
CIP2A as a Key Regulator for AKT Phosphorylation Has Partial Impact Determining Clinical Outcome in Breast Cancer

 , , ,
, , , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Cultures
2.2. Patient Samples
2.3. Western Blot Analysis
2.4. Ex Vivo Models
2.5. In Vivo Animal Model
2.6. Immunohistochemistry
2.7. Statistical Analysis
3. Results
3.1. Prevalence of p-AKT in Human Breast Cancer and Its Association with Clinical and Molecular Parameters
3.2. Clinical Significance of p-AKT in Human Breast Cancer
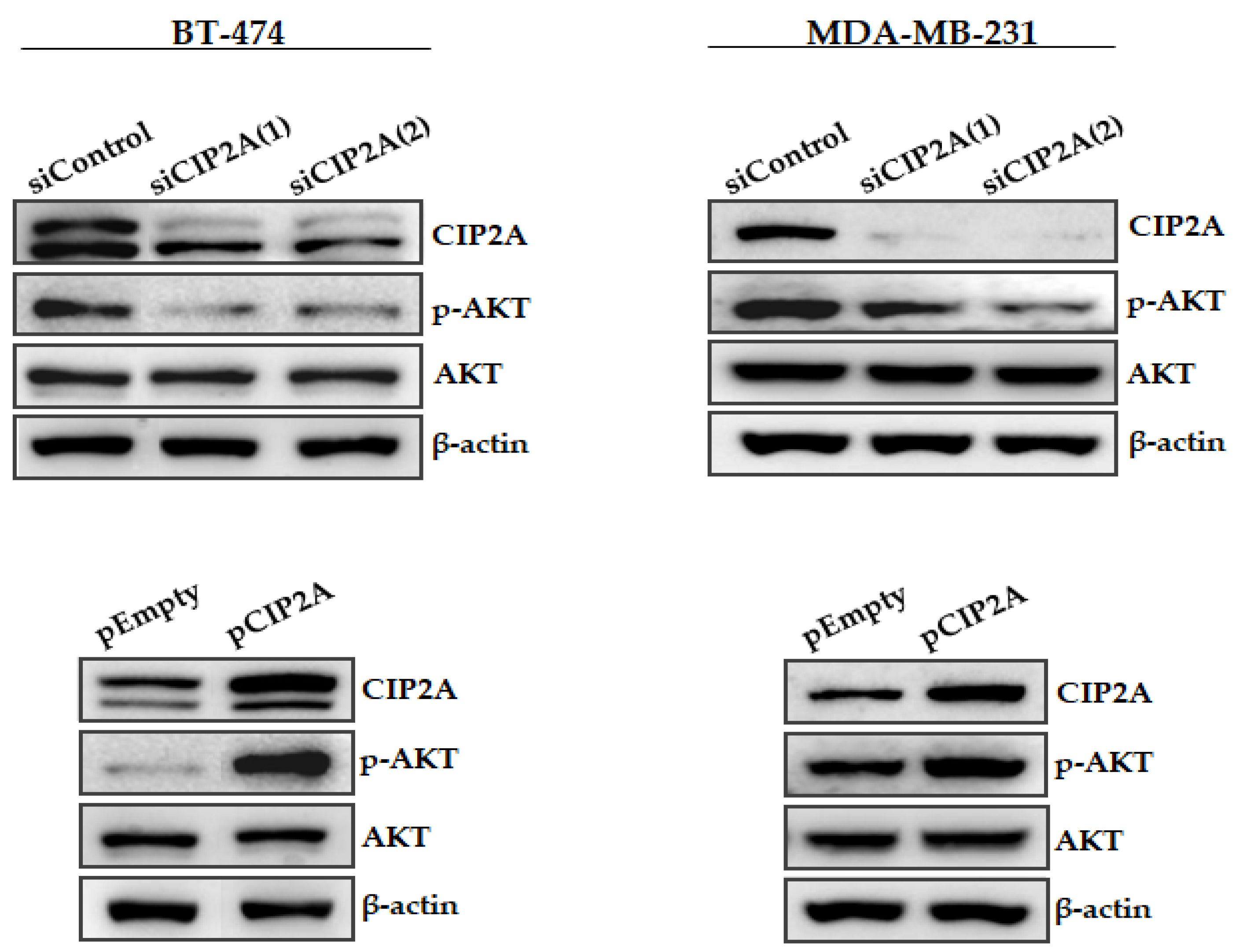
3.3. Modulation of CIP2A Expression Affects AKT Phosphorylation in Breast Cancer Cells
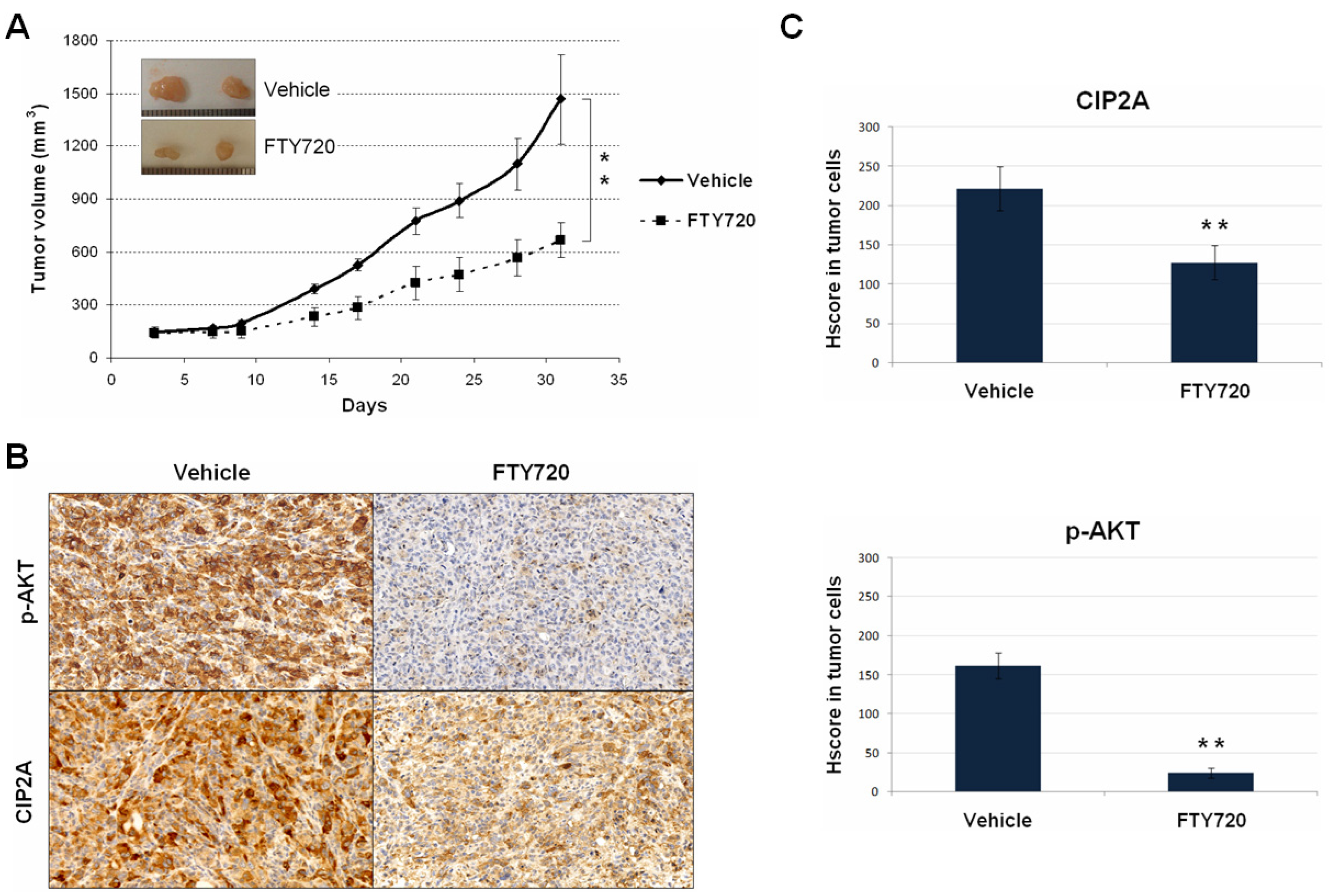
3.4. The Use of FTY720 Inhibits the CIP2A/AKT Axis Leading to Potent In Vivo Antitumor Effects
3.5. AKT Phosphorylation Status Determines Response to Doxorubicin Treatment
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics. CA Cancer J. Clin. 2011, 61, 69–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simpson, P.T.; Reis-Filho, J.S.; Gale, T.; Lakhani, S.R. Molecular evolution of breast cancer. J. Pathol. 2005, 205, 248–254. [Google Scholar] [PubMed]
- De Abreu, F.B.; Schwartz, G.N.; Wells, W.A.; Tsongalis, G.J. Personalized therapy for breast cancer. Clin. Genet. 2014, 86, 62–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Longley, D.B.; Johnston, P.G. Molecular mechanisms of drug resistance. J. Pathol. 2005, 205, 275–292. [Google Scholar]
- Coley, H.M. Mechanisms and strategies to overcome chemotherapy resistance in metastatic breast cancer. Cancer Treat. Rev. 2008, 34, 378–390. [Google Scholar] [CrossRef]
- Rajput, S.; Puvvada, N.; Kumar, B.N.; Sarkar, S.; Konar, S.; Bharti, R.; Dey, G.; Mazumdar, A.; Pathak, A.; Fisher, P.B.; et al. Overcoming Akt induced therapeutic resistance in breast cancer through siRNA and thymoquinone encapsulated multilamellar gold niosomes. Mol. Pharm. 2015, 12, 4214–4225. [Google Scholar] [CrossRef]
- Mayer, I.A.; Arteaga, C.L. The PI3K/AKT Pathway as a Target for Cancer Treatment. Annu. Rev. Med. 2015, 67, 11–28. [Google Scholar] [CrossRef]
- Yang, Z.Y.; Di, M.Y.; Yuan, J.Q.; Shen, W.X.; Zheng, D.Y.; Chen, J.Z.; Mao, C.; Tang, J.L. The prognostic value of phosphorylated Akt in breast cancer: A systematic review. Sci. Rep. 2015, 5, 7758. [Google Scholar] [CrossRef] [Green Version]
- Junttila, M.R.; Puustinen, P.; Niemelä, M.; Ahola, R.; Arnold, H.; Böttzauw, T.; Ala-aho, R.; Nielsen, C.; Ivaska, J.; Taya, Y.; et al. CIP2A inhibits PP2A in human malignancies. Cell 2007, 130, 51–62. [Google Scholar] [CrossRef] [Green Version]
