controlled, phase 3b trial. Lancet Respir. Med. 2021. [Google Scholar] [CrossRef]
- O’Shea, K.M.; O’Carroll, O.M.; Carroll, C.; Grogan, B.; Connolly, A.; O’Shaughnessy, L.; Nicholson, T.T.; Gallagher, C.G.; McKone, E.F. Efficacy of elexacaftor/tezacaftor/ivacaftor in patients with cystic fibrosis and advanced lung disease. Eur. Respir. J. 2021, 57, 2003079. [Google Scholar] [CrossRef] [PubMed]
- Burgel, P.R.; Durieu, I.; Chiron, R.; Ramel, S.; Danner-Boucher, I.; Prevotat, A.; Grenet, D.; Marguet, C.; Reynaud-Gaubert, M.; Macey, J.; et al. Rapid Improvement after Starting Elexacaftor-Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis and Advanced Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2021, 204, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Bermingham, B.; Rueschhoff, A.; Ratti, G.; Nesmith, A.; Goodwin, D.; Gray, S.; Flume, P.; Solomon, G.M.; Cohen, L.; Garcia, B. Short-term effect of elexacaftor-tezacaftor-ivacaftor on lung functions and transplant planning in cystic fibrosis patients with advanced lung disease. J. Cyst. Fibros. 2021, 20, 768–771. [Google Scholar] [CrossRef]
- Djavid, A.R.; Thompson, A.E.; Irace, A.L.; Gusman, E.; Altman, K.; DiMango, E.A.; Keating, C.L. Efficacy of Elexacaftor/Tezacaftor/Ivacaftor in Advanced Cystic Fibrosis Lung Disease. Ann. Am. Thorac. Soc. 2021, 18, 1924–1927. [Google Scholar] [CrossRef]
- Nichols, D.P.; Paynter, A.C.; Heltshe, S.L.; Donaldson, S.H.; Frederick, C.A.; Freedman, S.D.; Gelfond, D.; Hoffman, L.R.; Kelly, A.; Narkewicz, M.R.; et al. Clinical Effectiveness of Elexacaftor/Tezacftor/Ivacaftor in People with Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 2021. [Google Scholar] [CrossRef]
- Burgel, P.R.; Da Silva, J.; Paillasseur, J.L.; Martin, C. Optimism with Caution: Elexacaftor-Tezacaftor-Ivacaftor in Patients with Advanced Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2021, 204, 372–374. [Google Scholar] [CrossRef]
- Graeber, S.Y.; Vitzthum, C.; Pallenberg, S.T.; Naehrlich, L.; Stahl, M.; Rohrbach, A.; Drescher, M.; Minso, R.; Ringshausen, F.C.; Rueckes-Nilges, C.; et al. Effects of Elexacaftor/Tezacaftor/Ivacaftor Therapy on CFTR Function in Patients with Cystic Fibrosis and One or Two F508del Alleles. Am. J. Respir. Crit. Care Med. 2021. [Google Scholar] [CrossRef]
- Keogh, R.H.; Cosgriff, R.; Andrinopoulou, E.R.; Brownlee, K.G.; Carr, S.B.; Diaz-Ordaz, K.; Granger, E.; Jewell, N.P.; Lewin, A.; Leyrat, C.; et al. Projecting the impact of triple CFTR modulator therapy on intravenous antibiotic requirements in cystic fibrosis using patient registry data combined with treatment effects from randomised trials. Thorax 2021. [Google Scholar] [CrossRef]
- Carnovale, V.; Iacotucci, P.; Terlizzi, V.; Colangelo, C.; Medio, P.; Ferrillo, L.; De Gregorio, F.; Francalanci, M.; Taccetti, G.; Buonaurio, S.; et al. Effectiveness and safety of elexacaftor/tezacaftor/ivacaftor in patients with cystic fibrosis and advanced lung disease with the F508del/minimal function genotype. Respir. Med. 2021, 189, 106646. [Google Scholar] [CrossRef] [PubMed]
