

ABO-Incompatible Liver Transplantation under the Desensitization Protocol with Rituximab: Effect on Biliary Microbiota and Metabolites
 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. Sample Collection and Storage
2.3. Definition of the Sample Groups
2.4. DNA Extraction, Illumina MiSeq Sequencing and Bioinformatic Analysis
2.5. Analysis of Targeted Metabolomics
2.6. Data Analysis
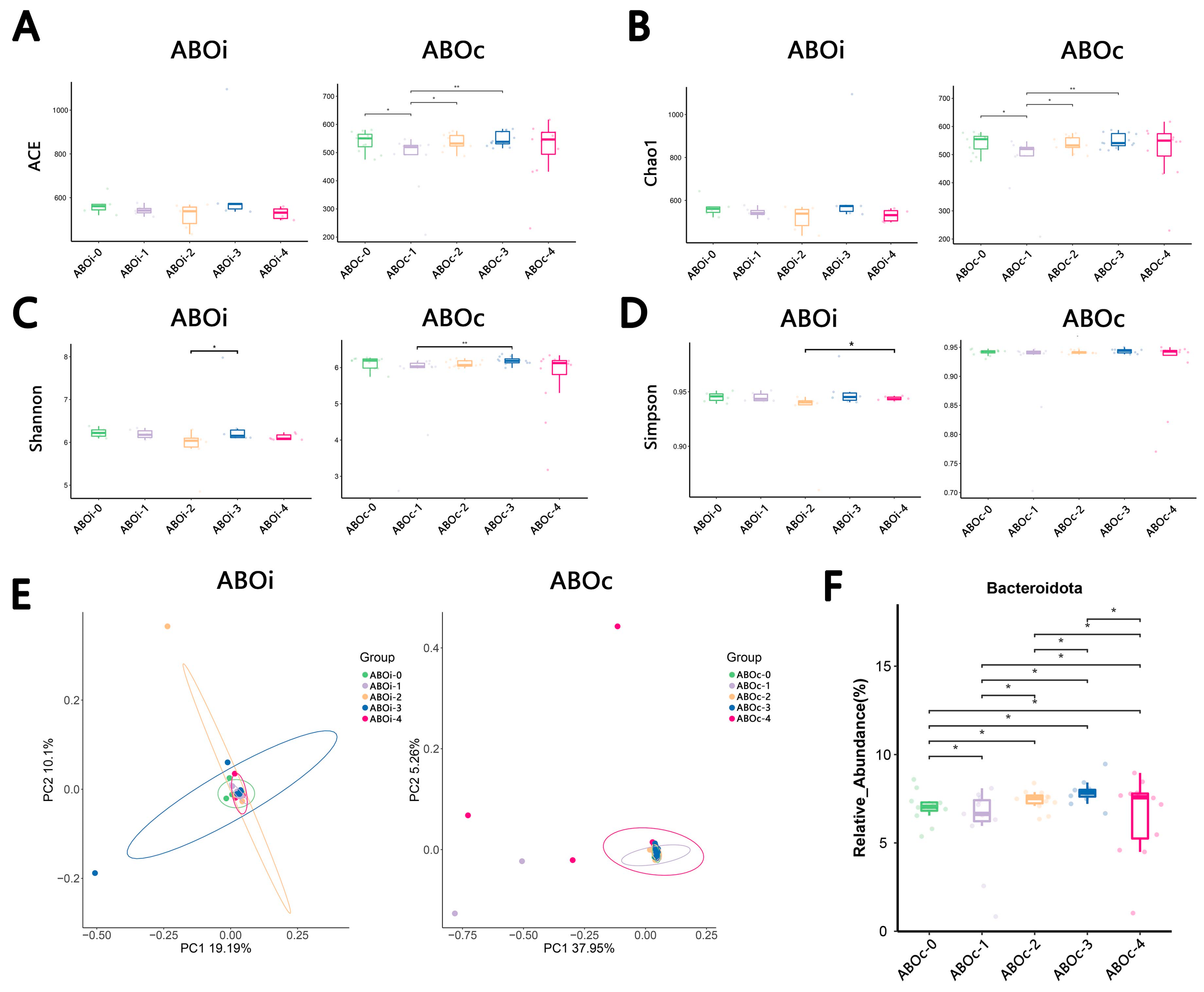
3. Results
3.1. Patient Characteristics and 16S rRNA Sequencing
3.2. Comparison between Biliary Microbiota of ABOi LT and ABOc LT at Diverse Times
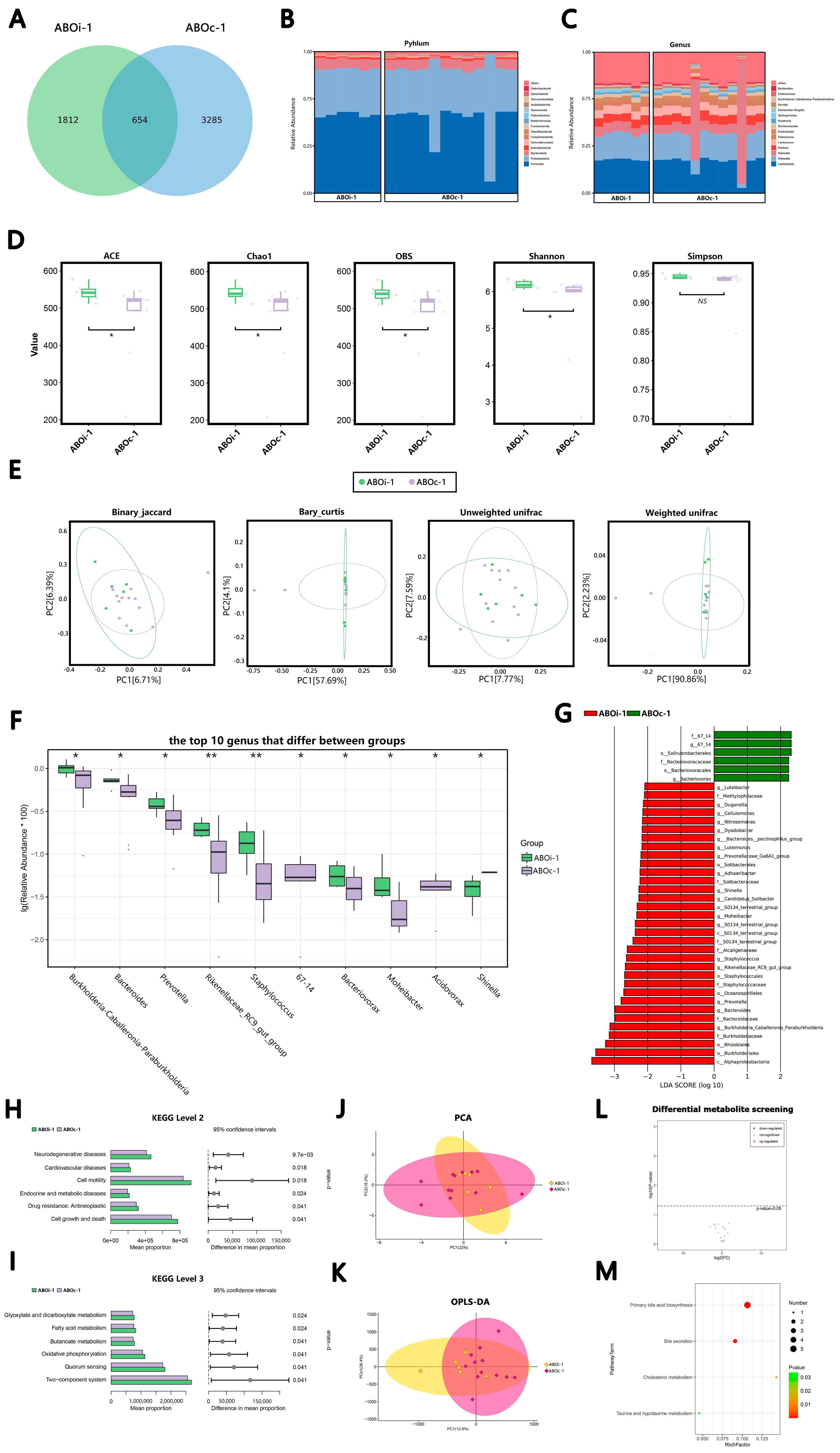
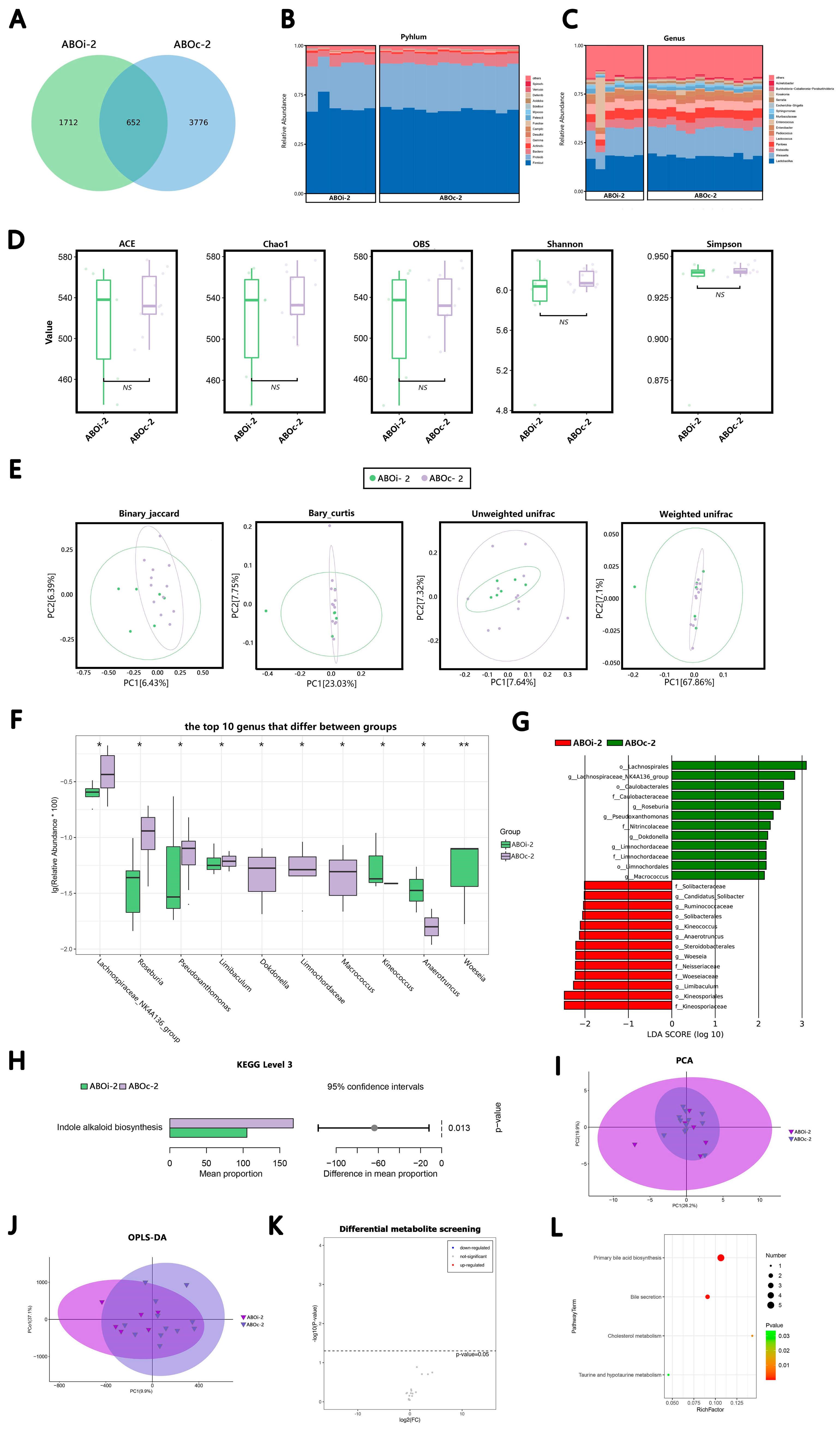
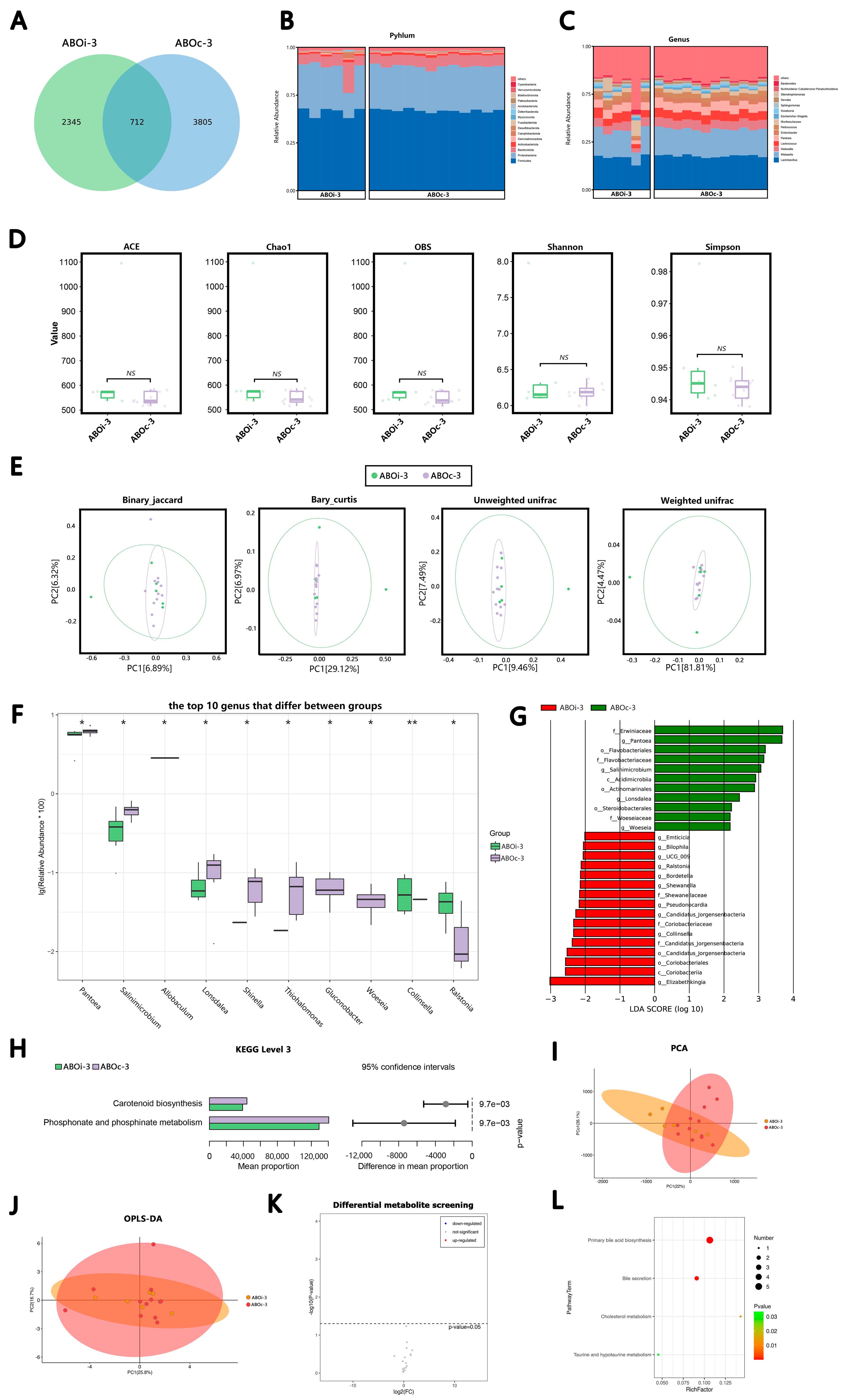
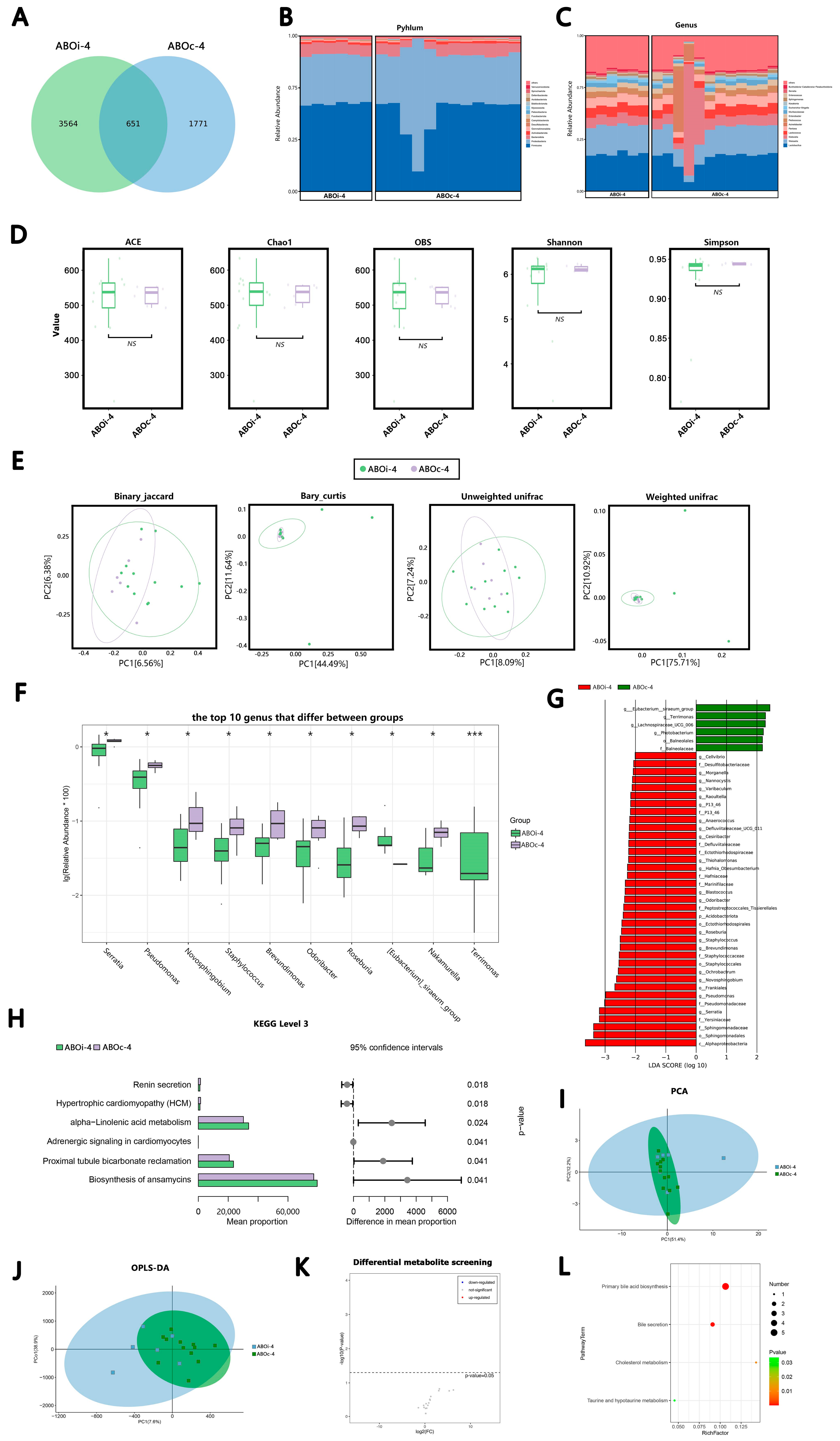
3.2.1. Two Days after LT
3.2.2. One Week after LT
3.2.3. Two Weeks after LT
3.2.4. One Month after LT
3.3. Changes in the Biliary Microbiota within One Month after LT
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Forner, A.; Reig, M.; Bruix, J. Hepatocellular carcinoma. Lancet 2018, 391, 1301–1314. [Google Scholar] [CrossRef]
- Gugenheim, J.; Samuel, D.; Reynes, M.; Bismuth, H. Liver transplantation across ABO blood group barriers. Lancet 1990, 336, 519–523. [Google Scholar] [CrossRef] [PubMed]
- Kawagishi, N.; Satomi, S. ABO-incompatible living donor liver transplantation: New insights into clinical relevance. Transplantation 2008, 85, 1523–1525. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.M.; Kwon, C.H.D.; Joh, J.W.; Han, S.; Yoo, J.; Kim, K.; Sinn, D.H.; Choi, G.S.; Gerber, D.A.; Egawa, H.; et al. ABO-incompatible Living Donor Liver Transplantation With Rituximab and Total Plasma Exchange Does Not Increase Hepatocellular Carcinoma Recurrence. Transplantation 2018, 102, 1695–1701. [Google Scholar] [CrossRef] [PubMed]
