
Acid Sphingomyelinase Deficiency: Sharing Experience of Disease Monitoring and Severity in France

 , ,
, ,
Abstract
:1. Introduction
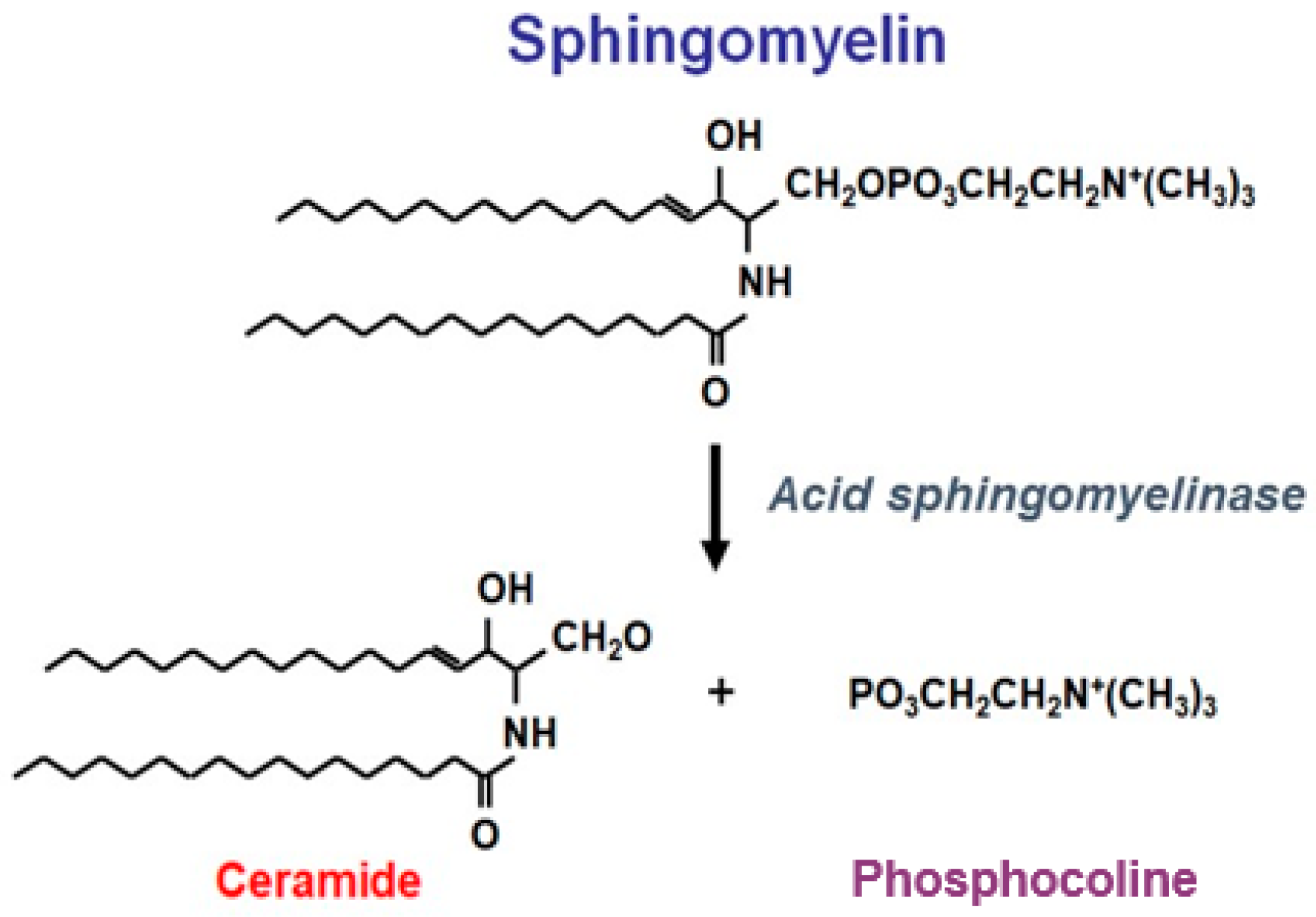
2. Clinical Study Evidence for the Use of Olipudase Alfa in the Non-Neurologic Manifestations of ASMD
3. Materials and Methods
4. French Expert Opinion: Monitoring of ASMD and Patient Severity Stratification
4.1. General Care of Patients with ASMD Type B Forms
4.2. Monitoring of Patients with ASMD Type B Forms
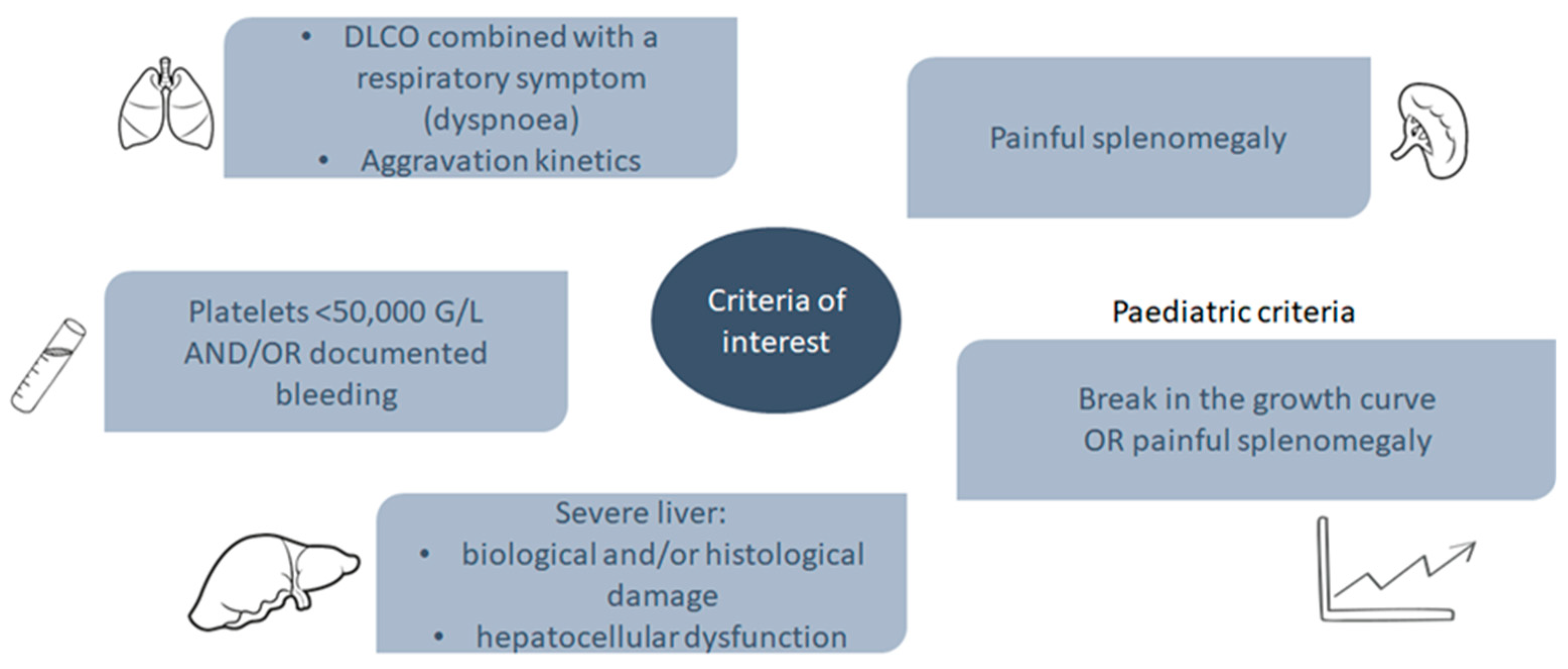
4.3. Patients with ASMD: Stratification by Disease Severity
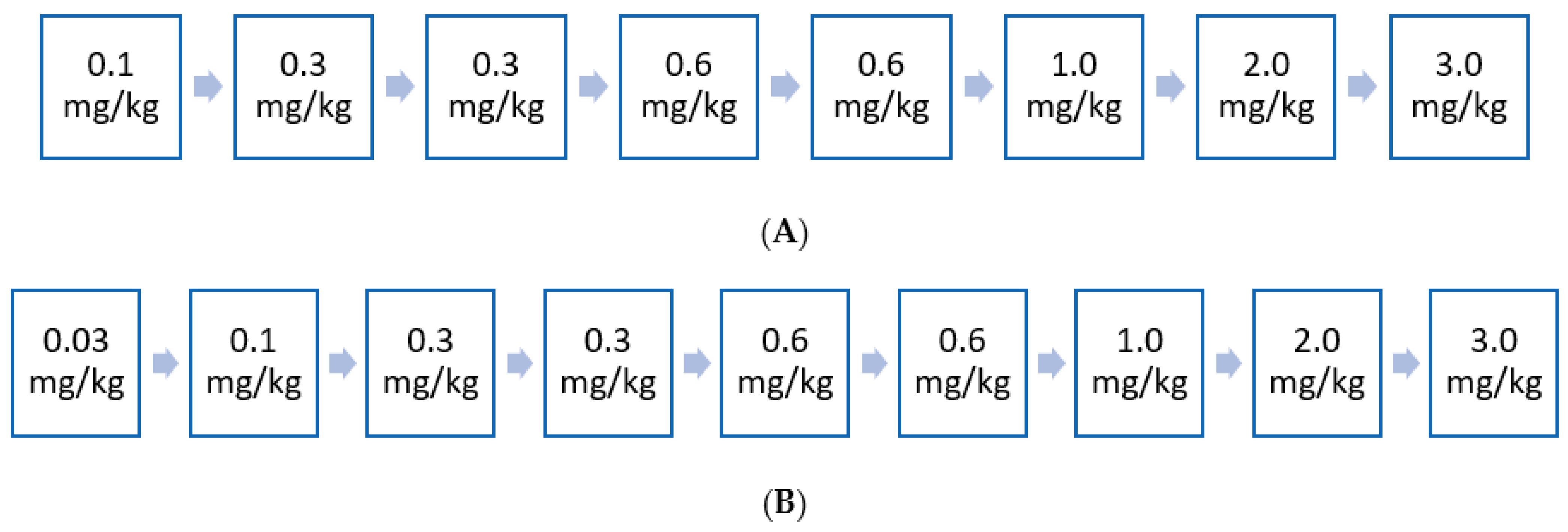
5. Early Access Experience to ERT in France
6. Discussion
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- McGovern, M.M.; Wasserstein, M.P.; Bembi, B.; Giugliani, R.; Mengel, K.E.; Vanier, M.T.; Zhang, Q.; Peterschmitt, M.J. Prospective study of the natural history of chronic acid sphingomyelinase deficiency in children and adults: Eleven years of observation. Orphanet J. Rare Dis. 2021, 16, 212. [Google Scholar] [CrossRef] [PubMed]
- Mathias, S.; Dressler, K.A.; Kolesnick, R.N. Characterization of a ceramide-activated protein kinase: Stimulation by tumor necrosis factor alpha. Proc. Natl. Acad. Sci. USA 1991, 88, 10009–10013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albeituni, S.; Stiban, J. Roles of ceramides and other sphingolipids in immune cell function and inflammation. Adv. Exp. Med. Biol. 2019, 1161, 169–191. [Google Scholar] [PubMed]
- Schütze, S.; Potthoff, K.; Machleidt, T.; Berkovic, D.; Wiegmann, K.; Krönke, M. TNF activates NF-kappa B by phosphatidylcholine-specific phospholipase C-induced “acidic” sphingomyelin breakdown. Cell 1992, 71, 765–776. [Google Scholar] [CrossRef]
- Wasserstein, M.P.; Desnick, R.J.; Schuchman, E.H.; Hossain, S.; Wallenstein, S.; Lamm, C.; McGovern, M.M. The natural history of type B Niemann-Pick disease: Results from a 10-year longitudinal study. Pediatrics 2004, 114, e672–e677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGovern, M.M.; Aron, A.; Brodie, S.E.; Desnick, R.J.; Wasserstein, M.P. Natural history of Type A Niemann-Pick disease: Possible endpoints for therapeutic trials. Neurology 2006, 66, 228–232. [Google Scholar] [CrossRef]
