


Biomarkers for Immune Checkpoint Inhibitors in Renal Cell Carcinoma


Abstract
1. Introduction
2. A Brier Primer on Anti-Tumor Immunity
3. Tumor Mutation Burden
4. Cytotoxic T Cells
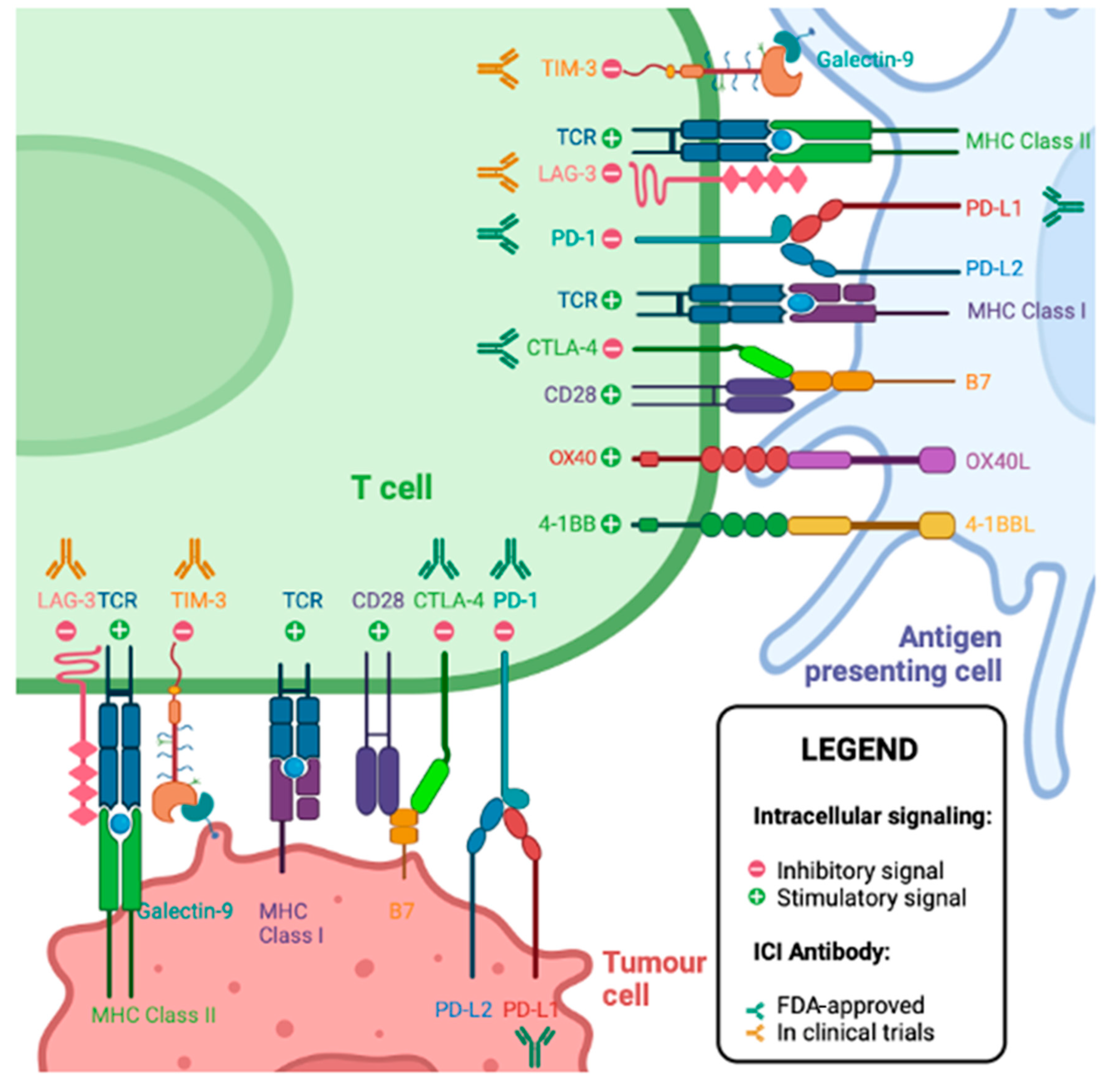
5. T Cell Checkpoint Molecules
6. Cytokines
7. Angiogenesis Factors
8. Gene Expression Profiles
9. Programmed Death-Ligand 1
10. PBRM-1 Loss
11. Future Directions
12. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Moch, H.; Amin, M.B.; Berney, D.M.; Comperat, E.M.; Gill, A.J.; Hartmann, A.; Menon, S.; Raspollini, M.R.; Rubin, M.A.; Srigley, J.R.; et al. The 2022 World Health Organization Classification of Tumours of the Urinary System and Male Genital Organs-Part A: Renal, Penile, and Testicular Tumours. Eur. Urol. 2022, 82, 458–468. [Google Scholar] [CrossRef] [PubMed]
- Choueiri, T.K.; Motzer, R.J. Systemic Therapy for Metastatic Renal-Cell Carcinoma. N. Engl. J. Med. 2017, 376, 354–366. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Jonasch, E.; Agarwal, N.; Alva, A.; Baine, M.; Beckermann, K.; Carlo, M.I.; Choueiri, T.K.; Costello, B.A.; Derweesh, I.H.; et al. Kidney Cancer, Version 3.2022, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2022, 20, 71–90. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Taube, J.M.; Anders, R.A.; Pardoll, D.M. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat. Rev. Cancer 2016, 16, 275–287. [Google Scholar] [CrossRef]
- Braun, D.A.; Bakouny, Z.; Hirsch, L.; Flippot, R.; Van Allen, E.M.; Wu, C.J.; Choueiri, T.K. Beyond conventional immune-checkpoint inhibition-novel immunotherapies for renal cell carcinoma. Nat. Rev. Clin. Oncol. 2021, 18, 199–214. [Google Scholar] [CrossRef]
- Motzer, R.J.; McDermott, D.F.; Escudier, B.; Burotto, M.; Choueiri, T.K.; Hammers, H.J.; Barthelemy, P.; Plimack, E.R.; Porta, C.; George, S.; et al. Conditional survival and long-term efficacy with nivolumab plus ipilimumab versus sunitinib in patients with advanced renal cell carcinoma. Cancer 2022, 128, 2085–2097. [Google Scholar] [CrossRef]
- Plimack, E.R.; Stus, V.; Gafanov, R.; Waddell, T.; Nosov, D.; Pouliot, F.; Alekseev, B.; Soulieres, D.; Melichar, B.; Vynnychenko, I.O.; et al. Pembrolizumab plus axitinib versus sunitinib as first-line therapy for advanced clear cell renal cell carcinoma: 5-year analysis of KEYNOTE-426. J. Clin. Oncol. 2023, 41, LBA4501. [Google Scholar] [CrossRef]
- Gebrael, G.; Sahu, K.K.; Agarwal, N.; Maughan, B.L. Update on combined immunotherapy for the treatment of advanced renal cell carcinoma. Hum. Vaccines Immunother. 2023, 19, 2193528. [Google Scholar] [CrossRef]