- Song, G.W.; Lee, S.G.; Hwang, S.; Kim, K.H.; Ahn, C.S.; Moon, D.B.; Ha, T.Y.; Jung, D.H.; Park, G.C.; Kim, W.J.; et al. ABO-Incompatible Adult Living Donor Liver Transplantation Under the Desensitization Protocol With Rituximab. Am. J. Transpl. 2016, 16, 157–170. [Google Scholar] [CrossRef] [Green Version]
- Raut, V.; Uemoto, S. Management of ABO-incompatible living-donor liver transplantation: Past and present trends. Surg. Today 2011, 41, 317–322. [Google Scholar] [CrossRef]
- Song, G.W.; Lee, S.G.; Hwang, S.; Kim, K.H.; Ahn, C.S.; Moon, D.B.; Ha, T.Y.; Jung, D.H.; Park, G.C.; Kang, S.H.; et al. Biliary stricture is the only concern in ABO-incompatible adult living donor liver transplantation in the rituximab era. J. Hepatol. 2014, 61, 575–582. [Google Scholar] [CrossRef]
- Liu, Y.; Sun, L.Y.; Zhu, Z.J.; Wei, L.; Qu, W.; Zeng, Z.G. Bile microbiota: New insights into biliary complications in liver transplant recipients. Ann. Transl. Med. 2020, 8, 354. [Google Scholar] [CrossRef]
- Han, J.; Wu, S.; Fan, Y.; Tian, Y.; Kong, J. Biliary Microbiota in Choledocholithiasis and Correlation With Duodenal Microbiota. Front. Cell. Infect. Microbiol. 2021, 11, 625589. [Google Scholar] [CrossRef]
- Choe, J.W.; Lee, J.M.; Hyun, J.J.; Lee, H.S. Analysis on Microbial Profiles & Components of Bile in Patients with Recurrent CBD Stones after Endoscopic CBD Stone Removal: A Preliminary Study. J. Clin. Med. 2021, 10, 3303. [Google Scholar] [CrossRef]