- McGovern, M.M.; Dionisi-Vici, C.; Giugliani, R.; Hwu, P.; Lidove, O.; Lukacs, Z.; Mengel, K.E.; Mistry, P.K.; Schuchman, E.H.; Wasserstein, M.P. Consensus recommendation for a diagnostic guideline for acid sphingomyelinase deficiency. Genet. Med. 2017, 19, 967–974. [Google Scholar] [CrossRef] [Green Version]
- Deodato, F.; Boenzi, S.; Taurisano, R.; Semeraro, M.; Sacchetti, E.; Carrozzo, R.; Dionisi-Vici, C. The impact of biomarkers analysis in the diagnosis of Niemann-Pick C disease and acid sphingomyelinase deficiency. Clin. Chim. Acta 2018, 486, 387–394. [Google Scholar] [CrossRef]
- McGovern, M.M.; Avetisyan, R.; Sanson, B.J.; Lidove, O. Disease manifestations and burden of illness in patients with acid sphingomyelinase deficiency (ASMD). Orphanet J. Rare Dis. 2017, 12, 41. [Google Scholar] [CrossRef]
- Kingma, S.D.; Bodamer, O.A.; Wijburg, F.A. Epidemiology and diagnosis of lysosomal storage disorders; challenges of screening. Best Pract. Res. Clin. Endocrinol. Metab. 2015, 29, 145–157. [Google Scholar] [CrossRef]
- Lidove, O.; Belmatoug, N.; Froissart, R.; Lavigne, C.; Durieu, I.; Mazodier, K.; Serratrice, C.; Douillard, C.; Goizet, C.; Cathebras, P.; et al. Acid sphingomyelinase deficiency (Niemann-Pick B disease) in adulthood: A retrospective multicentre study of 28 adult patients. Rev. Med. Interne 2017, 38, 291–299. [Google Scholar] [CrossRef]
- Cassiman, D.; Packman, S.; Bembi, B.; Ben Turkia, H.; Al-Sayed, M.; Schiff, M.; Imrie, J.; Mabe, P.; Takahashi, T.; Mengel, K.E.; et al. Cause of death in patients with chronic visceral and chronic neurovisceral acid sphingomyelinase deficiency (Niemann-Pick disease type B and B variant): Literature review and report of new cases. Mol. Genet. Metab. 2016, 118, 206–213. [Google Scholar] [CrossRef]