- Motzer, R.J.; Porta, C.; Eto, M.; Powles, T.; Grünwald, V.; Alekseev, B.; Rha, S.Y.; Merchan, J.R.; Goh, J.C.; Kapoor, A.; et al. Final prespecified overall survival (OS) analysis of CLEAR: 4-year follow-up of lenvatinib plus pembrolizumab (L+P) vs sunitinib (S) in patients (pts) with advanced renal cell carcinoma (aRCC). J. Clin. Oncol. 2023, 41, 4502-4502. [Google Scholar] [CrossRef]
- Li, H.; Sahu, K.K.; Maughan, B.L. Mechanism and Management of Checkpoint Inhibitor-Related Toxicities in Genitourinary Cancers. Cancers 2022, 14, 2460. [Google Scholar] [CrossRef]
- Jhunjhunwala, S.; Hammer, C.; Delamarre, L. Antigen presentation in cancer: Insights into tumour immunogenicity and immune evasion. Nat. Rev. Cancer 2021, 21, 298–312. [Google Scholar] [CrossRef]
- Chen, Y.; Song, Y.; Du, W.; Gong, L.; Chang, H.; Zou, Z. Tumor-associated macrophages: An accomplice in solid tumor progression. J. Biomed. Sci. 2019, 26, 78. [Google Scholar] [CrossRef]
- Sakaguchi, S.; Mikami, N.; Wing, J.B.; Tanaka, A.; Ichiyama, K.; Ohkura, N. Regulatory T Cells and Human Disease. Annu. Rev. Immunol. 2020, 38, 541–566. [Google Scholar] [CrossRef]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef]
- Scheper, W.; Kelderman, S.; Fanchi, L.F.; Linnemann, C.; Bendle, G.; de Rooij, M.A.J.; Hirt, C.; Mezzadra, R.; Slagter, M.; Dijkstra, K.; et al. Low and variable tumor reactivity of the intratumoral TCR repertoire in human cancers. Nat. Med. 2019, 25, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Kalbasi, A.; Ribas, A. Tumour-intrinsic resistance to immune checkpoint blockade. Nat. Rev. Immunol. 2020, 20, 25–39. [Google Scholar] [CrossRef]
- Urbantat, R.M.; Vajkoczy, P.; Brandenburg, S. Advances in Chemokine Signaling Pathways as Therapeutic Targets in Glioblastoma. Cancers 2021, 13, 2983. [Google Scholar] [CrossRef]
- Sun, C.; Mezzadra, R.; Schumacher, T.N. Regulation and Function of the PD-L1 Checkpoint. Immunity 2018, 48, 434–452. [Google Scholar] [CrossRef]
- Zagorulya, M.; Duong, E.; Spranger, S. Impact of anatomic site on antigen-presenting cells in cancer. J. Immunother. Cancer 2020, 8, e001204. [Google Scholar] [CrossRef] [PubMed]
- Bagchi, S.; Yuan, R.; Engleman, E.G. Immune Checkpoint Inhibitors for the Treatment of Cancer: Clinical Impact and Mechanisms of Response and Resistance. Annu. Rev. Pathol. 2021, 16, 223–249. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.D.; Coukos, G.; Holt, R.A.; Nelson, B.H. Targeting the undruggable: Immunotherapy meets personalized oncology in the genomic era. Ann. Oncol. 2015, 26, 2367–2374. [Google Scholar] [CrossRef]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef]
- Bos, R.; Marquardt, K.L.; Cheung, J.; Sherman, L.A. Functional differences between low- and high-affinity CD8+ T cells in the tumor environment. Oncoimmunology 2012, 1, 1239–1247. [Google Scholar] [CrossRef]