- Puustinen, P.; Rytter, A.; Mortensen, M.; Kohonen, P.; Moreira, J.M.; Jäättelä, M. CIP2A oncoprotein controls cell growth and autophagy through mTORC1 activation. J. Cell Biol. 2014, 204, 713–727. [Google Scholar] [CrossRef]
- Laine, A.; Sihto, H.; Come, C.; Rosenfeldt, M.T.; Zwolinska, A.; Niemelä, M.; Khanna, A.; Chan, E.K.; Kähäri, V.M.; Kellokumpu-Lehtinen, P.L.; et al. Senescence sensitivity of breast cancer cells is defined by positive feedback loop between CIP2A and E2F1. Cancer Discov. 2013, 3, 182–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rincón, R.; Cristóbal, I.; Zazo, S.; Arpí, O.; Menéndez, S.; Manso, R.; Lluch, A.; Eroles, P.; Rovira, A.; Albanell, J.; et al. PP2A inhibition determines poor outcome and doxorubicin resistance in early breast cancer and its activation shows promising therapeutic effects. Oncotarget 2015, 6, 4299–4314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.Y.; Hung, M.H.; Wang, D.S.; Chu, P.Y.; Su, J.C.; Teng, T.H.; Huang, C.T.; Chao, T.T.; Wang, C.Y.; Shiau, C.W.; et al. Tamoxifen induces apoptosis through cancerous inhibitor of protein phosphatase 2A-dependent phospho-Akt inactivation in estrogen receptor-negative human breast cancer cells. Breast Cancer Res. 2014, 16, 431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sangai, T.; Akcakanat, A.; Chen, H.; Tarco, E.; Wu, Y.; Do, K.A.; Miller, T.W.; Arteaga, C.L.; Mills, G.B.; Gonzalez-Angulo, A.M.; et al. Biomarkers of response to Akt inhibitor MK-2206 in breast cancer. Clin. Cancer Res. 2012, 18, 5816–5828. [Google Scholar] [CrossRef] [Green Version]
- Hudis, C.; Swanton, C.; Janjigian, Y.Y.; Lee, R.; Sutherland, S.; Lehman, R.; Chandarlapaty, S.; Hamilton, N.; Gajria, D.; Knowles, J.; et al. A phase 1 study evaluating the combination of an allosteric AKT inhibitor (MK-2206) and trastuzumab in patients with HER2-positive solid tumors. Breast Cancer Res. 2013, 15, R110. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Angulo, A.M.; Krop, I.; Akcakanat, A.; Chen, H.; Liu, S.; Li, Y.; Culotta, K.S.; Tarco, E.; Piha-Paul, S.; Moulder-Thompson, S.; et al. SU2C phase Ib study of paclitaxel and MK-2206 in advanced solid tumors and metastatic breast cancer. J. Natl. Cancer Inst. 2015, 107, dju493. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Peng, B.; Li, Y.; Lei, N.; Li, W.; Zhang, J.Y. p90/CIP2A mediates breast cancer cell proliferation and apoptosis. Mol. Biol. Rep. 2014, 41, 7471–7478. [Google Scholar] [CrossRef]
- Yin, J.; Chen, D.; Luo, K.; Lu, M.; Gu, Y.; Zeng, S.; Chen, X.; Song, Y.; Zhang, Z.; Zheng, G.; et al. Cip2a/miR-301a feedback loop promotes cell proliferation and invasion of triple-negative breast cancer. J. Cancer 2019, 10, 5964–5974. [Google Scholar] [CrossRef]