- Borie, R.; Crestani, B.; Guyard, A.; Lidove, O. Interstitial lung disease in lysosomal storage disorders. Eur. Respir. Rev. 2021, 30, 200363. [Google Scholar] [CrossRef]
- McGovern, M.M.; Wasserstein, M.P.; Giugliani, R.; Bembi, B.; Vanier, M.T.; Mengel, E.; Brodie, S.E.; Mendelson, D.; Skloot, G.; Desnick, R.J.; et al. A prospective, cross-sectional survey study of the natural history of Niemann-Pick disease type B. Pediatrics 2008, 122, e341–e349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuchman, E.H.; Desnick, R.J. Types A and B Niemann-Pick disease. Mol. Genet. Metab. 2017, 120, 27–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langeveld, M.; Hollak, C.E.M. Bone health in patients with inborn errors of metabolism. Rev. Endocr. Metab. Disord. 2018, 19, 81–92. [Google Scholar] [CrossRef] [Green Version]
- Eskes, E.C.B.; Sjourke, B.; Vaz, F.M.; Goorden, S.M.; van Kuilenburg, A.B.; Aerts, J.; Hollak, C.E. Biochemical and imaging parameters in acid sphingomyelinase deficiency: Potential utility as biomarkers. Mol. Genet. Metab. 2020, 130, 16–26. [Google Scholar] [CrossRef] [PubMed]
- McGovern, M.M.; Lippa, N.; Bagiella, E.; Schuchman, E.H.; Desnick, R.J.; Wasserstein, M.P. Morbidity and mortality in type B Niemann-Pick disease. Genet. Med. 2013, 15, 618–623. [Google Scholar] [CrossRef] [Green Version]
- Pokrzywinski, R.; Hareendran, A.; Nalysnyk, L.; Cowie, S.; Crowe, J.; Hopkin, J.; Joshi, D.; Pulikottil-Jacob, R. Impact and burden of acid sphingomyelinase deficiency from a patient and caregiver perspective. Sci. Rep. 2021, 11, 20972. [Google Scholar] [CrossRef]
- McGovern, M.M.; Pohl-Worgall, T.; Deckelbaum, R.J.; Simpson, W.; Mendelson, D.; Desnick, R.J.; Schuchman, E.H.; Wasserstein, M.P. Lipid abnormalities in children with types A and B Niemann Pick disease. J. Pediatr. 2004, 145, 77–81. [Google Scholar] [CrossRef]