- Pereira, P.; Aho, V.; Arola, J.; Boyd, S.; Jokelainen, K.; Paulin, L.; Auvinen, P.; Färkkilä, M. Bile microbiota in primary sclerosing cholangitis: Impact on disease progression and development of biliary dysplasia. PLoS ONE 2017, 12, e0182924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saab, M.; Mestivier, D.; Sohrabi, M.; Rodriguez, C.; Khonsari, M.R.; Faraji, A.; Sobhani, I. Characterization of biliary microbiota dysbiosis in extrahepatic cholangiocarcinoma. PLoS ONE 2021, 16, e0247798. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Jia, J.D.; Sun, L.Y.; Zhu, Z.J.; Zhang, J.R.; Wei, L.; Qu, W.; Zeng, Z.G. Characteristics of bile microbiota in liver transplant recipients with biliary injury. Int. J. Clin. Exp. Pathol. 2018, 11, 481–489. [Google Scholar] [PubMed]
- Kaufman, H.S.; Magnuson, T.H.; Lillemoe, K.D.; Frasca, P.; Pitt, H.A. The role of bacteria in gallbladder and common duct stone formation. Ann. Surg. 1989, 209, 584–591, discussion 591–592. [Google Scholar] [CrossRef]
- Theron, J.; Cloete, T.E. Molecular techniques for determining microbial diversity and community structure in natural environments. Crit. Rev. Microbiol. 2000, 26, 37–57. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Paramsothy, S.; Kamm, M.A.; Kaakoush, N.O.; Walsh, A.J.; van den Bogaerde, J.; Samuel, D.; Leong, R.W.L.; Connor, S.; Ng, W.; Paramsothy, R.; et al. Multidonor intensive faecal microbiota transplantation for active ulcerative colitis: A randomised placebo-controlled trial. Lancet 2017, 389, 1218–1228. [Google Scholar] [CrossRef]
- Kang, D.W.; Adams, J.B.; Gregory, A.C.; Borody, T.; Chittick, L.; Fasano, A.; Khoruts, A.; Geis, E.; Maldonado, J.; McDonough-Means, S.; et al. Microbiota Transfer Therapy alters gut ecosystem and improves gastrointestinal and autism symptoms: An open-label study. Microbiome 2017, 5, 10. [Google Scholar] [CrossRef]