- Tran, E.; Robbins, P.F.; Rosenberg, S.A. ‘Final common pathway’ of human cancer immunotherapy: Targeting random somatic mutations. Nat. Immunol. 2017, 18, 255–262. [Google Scholar] [CrossRef]
- Lowery, F.J.; Krishna, S.; Yossef, R.; Parikh, N.B.; Chatani, P.D.; Zacharakis, N.; Parkhurst, M.R.; Levin, N.; Sindiri, S.; Sachs, A.; et al. Molecular signatures of antitumor neoantigen-reactive T cells from metastatic human cancers. Science 2022, 375, 877–884. [Google Scholar] [CrossRef]
- Leidner, R.; Sanjuan Silva, N.; Huang, H.; Sprott, D.; Zheng, C.; Shih, Y.P.; Leung, A.; Payne, R.; Sutcliffe, K.; Cramer, J.; et al. Neoantigen T-Cell Receptor Gene Therapy in Pancreatic Cancer. N. Engl. J. Med. 2022, 386, 2112–2119. [Google Scholar] [CrossRef]
- Tran, E.; Robbins, P.F.; Lu, Y.C.; Prickett, T.D.; Gartner, J.J.; Jia, L.; Pasetto, A.; Zheng, Z.; Ray, S.; Groh, E.M.; et al. T-Cell Transfer Therapy Targeting Mutant KRAS in Cancer. N. Engl. J. Med. 2016, 375, 2255–2262. [Google Scholar] [CrossRef]
- Martin, S.D.; Brown, S.D.; Wick, D.A.; Nielsen, J.S.; Kroeger, D.R.; Twumasi-Boateng, K.; Holt, R.A.; Nelson, B.H. Low Mutation Burden in Ovarian Cancer May Limit the Utility of Neoantigen-Targeted Vaccines. PLoS ONE 2016, 11, e0155189. [Google Scholar] [CrossRef]
- Martin, S.D.; Wick, D.A.; Nielsen, J.S.; Little, N.; Holt, R.A.; Nelson, B.H. A library-based screening method identifies neoantigen-reactive T cells in peripheral blood prior to relapse of ovarian cancer. Oncoimmunology 2017, 7, e1371895. [Google Scholar] [CrossRef] [PubMed]
- Merino, D.M.; McShane, L.M.; Fabrizio, D.; Funari, V.; Chen, S.J.; White, J.R.; Wenz, P.; Baden, J.; Barrett, J.C.; Chaudhary, R.; et al. Establishing guidelines to harmonize tumor mutational burden (TMB): In silico assessment of variation in TMB quantification across diagnostic platforms: Phase I of the Friends of Cancer Research TMB Harmonization Project. J. Immunother. Cancer 2020, 8, e000147. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.C.; Selitsky, S.R.; Chai, S.; Armistead, P.M.; Vincent, B.G.; Serody, J.S. Alternative tumour-specific antigens. Nat. Rev. Cancer 2019, 19, 465–478. [Google Scholar] [CrossRef]
- Vega, D.M.; Yee, L.M.; McShane, L.M.; Williams, P.M.; Chen, L.; Vilimas, T.; Fabrizio, D.; Funari, V.; Newberg, J.; Bruce, L.K.; et al. Aligning tumor mutational burden (TMB) quantification across diagnostic platforms: Phase II of the Friends of Cancer Research TMB Harmonization Project. Ann. Oncol. 2021, 32, 1626–1636. [Google Scholar] [CrossRef]