- Miranda, S.R.; He, X.; Simonaro, C.M.; Gatt, S.; Dagan, A.; Desnick, R.J.; Schuchman, E.H. Infusion of recombinant human acid sphingomyelinase into niemann-pick disease mice leads to visceral, but not neurological, correction of the pathophysiology. FASEB J. 2000, 14, 1988–1995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, J.M.; Thompson, A.M.; Vitsky, A.; Hawes, M.; Chuang, W.L.; Pacheco, J.; Wilson, S.; McPherson, J.M.; Thurberg, B.L.; Karey, K.P.; et al. Nonclinical safety assessment of recombinant human acid sphingomyelinase (rhASM) for the treatment of acid sphingomyelinase deficiency: The utility of animal models of disease in the toxicological evaluation of potential therapeutics. Mol. Genet. Metab. 2015, 114, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Wasserstein, M.P.; Jones, S.A.; Soran, H.; Diaz, G.A.; Lippa, N.; Thurberg, B.L.; Culm-Merdek, K.; Shamiyeh, E.; Inguilizian, H.; Cox, G.F.; et al. Successful within-patient dose escalation of olipudase alfa in acid sphingomyelinase deficiency. Mol. Genet. Metab. 2015, 116, 88–97. [Google Scholar] [CrossRef]
- Wasserstein, M.; Arash-Kaps, L.; Barbato, A.; Gallagher, R.C.; Giugliani, R.; Guelbert, N.B.; Hollak, C.; Ikezoe, T.; Lachmann, R.; Lidove, O.; et al. Adults with chronic acid sphingomyelinase deficiency show significant visceral, pulmonary, and hematologic improvements after enzyme replacement therapy with olipudase alfa: 1-year results of the ASCEND placebo-controlled trial. Mol. Genet. Metab. 2021, 132, S110. [Google Scholar] [CrossRef]
- McGovern, M.M.; Wasserstein, M.P.; Kirmse, B.; Duvall, W.L.; Schiano, T.; Thurberg, B.L.; Richards, S.; Cox, G.F. Novel first-dose adverse drug reactions during a phase I trial of olipudase alfa (recombinant human acid sphingomyelinase) in adults with Niemann–Pick disease type B (acid sphingomyelinase deficiency). Genet. Med. 2016, 18, 34–40. [Google Scholar] [CrossRef] [Green Version]
- Wasserstein, M.P.; Diaz, G.A.; Lachmann, R.H.; Jouvin, M.-H.; Nandy, I.; Ji, A.J.; Puga, A.C. Olipudase alfa for treatment of acid sphingomyelinase deficiency (ASMD): Safety and efficacy in adults treated for 30 months. J. Inherit. Metab. Dis. 2018, 41, 829–838. [Google Scholar] [CrossRef] [Green Version]