- Zhao, L.; Zhang, F.; Ding, X.; Wu, G.; Lam, Y.Y.; Wang, X.; Fu, H.; Xue, X.; Lu, C.; Ma, J.; et al. Gut bacteria selectively promoted by dietary fibers alleviate type 2 diabetes. Science 2018, 359, 1151–1156. [Google Scholar] [CrossRef]
- Liwinski, T.; Zenouzi, R.; John, C.; Ehlken, H.; Rühlemann, M.C.; Bang, C.; Groth, S.; Lieb, W.; Kantowski, M.; Andersen, N.; et al. Alterations of the bile microbiome in primary sclerosing cholangitis. Gut 2020, 69, 665–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, B.; Zhou, B.; Dong, C.; Zhang, R.; Xie, D.; Tian, Y.; Yang, L. Lactobacillus reuteri Alleviates Gastrointestinal Toxicity of Rituximab by Regulating the Proinflammatory T Cells in vivo. Front. Microbiol. 2021, 12, 645500. [Google Scholar] [CrossRef] [PubMed]
- Dinges, M.M.; Orwin, P.M.; Schlievert, P.M. Exotoxins of Staphylococcus aureus. Clin. Microbiol. Rev. 2000, 13, 16–34, table of contents. [Google Scholar] [CrossRef] [PubMed]
- Pennekamp, F.; Pontarp, M.; Tabi, A.; Altermatt, F.; Alther, R.; Choffat, Y.; Fronhofer, E.A.; Ganesanandamoorthy, P.; Garnier, A.; Griffiths, J.I.; et al. Biodiversity increases and decreases ecosystem stability. Nature 2018, 563, 109–112. [Google Scholar] [CrossRef] [Green Version]
- Turnbaugh, P.J.; Hamady, M.; Yatsunenko, T.; Cantarel, B.L.; Duncan, A.; Ley, R.E.; Sogin, M.L.; Jones, W.J.; Roe, B.A.; Affourtit, J.P.; et al. A core gut microbiome in obese and lean twins. Nature 2009, 457, 480–484. [Google Scholar] [CrossRef] [Green Version]
- Dethlefsen, L.; Huse, S.; Sogin, M.L.; Relman, D.A. The pervasive effects of an antibiotic on the human gut microbiota, as revealed by deep 16S rRNA sequencing. PLoS Biol. 2008, 6, e280. [Google Scholar] [CrossRef]
- Song, W.; Sun, L.Y.; Zhu, Z.J.; Wei, L.; Qu, W.; Zeng, Z.G.; Yang, Y.S. Characteristics of Gut Microbiota in Children With Biliary Atresia After Liver Transplantation. Front. Physiol. 2021, 12, 704313. [Google Scholar] [CrossRef]