- Salvatore, D.; Terlizzi, V.; Francalanci, M.; Taccetti, G.; Messore, B.; Biglia, C.; Pisi, G.; Calderazzo, M.A.; Caloiero, M.; Pizzamiglio, G.; et al. Ivacaftor improves lung disease in patients with advanced CF carrying CFTR mutations that confer residual function. Respir. Med. 2020, 171, 106073. [Google Scholar] [CrossRef] [PubMed]
- Bonhoure, A.; Boudreau, V.; Litvin, M.; Colomba, J.; Bergeron, C.; Mailhot, M.; Tremblay, F.; Lavoie, A.; Rabasa-Lhoret, R. Overweight, obesity and significant weight gain in adult patients with cystic fibrosis association with lung function and cardiometabolic risk factors. Clin. Nutr. 2020, 39, 2910–2916. [Google Scholar] [CrossRef] [PubMed]
- Gramegna, A.; Aliberti, S.; Contarini, M.; Savi, D.; Sotgiu, G.; Majo, F.; Saderi, L.; Lucidi, V.; Amati, F.; Pappalettera, M.; et al. Overweight and obesity in adults with cystic fibrosis: An Italian multicenter cohort study. J. Cyst. Fibros. 2021, 21, 111–114. [Google Scholar] [CrossRef] [PubMed]
- Gabel, M.E.; Fox, C.K.; Grimes, R.; Lowman, J.D.; McDonald, C.M.; Stallings, V.A.; Michel, S.H. Overweight and Cystic Fibrosis: An Unexpected Challenge. Pediatr. Pulmonol. 2021. [Google Scholar] [CrossRef]
- Shteinberg, M.; Taylor-Cousar, J.L. Impact of CFTR modulator use on outcomes in people with severe cystic fibrosis lung disease. Eur. Respir. Rev. 2020, 29, 190112. [Google Scholar] [CrossRef] [Green Version]
- Hisert, K.B.; Heltshe, S.L.; Pope, C.; Jorth, P.; Wu, X.; Edwards, R.M.; Radey, M.; Accurso, F.J.; Wolter, D.J.; Cooke, G.; et al. Restoring cystic fibrosis transmembrane conductance regulator function reduces airway bacteria and inflammation in people with cystic fibrosis and chronic lung infections. Am. J. Respir. Crit. Care Med. 2017, 195, 1617–1628. [Google Scholar] [CrossRef]
- Harris, J.K.; Wagner, B.D.; Zemanick, E.T.; Robertson, C.E.; Stevens, M.J.; Heltshe, S.L.; Rowe, S.M.; Sagel, S.D. Changes in Airway Microbiome and Inflammation with Ivacaftor Treatment in Patients with Cystic Fibrosis and the G551D Mutation. Ann. Am. Thorac. Soc. 2020, 17, 212–220. [Google Scholar] [CrossRef]
- Graeber, S.Y.; Boutin, S.; Wielpütz, M.O.; Joachim, C.; Frey, D.L.; Wege, S.; Sommerburg, O.; Kauczor, H.U.; Stahl, M.; Dalpke, A.H.; et al. Effects of Lumacaftor-Ivacaftor on Lung Clearance Index, Magnetic Resonance Imaging, and Airway Microbiome in F508del Homozygous Patients with Cystic Fibrosis. Ann. Am. Thorac. Soc. 2021, 18, 971–980. [Google Scholar] [CrossRef]
- Barry, P.J.; Mall, M.A.; Álvarez, A.; Colombo, C.; de Winter-de Groot, K.M.; Fajac, I.; McBennett, K.A.; McKone, E.F.; Ramsey, B.W.; Sutharsan, S.; et al. Triple Therapy for Cystic Fibrosis F508del-Gating and -Residual Function Genotypes. N. Engl. J. Med. 2021, 385, 815–825. [Google Scholar] [CrossRef] [PubMed]
- Stanojevic, S.; Vukovojac, K.; Sykes, J.; Ratjen, F.; Tullis, E.; Stephenson, A.L. Projecting the impact of delayed access to elexacaftor/tezacaftor/ivacaftor for people with cystic fibrosis. J. Cyst. Fibros. 2021, 20, 243–249. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).