- Diaz, G.A.; Jones, S.A.; Scarpa, M.; Mengel, K.E.; Giugliani, R.; Guffon, N.; Batsu, I.; Fraser, P.A.; Li, J.; Zhang, Q.; et al. One-year results of a clinical trial of olipudase alfa enzyme replacement therapy in pediatric patients with acid sphingomyelinase deficiency. Genet. Med. 2021, 23, 1543–1550. [Google Scholar] [CrossRef]
- Thurberg, B.L.; Wasserstein, M.P.; Jones, S.A.; Schiano, T.D.; Cox, G.F.; Puga, A.C. Clearance of hepatic sphingomyelin by olipudase alfa is associated with improvement in lipid profiles in acid sphingomyelinase deficiency. Am. J. Surg. Pathol. 2016, 40, 1232–1242. [Google Scholar] [CrossRef] [Green Version]
- Thurberg, B.L.; Diaz, G.A.; Lachmann, R.H.; Schiano, T.; Wasserstein, M.P.; Ji, A.J.; Zaher, A.; Peterschmitt, M.J. Long-term efficacy of olipudase alfa in adults with acid sphingomyelinase deficiency (ASMD): Further clearance of hepatic sphingomyelin is associated with additional improvements in pro- and anti-atherogenic lipid profiles after 42 months of treatment. Mol. Genet. Metab. 2020, 131, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Garside, B.; Ho, J.H.; Kwok, S.; Liu, Y.; Dhage, S.; Donn, R.; Iqbal, Z.; Jones, S.A.; Soran, H. Changes in PCSK 9 and apolipoprotein B100 in Niemann-Pick disease after enzyme replacement therapy with olipudase alfa. Orphanet J. Rare Dis. 2021, 16, 107. [Google Scholar] [CrossRef]
- Wasserstein, M.; Arash-Kaps, L.; Barbato, A.; Gallagher, R.; Giugliani, R.; Guelbert, N.; Hollak, C.; Ikezoe, T.; Lachmann, R.; Lidove, O.; et al. One-year results of the placebo-controlled ASCEND trial of olipudase alfa enzyme replacement therapy in adults with chronic acid sphingomyelinase deficiency [OP093]. Mol. Genet. Metab. 2021, 132, S64–S65. [Google Scholar] [CrossRef]
- Sanofi-Genzyme. Press Release. Sanofi: FDA Grants Breakthrough Therapy Designation for Genzyme’s Olipudase Alfa. Available online: https://www.sanofi.com/en/media-room/press-releases/2015/2015-06-04-07-00-00 (accessed on 22 September 2021).