- Wong, J.M.; de Souza, R.; Kendall, C.W.; Emam, A.; Jenkins, D.J. Colonic health: Fermentation and short chain fatty acids. J. Clin. Gastroenterol. 2006, 40, 235–243. [Google Scholar] [CrossRef]
- Vince, A.J.; McNeil, N.I.; Wager, J.D.; Wrong, O.M. The effect of lactulose, pectin, arabinogalactan and cellulose on the production of organic acids and metabolism of ammonia by intestinal bacteria in a faecal incubation system. Br. J. Nutr. 1990, 63, 17–26. [Google Scholar] [CrossRef]
- Zoetendal, E.G.; Ben-Amor, K.; Harmsen, H.J.; Schut, F.; Akkermans, A.D.; de Vos, W.M. Quantification of uncultured Ruminococcus obeum-like bacteria in human fecal samples by fluorescent in situ hybridization and flow cytometry using 16S rRNA-targeted probes. Appl. Env. Microbiol. 2002, 68, 4225–4232. [Google Scholar] [CrossRef]
- Walterson, A.M.; Stavrinides, J. Pantoea: Insights into a highly versatile and diverse genus within the Enterobacteriaceae. FEMS Microbiol. Rev. 2015, 39, 968–984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fender, J.E.; Bender, C.M.; Stella, N.A.; Lahr, R.M.; Kalivoda, E.J.; Shanks, R.M. Serratia marcescens quinoprotein glucose dehydrogenase activity mediates medium acidification and inhibition of prodigiosin production by glucose. Appl. Env. Microbiol. 2012, 78, 6225–6235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, J.W.; Ren, Z.G.; Lu, H.F.; Zhang, H.; Li, A.; Cui, G.Y.; Jia, J.J.; Xie, H.Y.; Chen, X.H.; He, Y.; et al. Optimal immunosuppressor induces stable gut microbiota after liver transplantation. World J. Gastroenterol. 2018, 24, 3871–3883. [Google Scholar] [CrossRef] [PubMed]