- Esposito Abate, R.; Cheetham, M.H.; Fairley, J.A.; Pasquale, R.; Sacco, A.; Nicola, W.; Deans, Z.C.; Patton, S.J.; Normanno, N. External quality assessment (EQA) for tumor mutational burden: Results of an international IQN path feasibility pilot scheme. Virchows Arch. 2023, 482, 347–355. [Google Scholar] [CrossRef]
- Fridland, S.; Choi, J.; Nam, M.; Schellenberg, S.J.; Kim, E.; Lee, G.; Yoon, N.; Chae, Y.K. Assessing tumor heterogeneity: Integrating tissue and circulating tumor DNA (ctDNA) analysis in the era of immuno-oncology-blood TMB is not the same as tissue TMB. J. Immunother. Cancer 2021, 9, e002551. [Google Scholar] [CrossRef]
- Lin, M.; Whitmire, S.; Chen, J.; Farrel, A.; Shi, X.; Guo, J.T. Effects of short indels on protein structure and function in human genomes. Sci. Rep. 2017, 7, 9313. [Google Scholar] [CrossRef]
- Wu, H.X.; Wang, Z.X.; Zhao, Q.; Chen, D.L.; He, M.M.; Yang, L.P.; Wang, Y.N.; Jin, Y.; Ren, C.; Luo, H.Y.; et al. Tumor mutational and indel burden: A systematic pan-cancer evaluation as prognostic biomarkers. Ann. Transl. Med. 2019, 7, 640. [Google Scholar] [CrossRef]
- Turajlic, S.; Litchfield, K.; Xu, H.; Rosenthal, R.; McGranahan, N.; Reading, J.L.; Wong, Y.N.S.; Rowan, A.; Kanu, N.; Al Bakir, M.; et al. Insertion-and-deletion-derived tumour-specific neoantigens and the immunogenic phenotype: A pan-cancer analysis. Lancet Oncol. 2017, 18, 1009–1021. [Google Scholar] [CrossRef]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef]
- Yarchoan, M.; Hopkins, A.; Jaffee, E.M. Tumor Mutational Burden and Response Rate to PD-1 Inhibition. N. Engl. J. Med. 2017, 377, 2500–2501. [Google Scholar] [CrossRef] [PubMed]
- Samstein, R.M.; Lee, C.H.; Shoushtari, A.N.; Hellmann, M.D.; Shen, R.; Janjigian, Y.Y.; Barron, D.A.; Zehir, A.; Jordan, E.J.; Omuro, A.; et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat. Genet. 2019, 51, 202–206. [Google Scholar] [CrossRef] [PubMed]
- Snyder, A.; Makarov, V.; Merghoub, T.; Yuan, J.; Zaretsky, J.M.; Desrichard, A.; Walsh, L.A.; Postow, M.A.; Wong, P.; Ho, T.S.; et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N. Engl. J. Med. 2014, 371, 2189–2199. [Google Scholar] [CrossRef]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef]
- Ready, N.; Hellmann, M.D.; Awad, M.M.; Otterson, G.A.; Gutierrez, M.; Gainor, J.F.; Borghaei, H.; Jolivet, J.; Horn, L.; Mates, M.; et al. First-Line Nivolumab Plus Ipilimumab in Advanced Non-Small-Cell Lung Cancer (CheckMate 568): Outcomes by Programmed Death Ligand 1 and Tumor Mutational Burden as Biomarkers. J. Clin. Oncol. 2019, 37, 992–1000. [Google Scholar] [CrossRef]
- Rizvi, N.A.; Cho, B.C.; Reinmuth, N.; Lee, K.H.; Luft, A.; Ahn, M.J.; van den Heuvel, M.M.; Cobo, M.; Vicente, D.; Smolin, A.; et al. Durvalumab with or without Tremelimumab vs Standard Chemotherapy in First-Line Treatment of Metastatic Non-Small Cell Lung Cancer: The MYSTIC Phase 3 Randomized Clinical Trial. JAMA Oncol. 2020, 6, 661–674. [Google Scholar] [CrossRef]