- European Medicines Agency. EU/3/01/056: Orphan Designation for the Treatment of Niemann-Pick Disease. Available online: https://www.ema.europa.eu/en/medicines/human/orphan-designations/eu301056 (accessed on 21 September 2021).
- Vanier, M.T. Niemann-Pick diseases. Handb. Clin. Neurol. 2013, 113, 1717–1721. [Google Scholar]
- Bell, E.C.; Cox, N.S.; Goh, N.; Glaspole, I.; Westall, G.P.; Watson, A.; Holland, A.E. Oxygen therapy for interstitial lung disease: A systematic review. Eur. Respir. Rev. 2017, 26, 160080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dowman, L.; Hill, C.J.; May, A.; Holland, A.E. Pulmonary rehabilitation for interstitial lung disease. Cochrane Database Syst. Rev. 2021, 2, CD006322. [Google Scholar] [CrossRef] [PubMed]
- Mauhin, W.; Levade, T.; Vanier, M.T.; Froissart, R.; Lidove, O. Prevalence of cancer in acid sphingomyelinase deficiency. J. Clin. Med. 2021, 10, 5029. [Google Scholar] [CrossRef]
- Ordieres-Ortega, L.; Galeano-Valle, F.; Mallén-Pérez, M.; Muñoz-Delgado, C.; Apaza-Chavez, J.E.; Menárguez-Palanca, F.J.; Alvarez-Sala Walther, L.A.; Demelo-Rodríguez, P. Niemann-Pick disease type-B: A unique case report with compound heterozygosity and complicated lipid management. BMC Med. Genet. 2020, 21, 94. [Google Scholar] [CrossRef] [PubMed]
- Arenz, C. Small molecule inhibitors of acid sphingomyelinase. Cell Physiol. Biochem. 2010, 26, 1–8. [Google Scholar] [CrossRef]
- Bonanni, P.; Grazzini, M.; Niccolai, G.; Paolini, D.; Varone, O.; Bartoloni, A.; Bartalesi, F.; Santini, M.G.; Baretti, S.; Bonito, C.; et al. Recommended vaccinations for asplenic and hyposplenic adult patients. Hum. Vaccin. Imunother. 2017, 13, 359–368. [Google Scholar] [CrossRef] [Green Version]
- Lee, G.M. Preventing infections in children and adults with asplenia. Hematol. Am. Soc. Hematol. Educ. Program 2020, 2020, 328–335. [Google Scholar] [CrossRef]
- Gao, Y.D.; Ding, M.; Dong, X.; Zhang, J.-J.; Azkur, A.K.; Azkur, D.; Gan, H.; Sun, Y.-L.; Fu, W.; Li, W.; et al. Risk factors for severe and critically ill COVID-19 patients: A review. Allergy 2021, 76, 428–455. [Google Scholar] [CrossRef]
- Vanier, M.T. Prenatal diagnosis of Niemann-Pick diseases types A, B and C. Prenat. Diagn. 2002, 22, 630–632. [Google Scholar] [CrossRef] [PubMed]
- Wasserstein, M.P.; Schulman, E.H. Acid sphingomyelinase deficiency. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Mirzaa, G., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2006; pp. 1993–2021. [Google Scholar]
- Agence Nationale de Sécurité du Médicament et des Produits de Santé (ANSM). Autorisations Temporaires d’Utilisation (ATU). Available online: https://archiveansm.integra.fr/Activites/Autorisations-temporaires-d-utilisation-ATU/Qu-est-ce-qu-une-autorisation-temporaire-d-utilisation/(offset)/1 (accessed on 18 July 2021).