- Liu, H.; Qiu, H.; Song, Y.; Liu, Y.; Wang, H.; Lu, M.; Deng, M.; Gu, Y.; Yin, J.; Luo, K.; et al. Cip2a promotes cell cycle progression in triple-negative breast cancer cells by regulating the expression and nuclear export of p27Kip1. Oncogene 2017, 36, 1952–1964. [Google Scholar] [CrossRef]
- Côme, C.; Laine, A.; Chanrion, M.; Edgren, H.; Mattila, E.; Liu, X.; Jonkers, J.; Ivaska, J.; Isola, J.; Darbon, J.M.; et al. CIP2A is associated with human breast cancer aggressivity. Clin. Cancer Res. 2009, 15, 5092–5100. [Google Scholar] [CrossRef] [Green Version]
- Tseng, L.M.; Liu, C.Y.; Chang, K.C.; Chu, P.Y.; Shiau, C.W.; Chen, K.F. CIP2A is a target of bortezomib in human triple negative breast cancer cells. Breast Cancer Res. 2012, 14, R68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Q.; Zhao, M.; Parris, A.B.; Xing, Y.; Yang, X. Genistein targets the cancerous inhibitor of PP2A to induce growth inhibition and apoptosis in breast cancer cells. Int. J. Oncol. 2016, 49, 1203–1210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, Y.A.; Park, J.S.; Park, M.Y.; Oh, K.S.; Lee, M.S.; Lim, J.S.; Kim, K.I.; Kim, K.Y.; Kwon, J.; Yoon, Y.D.; et al. Increase in CIP2A expression is associated with doxorubicin resistance. FEBS Lett. 2011, 585, 755–760. [Google Scholar] [CrossRef] [Green Version]
- Janghorban, M.; Farrell, A.S.; Allen-Petersen, B.L.; Pelz, C.; Daniel, C.J.; Oddo, J.; Langer, E.M.; Christensen, D.J.; Sears, R.C. Targeting c-MYC by antagonizing PP2A inhibitors in breast cancer. Proc. Natl. Acad. Sci. USA 2014, 111, 9157–9162. [Google Scholar] [CrossRef] [Green Version]
- Osman, N.T.; Khalaf, M.; Ibraheem, S. Assessment of CIP2A and ROCK-I expression and their prognostic value in breast cancer. Pol. J. Pathol. 2020, 71, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Feng, T.T.; Guo, Y.; Yu, X.; Huang, Q.; Zhang, L.; Tang, W.; Liu, Y. Expression of cancerous inhibitor of protein phosphatase 2A in human triple negative breast cancer correlates with tumor survival, invasion and autophagy. Oncol. Lett. 2016, 12, 5370–5376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cristóbal, I.; Zazo, S.; Torrejón, B.; Pedregal, M.; Madoz-Gúrpide, J.; Lluch, A.; Eroles, P.; Rovira, A.; Albanell, J.; García-Foncillas, J.; et al. CIP2A confirms its prognostic value in triple-negative breast cancer. Oncogene 2017, 36, 3357–3358. [Google Scholar] [CrossRef]
- Baldacchino, S.; Wastall, L.M.; Saliba, C.; Hughes, T.A.; Scerri, C.; Berwick, A.; Speirs, V.; Hanby, A.M.; Grech, G. CIP2A expression predicts recurrences of tamoxifen-treated breast cancer. Tumour Biol. 2017, 39, 1010428317722064. [Google Scholar] [CrossRef] [Green Version]