- Bajaj, J.S.; Khoruts, A. Microbiota changes and intestinal microbiota transplantation in liver diseases and cirrhosis. J. Hepatol. 2020, 72, 1003–1027. [Google Scholar] [CrossRef] [Green Version]
- Kriss, M.; Verna, E.C.; Rosen, H.R.; Lozupone, C.A. Functional Microbiomics in Liver Transplantation: Identifying Novel Targets for Improving Allograft Outcomes. Transplantation 2019, 103, 668–678. [Google Scholar] [CrossRef]
- Molinero, N.; Ruiz, L.; Milani, C.; Gutiérrez-Díaz, I.; Sánchez, B.; Mangifesta, M.; Segura, J.; Cambero, I.; Campelo, A.B.; García-Bernardo, C.M.; et al. The human gallbladder microbiome is related to the physiological state and the biliary metabolic profile. Microbiome 2019, 7, 100. [Google Scholar] [CrossRef] [Green Version]
- D’Amico, F.; Bertacco, A.; Finotti, M.; Di Renzo, C.; Rodriguez-Davalos, M.I.; Gondolesi, G.E.; Cillo, U.; Mulligan, D.; Geibel, J. Bile Microbiota in Liver Transplantation: Proof of Concept Using Gene Amplification in a Heterogeneous Clinical Scenario. Front. Surg. 2021, 8, 621525. [Google Scholar] [CrossRef]
- Choi, S.J.; Kim, Y.; Jeon, J.; Gwak, H.J.; Kim, M.; Kang, K.; Kim, Y.; Jeong, J.; Jung, Y.K.; Lee, K.G.; et al. Association of Microbial Dysbiosis with Gallbladder Diseases Identified by Bile Microbiome Profiling. J. Korean Med. Sci. 2021, 36, e189. [Google Scholar] [CrossRef]
- Lyu, Z.; Yu, T.; Zhang, L.; Xu, X.; Zhang, Y.; Li, J.; Li, Z.; Zhang, W.; Hou, S. Analysis of the relationship between bile duct and duodenal microbiota reveals that potential dysbacteriosis is the main cause of primary common bile duct stones. Synth. Syst. Biotechnol. 2021, 6, 414–428. [Google Scholar] [CrossRef]