- Jones, S.A.; McGovern, M.; Lidove, O.; Giugliani, R.; Mistry, P.K.; Dionisi-Vici, C.; Munoz-Rojas, M.V.; Nalysnyk, L.; Schecter, A.D.; Wasserstein, M. Clinical relevance of endpoints in clinical trials for acid sphingomyelinase deficiency enzyme replacement therapy. Mol Genet Metab. 2020, 131, 116–123. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Historical Classification | Recommended Nomenclature |
|---|---|
| Niemann–Pick disease type A (NPD A) | Infantile neurovisceral ASMD (ASMD Type A) |
| Intermediate or variant phenotype (NPD A/B) | Chronic neurovisceral ASMD (ASMD Type A/B) |
| Niemann–Pick disease type B (NPD B) | Chronic visceral ASMD (ASMD Type B) |
| Exam | Baseline Visit | Follow-Up Visit in the First 6 Months | Follow-Up Visit Every 3 Months for the First Year | Follow-Up Visit Every Year |
|---|---|---|---|---|
| Pulmonary assessment | ||||
| Respiratory functional exploration (DLco—not before 6 years) | X | X | X | |
| CT: thoracic | X | X a | ||
| Hepato-splenic assessment | ||||
| Splenic and hepatic volume (ultrasound +/− abdominal MRI in adults; ultrasound in children) | X | X | X | |
| Portal hypertension (abdominal ultrasound with Doppler) | X | X | X | |
| Transaminases, alkaline phosphatase, bilirubin | X | X | X | |
| Haematological assessment | ||||
| CBC, platelets, haemostasis assessment (PT, factor V, fibrin, APTT, ferritin) | X | X | X | |
| Lipid blood-test including total, LDL and HDL cholesterol and triglycerides | X | X | X |
| Test | Monitoring Frequency | Additional Points |
|---|---|---|
| Lung assessment | ||
| PFT | PFT possible in children from 5–6 years Need to monitor DLco (annually or at clinician’s discretion) following olipudase alfa treatment initiation | |
| Blood gases | - | Non-predictive test Not routinely recommended, especially in children, and depending on symptomatology in adults |
| CT scan | At baseline visit and at follow-up visit annually, unless normal (then assess every 5 years) | Radiation examination in a population at risk of cancer In paediatric patients <4–5 years, sedation or general anaesthesia is required (depending on the child’s behaviour) |
| Hepato-splenic assessment | ||
| Liver blood tests | At baseline visit, at follow-up visit in first 6 months, and at follow-up visit annually | Assess GGT, transaminases, ALP, bilirubin, PT, CRP, Factor V |
| Abdominal ultrasound with doppler | At baseline visit, at follow-up visit in first 6 months, and at follow-up visit annually | Early detection of steatosis, PHT, cirrhosis or nodule Possible in children < 5 years of age |
| Abdominal MRI (adult patients) | At initial assessment and then every 2 years | Higher sensitivity for nodule detection Possible in children > 5 years of age |
| Fibroscan | - | Not recommended as not validated for ASMD |
| Liver biopsy | To consider if hepatocarcinoma is suspected | No correlation with biological tests (transaminases usually < 5 N). Characterisation of nodule may require alpha-foetoprotein, liver ultrasound, and MRI |
| Alpha-fetoprotein | - | No recommendation for systematic assessment |
| Haematological assessment | ||
| CBC, platelets, haemostasis test (PT, Factor V, fibrin, aPTT), ferritin | Assessment at baseline visit, at follow-up visit every 3 months during first year, and at follow-up visit annually | - |
| Protein electrophoresis | At initial assessment and then annually | Risk of hyper or hypogammaglobulinemia and risk of MGUS No need to perform any immunoelectrophoresis |
| Serum albumin | At initial assessment then every 2–3 years if the initial assay is normal | - |
| Bone assessment and growth evaluation | ||
| Growth curve | Assessment at baseline visit and at follow-up visit every 6 months | - |
| Phosphocalcium balance | Assessment at baseline visit and at follow-up visit every 6 months | Assessment to include blood calcium and phosphorus, vitamin D, creatinine, proteinuria, urine calcium and sodium, and creatininuria Patients are at risk of cholestasis |
| Absorptiometry | Assessment at baseline visit and then every 5 years if normal or every 3 years if anormal | Possible in children aged from 5–6 years old |
| Resorption markers | Optional | To be performed if osteoporosis is known |
| Cardiovascular assessment and lipid profile | ||
| Echocardiography | At initial assessment and then every 2 years | |
| Coronary computed tomographic angiography | Discuss coronary computed tomographic angiography depending on lipid profile and other cardiovascular risk factors | No monitoring data in ASMD type B disease |