- Cai, F.; Zhang, L.; Xiao, X.; Duan, C.; Huang, Q.; Fan, C.; Li, J.; Liu, X.; Li, S.; Liu, Y. Cucurbitacin B reverses multidrug resistance by targeting CIP2A to reactivate protein phosphatase 2A in MCF-7/adriamycin cells. Oncol. Rep. 2016, 36, 1180–1186. [Google Scholar] [CrossRef]
- Nishio, E.; Hayashi, T.; Akaza, M.; Hisatomi, Y.; Hikichi, M.; Fujii, T.; Utsumi, T.; Harada, N.; Shimono, Y. Upregulation of CIP2A in estrogen depletion-resistant breast cancer cells treated with low-dose everolimus. FEBS Open Bio 2020, 10, 2072–2080. [Google Scholar] [CrossRef]
- Liu, C.Y.; Hu, M.H.; Hsu, C.J.; Huang, C.T.; Wang, D.S.; Tsai, W.C.; Chen, Y.T.; Lee, C.H.; Chu, P.Y.; Hsu, C.C.; et al. Lapatinib inhibits CIP2A/PP2A/p-Akt signaling and induces apoptosis in triple negative breast cancer cells. Oncotarget 2016, 7, 9135–9149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Q.; Qin, S.; Yuan, X.; Zhang, L.; Ji, J.; Liu, X.; Ma, W.; Zhang, Y.; Liu, P.; Sun, Z.; et al. Arctigenin inhibits triple-negative breast cancers by targeting CIP2A to reactivate protein phosphatase 2A. Oncol. Rep. 2017, 38, 598–606. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.Y.; Huang, T.T.; Huang, C.T.; Hu, M.H.; Wang, D.S.; Wang, W.L.; Tsai, W.C.; Lee, C.H.; Lau, K.Y.; Yang, H.P.; et al. EGFR-independent Elk1/CIP2A signalling mediates apoptotic effect of an erlotinib derivative TD52 in triple-negative breast cancer cells. Eur. J. Cancer 2017, 72, 112–123. [Google Scholar] [CrossRef] [PubMed]
- Cristóbal, I.; Madoz-Gúrpide, J.; Manso, R.; González-Alonso, P.; Rojo, F.; García-Foncillas, J. Potential anti-tumor effects of FTY720 associated with PP2A activation: A brief review. Curr. Med. Res. Opin. 2016, 32, 1137–1141. [Google Scholar] [CrossRef]
- Cristóbal, I.; Manso, R.; Rincón, R.; Caramés, C.; Senin, C.; Borrero, A.; Martínez-Useros, J.; Rodriguez, M.; Zazo, S.; Aguilera, O.; et al. PP2A inhibition is a common event in colorectal cancer and its restoration using FTY720 shows promising therapeutic potential. Mol. Cancer Ther. 2014, 13, 938–947. [Google Scholar] [CrossRef] [Green Version]
- Cristóbal, I.; González-Alonso, P.; Daoud, L.; Solano, E.; Torrejón, B.; Manso, R.; Madoz-Gúrpide, J.; Rojo, F.; García-Foncillas, J. Activation of the Tumor Suppressor PP2A Emerges as a Potential Therapeutic Strategy for Treating Prostate Cancer. Mar. Drugs 2015, 13, 3276–3286. [Google Scholar] [CrossRef] [Green Version]
- Elston, C.W.; Ellis, I.O. Pathological prognostic factors in breast cancer. I. The value of histological grade in breast cancer: Experience from a large study with long-term follow-up. Histopathology 1991, 19, 403–410. [Google Scholar] [CrossRef]