- Moore, E.W.; Celic, L.; Ostrow, J.D. Interactions between ionized calcium and sodium taurocholate: Bile salts are important buffers for prevention of calcium-containing gallstones. Gastroenterology 1982, 83, 1079–1089. [Google Scholar] [CrossRef]
- Berman, M.D.; Carey, M.C. Metastable and equilibrium phase diagrams of unconjugated bilirubin IXα as functions of pH in model bile systems: Implications for pigment gallstone formation. Am. J. Physiol. Liver Physiol. 2015, 308, G42–G55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kochhar, G.; Parungao, J.M.; Hanouneh, I.A.; Parsi, M.A. Biliary complications following liver transplantation. World J. Gastroenterol. 2013, 19, 2841–2846. [Google Scholar] [CrossRef] [PubMed]
- López-Andújar, R.; Orón, E.M.; Carregnato, A.F.; Suárez, F.V.; Herraiz, A.M.; Rodríguez, F.S.; Carbó, J.J.; Ibars, E.P.; Sos, J.E.; Suárez, A.R.; et al. T-tube or no T-tube in cadaveric orthotopic liver transplantation: The eternal dilemma: Results of a prospective and randomized clinical trial. Ann. Surg. 2013, 258, 21–29. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ABOi LT Group | ABOc LT Group | p-Value | |
|---|---|---|---|
| Patients, n | 6 | 12 | NA |
| Male, n (%) | 6 (100%) | 11 (91.7%) | 1.000 |
| Age (years) | 48 (32–59) | 46 (24–56) | 0.424 |
| BMI (kg/m2) | 21.75 (18.9–26.3) | 22.31 (16.9–26.1) | 0.574 |
| Previous abdominal surgery, n (%) | 1 (16.7%) | 2 (16.7%) | 1.000 |
| WBC (109/L) | 11.5 (7.1–11.3) | 4.8 (0.9–12.5) | 0.007 |
| Hb (g/L) | 95 (84–106) | 86 (62–111) | 0.146 |
| Blood Plt (109/L) | 128 (51–212) | 46 (19–97) | 0.003 |
| CRP (g/dL) | 27.2 (21.7–51.2) | 8.7 (<5–20.7) | 0.004 |
| ALB (U/L) | 33.9 (28.3–37.9) | 33.8 (30–42.2) | 0.639 |
| ALT (U/L) | 73 (8–200) | 38 (11–130) | 0.190 |
| AST (U/L) | 150 (30–534) | 54 (18–90) | 0.083 |