| Lipid profile | Assessment at baseline visit, at follow-up visit every 3 months during first year, and at follow-up visit annually | Unproven benefit of statins in primary prevention |
| Neurological assessment a | ||
| Peripheral | ||
| Clinical examination | Monitoring frequency: at initial assessment and then annually | |
| EMG | Monitoring frequency: to be considered if there are clinical call points | |
| Central nervous system | ||
| Brain MRI | Monitoring frequency in adults: to be considered at initial assessment according to the clinical context and the neuropsychological tests Monitoring frequency in children: to be carried out at initial assessment if the diagnosis is made at a very early stage of childhood | |
| Ophthalmic assessment | ||
| Ocular fundus | At initial assessment | The macular cherry-red spot is present in all patients with form A, and has been reported in one third of patients with form B [20] |
| Visual acuity | At initial assessment | Loss of visual acuity in infantile type (no effect in chronic visceral ASMD) |
| Other | ||
| Dermatological examination | At initial assessment and then annually | Necessary in order to assess the presence of lymphoedema (eyelid infiltration) |
| TSH assay (in combination with anti-TPO antibodies) | At initial assessment and then annually | Necessary for the investigation of thyroid autoimmunity |
| Biomarkers | ||
| Chitotriosidase | At initial assessment, follow-up every 3 months for the first year, and then every year | Chitotriosidase activity is absent in 6–8% of individuals in the general population |
| Specialised assays, contact reference laboratories | ||
| Lysosphingomyelin + Lysosphingomyelin isoform 509 | At initial assessment, follow-up every 3 months for the first year, and then every year | Specialised assays, contact reference laboratories |
| Group | Definition |
|---|---|
| Group outside the early access spectrum application | ASMD types B and A/B (NPD B or A/B) with a short-term vital prognosis (e.g., cancer) |
| Group 1: Patients with severe organ involvement | DLco a <50% and/ or dyspnoea Platelets <50 G/L ± recurrent bleeding or bruising Abdominal pain Urgent need to start treatment Break in growth curve (≥2 standard deviation) |
| Group 2: Patients in therapeutic trials | Patient continues to receive treatment as part of clinical trial or extension study |
| Group 3: Patients with moderate organ involvement | 50% < DLco < 70% 50 G/L < Platelets < 100 G/L without bleeding or bruising Failure to thrive/break in growth curve (>1 standard deviation) |
| Group 4: Patients with mild organ involvement | DLco > 70% Platelets > 100 G/L Abnormal thoracic imagery and/or hepatosplenomegaly |
| Group 5: Patients with no symptoms | DLCO > 70% Platelets > 100 G/L No symptom Normal thoracic imagery No hepato-splenomegaly |
| Criteria | ASCEND (Adults) | ASCEND Peds |
|---|---|---|
| Lung | Inclusion criteria:
| Exclusion criteria:
|
| Spleen | Inclusion criteria:
| Inclusion criteria:
|
| Liver | Exclusion criteria:
| Exclusion criteria:
|
| Blood | Exclusion criteria:
| Exclusion criteria:
|
| Bone/growth retardationBone damageDelayed growth |
|
|
| Central nervous system |
| Exclusion criteria:
|
| Cardiovascular | Exclusion criteria:
| Exclusion criteria:
|
| Pain/fatigue | Inclusion criteria:
|
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mauhin, W.; Borie, R.; Dalbies, F.; Douillard, C.; Guffon, N.; Lavigne, C.; Lidove, O.; Brassier, A. Acid Sphingomyelinase Deficiency: Sharing Experience of Disease Monitoring and Severity in France. J. Clin. Med. 2022, 11, 920. https://doi.org/10.3390/jcm11040920
Mauhin W, Borie R, Dalbies F, Douillard C, Guffon N, Lavigne C, Lidove O, Brassier A. Acid Sphingomyelinase Deficiency: Sharing Experience of Disease Monitoring and Severity in France. Journal of Clinical Medicine. 2022; 11(4):920. https://doi.org/10.3390/jcm11040920
Chicago/Turabian StyleMauhin, Wladimir, Raphaël Borie, Florence Dalbies, Claire Douillard, Nathalie Guffon, Christian Lavigne, Olivier Lidove, and Anaïs Brassier. 2022. "Acid Sphingomyelinase Deficiency: Sharing Experience of Disease Monitoring and Severity in France" Journal of Clinical Medicine 11, no. 4: 920. https://doi.org/10.3390/jcm11040920
APA StyleMauhin, W., Borie, R., Dalbies, F., Douillard, C., Guffon, N., Lavigne, C., Lidove, O., & Brassier, A. (2022). Acid Sphingomyelinase Deficiency: Sharing Experience of Disease Monitoring and Severity in France. Journal of Clinical Medicine, 11(4), 920. https://doi.org/10.3390/jcm11040920