- Hammond, M.E.; Hayes, D.F.; Dowsett, M.; Allred, D.C.; Hagerty, K.L.; Badve, S.; Fitzgibbons, P.L.; Francis, G.; Goldstein, N.S.; Hayes, M.; et al. American Society of Clinical Oncology/College of American Pathologists guideline recommendations for immunohistochemical testing of estrogen and progesterone receptors in breast cancer. J. Clin. Oncol. 2010, 28, 2784–2795. [Google Scholar] [CrossRef] [Green Version]
- Wolff, A.C.; Hammond, M.E.; Hicks, D.G.; Dowsett, M.; McShane, L.M.; Allison, K.H.; Allred, D.C.; Bartlett, J.M.; Bilous, M.; Fitzgibbons, P.; et al. Recommendations for human epidermal growth factor receptor 2 testing in breast cancer: American Society of Clinical Oncology/College of American Pathologists clinical practice guideline update. J. Clin. Oncol. 2013, 31, 3997–4013. [Google Scholar] [CrossRef]
- Dowsett, M.; Nielsen, T.O.; A’Hern, R.; Bartlett, J.; Coombes, R.C.; Cuzick, J.; Ellis, M.; Henry, N.L.; Hugh, J.C.; Lively, T.; et al. Assessment of Ki67 in breast cancer: Recommendations from the International Ki67 in Breast Cancer working group. J. Natl. Cancer Inst. 2011, 103, 1656–1664. [Google Scholar] [CrossRef] [Green Version]
- Di Nicolantonio, F.; Knight, L.A.; Whitehouse, P.A.; Mercer, S.J.; Sharma, S.; Charlton, P.A.; Norris, D.; Cree, I.A. The ex vivo characterization of XR5944 (MLN944) against a panel of human clinical tumor samples. Mol. Cancer Ther. 2004, 3, 1631–1637. [Google Scholar] [PubMed]
- Serra, V.; Scaltriti, M.; Prudkin, L.; Eichhorn, P.J.; Ibrahim, Y.H.; Chandarlapaty, S.; Markman, B.; Rodriguez, O.; Guzman, M.; Rodriguez, S.; et al. PI3K inhibition results in enhanced HER signaling and acquired ERK dependency in HER2-overexpressing breast cancer. Oncogene 2011, 30, 2457–2547. [Google Scholar] [CrossRef] [PubMed]
- Azuma, H.; Takahara, S.; Ichimaru, N.; Wang, J.D.; Itoh, Y.; Otsuki, Y.; Morimoto, J.; Fukui, R.; Hoshiga, M.; Ishihara, T.; et al. Marked prevention of tumor growth and metastasis by a novel immunosuppressive agent, FTY720, in mouse breast cancer models. Cancer Res. 2002, 62, 1410–1419. [Google Scholar] [PubMed]
- Rojo, F.; Gonzalez-Navarrete, I.; Bragado, R.; Dalmases, A.; Menéndez, S.; Cortes-Sempere, M.; Suárez, C.; Oliva, C.; Servitja, S.; Rodriguez-Fanjul, V.; et al. Mitogen-activated protein kinase phosphatase-1 in human breast cancer independently predicts prognosis and is repressed by doxorubicin. Clin. Cancer Res. 2009, 15, 3530–3539. [Google Scholar] [CrossRef] [Green Version]
- Generali, D.; Buffa, F.M.; Berruti, A.; Brizzi, M.P.; Campo, L.; Bonardi, S.; Bersiga, A.; Allevi, G.; Milani, M.; Aguggini, S.; et al. Phosphorylated ERalpha, HIF-1alpha, and MAPK signaling as predictors of primary endocrine treatment response and resistance in patients with breast cancer. J. Clin. Oncol. 2009, 27, 227–234. [Google Scholar] [CrossRef]