| GGT (U/L) | 137 (18–438) | 40 (15–107) | 0.223 |
| ALP (U/L) | 220 (45–798) | 107 (44–186) | 0.606 |
| TBIL (μmol/L) | 263 (37–594) | 276 (21–638) | 0.851 |
| Cr (μmol/L) | 112 (39–346) | 80 (56–138) | 0.925 |
| MELD score | 28 (18–36) | 26 (10–37) | 0.673 |
| Warm ischemia time (min) | 3.3 (3–4) | 3.3 (3–5) | 0.811 |
| Cold ischemia time (min) | 62.8 (42–79) | 42.3 (29–58) | 0.006 |
| Operation time (min) | 343 (229–420) | 287 (207–366) | 0.061 |
| Post-transplantation ICU stay time (days) | 16 (10–24) | 14 (5–36) | 0.477 |
| Donor age (years) | 50 (36–64) | 45 (11–59) | 0.605 |
| Donor male, n (%) | 5 (83.3%) | 11 (91.7%) | 1.000 |
| Donor BMI (kg/m2) | 24.72 (20.8–30.4) | 22.83 (14.7–27.4) | 0.454 |
| GRWR (%) | 1.59 (1.18–2.05) | 1.70 (1.05–2.40) | 0.708 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiao, M.; Wan, Z.; Lin, X.; Wang, D.; Chen, Z.; Gu, Y.; Ding, S.; Zheng, S.; Li, Q. ABO-Incompatible Liver Transplantation under the Desensitization Protocol with Rituximab: Effect on Biliary Microbiota and Metabolites. J. Clin. Med. 2023, 12, 141. https://doi.org/10.3390/jcm12010141
Xiao M, Wan Z, Lin X, Wang D, Chen Z, Gu Y, Ding S, Zheng S, Li Q. ABO-Incompatible Liver Transplantation under the Desensitization Protocol with Rituximab: Effect on Biliary Microbiota and Metabolites. Journal of Clinical Medicine. 2023; 12(1):141. https://doi.org/10.3390/jcm12010141
Chicago/Turabian StyleXiao, Min, Zhenmiao Wan, Xin Lin, Di Wang, Zhitao Chen, Yangjun Gu, Songming Ding, Shusen Zheng, and Qiyong Li. 2023. "ABO-Incompatible Liver Transplantation under the Desensitization Protocol with Rituximab: Effect on Biliary Microbiota and Metabolites" Journal of Clinical Medicine 12, no. 1: 141. https://doi.org/10.3390/jcm12010141