- McShane, L.M.; Altman, D.G.; Sauerbrei, W.; Taube, S.E.; Gion, M.; Clark, G.M. Statistics Subcommittee of the NCI-EORTC Working Group on Cancer Diagnostics. Reporting recommendations for tumor marker prognostic studies. J. Clin. Oncol. 2005, 23, 9067–9072. [Google Scholar] [CrossRef] [Green Version]
- De, P.; Carlson, J.; Leyland-Jones, B.; Dey, N. Oncogenic nexus of cancerous inhibitor of protein phosphatase 2A (CIP2A): An oncoprotein with many hands. Oncotarget 2014, 5, 4581–4602. [Google Scholar] [CrossRef] [Green Version]
- Khanna, A.; Pimanda, J.E.; Westermarck, J. Cancerous inhibitor of protein phosphatase 2A, an emerging human oncoprotein and a potential cancer therapy target. Cancer Res. 2013, 73, 6548–6553. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.F.; Liu, C.Y.; Lin, Y.C.; Yu, H.C.; Liu, T.H.; Hou, D.R.; Chen, P.J.; Cheng, A.L. CIP2A mediates effects of bortezomib on phospho-Akt and apoptosis in hepatocellular carcinoma cells. Oncogene 2010, 29, 6257–6626. [Google Scholar]
- Wei, L.; Qu, W.; Sun, J.; Wang, X.; Lv, L.; Xie, L.; Song, X. Knockdown of cancerous inhibitor of protein phosphatase 2A may sensitize NSCLC cells to cisplatin. Cancer Gene Ther. 2014, 21, 194–199. [Google Scholar] [CrossRef]
- Huang, J.; Jia, J.; Tong, Q.; Liu, J.; Qiu, J.; Sun, R.; Yao, L.; Yang, C. Knockdown of cancerous inhibitor of protein phosphatase 2A may sensitize metastatic castration-resistant prostate cancer cells to cabazitaxel chemotherapy. Tumour Biol. 2015, 36, 1589–1594. [Google Scholar] [CrossRef] [PubMed]
- Xing, M.L.; Lu, Y.F.; Wang, D.F.; Zou, X.Y.; Zhang, S.X.; Yun, Z. Clinical significance of sCIP2A levels in breast cancer. Eur. Rev. Med. Pharmacol. Sci. 2016, 20, 82–91. [Google Scholar] [PubMed]
- Zhao, M.; Howard, E.W.; Parris, A.B.; Guo, Z.; Zhao, Q.; Ma, Z.; Xing, Y.; Liu, B.; Edgerton, S.M.; Thor, A.; et al. Activation of cancerous inhibitor of PP2A (CIP2A) contributes to lapatinib resistance through induction of CIP2A-Akt feedback loop in ErbB2-positive breast cancer cells. Oncotarget 2017, 8, 58847–58864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.H.; Chu, P.Y.; Chen, J.L.; Huang, C.T.; Lee, C.H.; Lau, K.Y.; Wang, W.L.; Wang, Y.L.; Lien, P.J.; Tseng, L.-M.; et al. SET Overexpression is Associated with Worse Recurrence-Free Survival in Patients with Primary Breast Cancer Receiving Adjuvant Tamoxifen Treatment. J. Clin. Med. 2018, 7, 245. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.Y.; Huang, T.T.; Chen, Y.T.; Chen, J.L.; Chu, P.Y.; Huang, C.T.; Wang, W.L.; Lau, K.Y.; Dai, M.S.; Shiau, C.W. Targeting SET to restore PP2A activity disrupts an oncogenic CIP2A-feedforward loop and impairs triple negative breast cancer progression. EBioMedicine 2019, 40, 263–275. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Prever, L.; Hirsch, E.; Gulluni, F. Targeting PI3K/AKT/mTOR Signaling Pathway in Breast Cancer. Cancers 2021, 13, 3517. [Google Scholar] [CrossRef]
- Zhu, Z.; Wei, Z. CIP2A silencing alleviates doxorubicin resistance in MCF7/ADR cells through activating PP2A and autophagy. Clin. Transl. Oncol. 2021, 23, 1542–1548. [Google Scholar] [CrossRef]
- Guo, B.; Wu, S.; Zhu, X.; Zhang, L.; Deng, J.; Li, F.; Wang, Y.; Zhang, S.; Wu, R.; Lu, J.; et al. Micropeptide CIP2A-BP encoded by LINC00665 inhibits triple-negative breast cancer progression. EMBO J. 2020, 39, e102190. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. Cases | No. Low p-AKT (%) | No. High p-AKT (%) | p-Value | |||
|---|---|---|---|---|---|---|
| p-AKT | 220 | 140 | (63.6) | 80 | (36.4) | |
| T | 220 | 140 | 80 | 0.149 | ||
| 1 | 107 | 68 | (63.6) | 39 | (36.4) | |
| 2 | 89 | 52 | (58.4) | 37 | (41.6) | |
| 3 | 22 | 18 | (81.8) | 4 | (18.2) | |
| 4 | 2 | 2 | (100) | 0 | (0) | |
| N | 220 | 140 | 80 | 0.075 | ||
| 0 | 128 | 82 | (64.1) | 46 | (35.9) | |
| 1 | 49 | 26 | (53.1) | 23 | (46.9) | |
| 2 | 25 | 21 | (84) | 4 | (16) | |
| 3 | 18 | 11 | (61.1) | 7 | (38.9) | |
| Stage | 218 | 140 | 78 | 0.757 | ||
| 1 | 80 | 51 | (63.8) | 29 | (36.2) | |
| 2 | 96 | 60 | (62.5) | 36 | (37.5) | |
| 3 | 42 | 29 | (69) | 13 | (31) | |
| Grade | 220 | 140 | 80 | 0.060 | ||
| 1 | 33 | 15 | (45.5) | 17 | (54.5) | |
| 2 | 103 | 68 | (66) | 30 | (34) | |
| 3 | 84 | 57 | (67.9) | 26 | (32.1) | |
| Morphological type | 98 | 52 | 46 | 0.063 | ||
| IDC | 93 | 47 | (50.5) | 46 | (49.5) | |
| ILC | 5 | 5 | (100) | 0 | (0) | |
| ER | 220 | 140 | 80 | 0.049 | ||
| Negative | 83 | 46 | (55.4) | 37 | (44.6) | |
| Positive | 137 | 94 | (68.6) | 43 | (31.4) | |
| PR | 220 | 140 | 80 | 0.398 | ||
| Negative | 99 | 60 | (60.6) | 39 | (39.4) | |
| Positive | 121 | 80 | (66.1) | 41 | (33.9) | |
| HER2 | 220 | 140 | 80 | 0.252 | ||
| Negative | 149 | 91 | (61.1) | 58 | (38.9) | |
| Positive | 71 | 49 | (69) | 22 | (31) | |
| Hormonal status | 213 | 134 | 79 | 0.079 | ||
| Premenopausal | 58 | 42 | (72.4) | 16 | (27.6) | |
| Postmenopausal | 155 | 92 | (59.4) | 63 | (40.6) | |
| Relapse | 220 | 140 | 80 | 0.317 | ||
| No | 160 | 105 | (65.6) | 55 | (34.4) | |
| Yes | 60 | 35 | (58.3) | 25 | (41.7) | |
| Ki-67 | 220 | 140 | 80 | 0.646 | ||
| Low | 147 | 92 | (62.6) | 55 | (37.4) | |
| High | 73 | 48 | (65.8) | 25 | (34.2) | |
| Molecular subtype | 220 | 140 | 80 | 0.192 | ||
| Luminal | 95 | 62 | (65.3) | 33 | (34.7) | |
| HER2-positive | 71 | 49 | (69) | 22 | (31) | |
| Triple-negative | 54 | 29 | (53.7) | 25 | (46.3) | |
| CIP2A | 220 | 140 | 80 | <0.001 | ||
| Negative | 180 | 133 | (73.9) | 47 | (26.1) | |
| Positive | 40 | 7 | (17.5) | 33 | (82.5) | |
| Univariate OS Analysis | Multivariate OS Cox Analysis | |||||||
|---|---|---|---|---|---|---|---|---|
| HR | 95% CI | p-Value | HR | 95% CI | p-Value | |||
| Lower | Upper | Lower | Upper | |||||
| Stage | 0.003 | 0.574 | ||||||
| 1–2 | 1.000 | 1.000 | ||||||
| 3 | 1.951 | 1.262 to 3.017 | 0.800 | 0.367 to 1.742 | ||||
| Grade | 0.029 | 0.032 | ||||||
| 1–2 | 1.000 | 1.000 | ||||||
| 3 | 1.742 | 1.060 to 2.864 | 1.845 | 1.053 to 3.231 | ||||
| T | 0.001 | 0.045 | ||||||
| 1–2 | 1.000 | 1.000 | ||||||
| 3–4 | 1.938 | 1.297 to 2.897 | 1.884 | 1.014 to 3.502 | ||||
| N | <0.001 | 0.037 | ||||||
| − | 1.000 | 1.000 | ||||||
| + | 1.656 | 1.270 to 2.160 | 1.454 | 1.023 to 2.067 | ||||
| CIP2A | 0.019 | 0.746 | ||||||
| Low | 1.000 | 1.000 | ||||||
| High | 2.154 | 1.132 to 4.099 | 0.872 | 0.381 to 1.995 | ||||
| p-AKT | 0.002 | 0.003 | ||||||
| Low | 1.000 | 1.000 | ||||||
| High | 2.587 | 1.406 to 4.759 | 3.251 | 1.477 to 7.158 | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luque, M.; Cristóbal, I.; Sanz-Álvarez, M.; Santos, A.; Zazo, S.; Eroles, P.; Arpí, O.; Rovira, A.; Albanell, J.; Madoz-Gúrpide, J.; et al. CIP2A as a Key Regulator for AKT Phosphorylation Has Partial Impact Determining Clinical Outcome in Breast Cancer. J. Clin. Med. 2022, 11, 1610. https://doi.org/10.3390/jcm11061610
Luque M, Cristóbal I, Sanz-Álvarez M, Santos A, Zazo S, Eroles P, Arpí O, Rovira A, Albanell J, Madoz-Gúrpide J, et al. CIP2A as a Key Regulator for AKT Phosphorylation Has Partial Impact Determining Clinical Outcome in Breast Cancer. Journal of Clinical Medicine. 2022; 11(6):1610. https://doi.org/10.3390/jcm11061610
Chicago/Turabian StyleLuque, Melani, Ion Cristóbal, Marta Sanz-Álvarez, Andrea Santos, Sandra Zazo, Pilar Eroles, Oriol Arpí, Ana Rovira, Joan Albanell, Juan Madoz-Gúrpide, and et al. 2022. "CIP2A as a Key Regulator for AKT Phosphorylation Has Partial Impact Determining Clinical Outcome in Breast Cancer" Journal of Clinical Medicine 11, no. 6: 1610. https://doi.org/10.3390/jcm11061610
APA StyleLuque, M., Cristóbal, I., Sanz-Álvarez, M., Santos, A., Zazo, S., Eroles, P., Arpí, O., Rovira, A., Albanell, J., Madoz-Gúrpide, J., García-Foncillas, J., & Rojo, F. (2022). CIP2A as a Key Regulator for AKT Phosphorylation Has Partial Impact Determining Clinical Outcome in Breast Cancer. Journal of Clinical Medicine, 11(6), 1610. https://doi.org/10.3390/jcm11061610