- Marabelle, A.; Fakih, M.; Lopez, J.; Shah, M.; Shapira-Frommer, R.; Nakagawa, K.; Chung, H.C.; Kindler, H.L.; Lopez-Martin, J.A.; Miller, W.H., Jr.; et al. Association of tumour mutational burden with outcomes in patients with advanced solid tumours treated with pembrolizumab: Prospective biomarker analysis of the multicohort, open-label, phase 2 KEYNOTE-158 study. Lancet Oncol. 2020, 21, 1353–1365. [Google Scholar] [CrossRef]
- Cristescu, R.; Aurora-Garg, D.; Albright, A.; Xu, L.; Liu, X.Q.; Loboda, A.; Lang, L.; Jin, F.; Rubin, E.H.; Snyder, A.; et al. Tumor mutational burden predicts the efficacy of pembrolizumab monotherapy: A pan-tumor retrospective analysis of participants with advanced solid tumors. J. Immunother. Cancer 2022, 10, e003091. [Google Scholar] [CrossRef]
- Marcus, L.; Fashoyin-Aje, L.A.; Donoghue, M.; Yuan, M.; Rodriguez, L.; Gallagher, P.S.; Philip, R.; Ghosh, S.; Theoret, M.R.; Beaver, J.A.; et al. FDA Approval Summary: Pembrolizumab for the Treatment of Tumor Mutational Burden-High Solid Tumors. Clin. Cancer Res. 2021, 27, 4685–4689. [Google Scholar] [CrossRef]
- Chalmers, Z.R.; Connelly, C.F.; Fabrizio, D.; Gay, L.; Ali, S.M.; Ennis, R.; Schrock, A.; Campbell, B.; Shlien, A.; Chmielecki, J.; et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017, 9, 34. [Google Scholar] [CrossRef] [PubMed]
- Tucker, M.D.; Rini, B.I. Predicting Response to Immunotherapy in Metastatic Renal Cell Carcinoma. Cancers 2020, 12, 2662. [Google Scholar] [CrossRef]
- Wood, M.A.; Weeder, B.R.; David, J.K.; Nellore, A.; Thompson, R.F. Burden of tumor mutations, neoepitopes, and other variants are weak predictors of cancer immunotherapy response and overall survival. Genome Med. 2020, 12, 33. [Google Scholar] [CrossRef]
- Labriola, M.K.; Zhu, J.; Gupta, R.T.; McCall, S.; Jackson, J.; Kong, E.F.; White, J.R.; Cerqueira, G.; Gerding, K.; Simmons, J.K.; et al. Characterization of tumor mutation burden, PD-L1 and DNA repair genes to assess relationship to immune checkpoint inhibitors response in metastatic renal cell carcinoma. J. Immunother. Cancer 2020, 8, e000319. [Google Scholar] [CrossRef]
- Motzer, R.J.; Choueiri, T.K.; McDermott, D.F.; Powles, T.; Vano, Y.A.; Gupta, S.; Yao, J.; Han, C.; Ammar, R.; Papillon-Cavanagh, S.; et al. Biomarker analysis from CheckMate 214: Nivolumab plus ipilimumab versus sunitinib in renal cell carcinoma. J. Immunother. Cancer 2022, 10, e004316. [Google Scholar] [CrossRef]
- McDermott, D.F.; Huseni, M.A.; Atkins, M.B.; Motzer, R.J.; Rini, B.I.; Escudier, B.; Fong, L.; Joseph, R.W.; Pal, S.K.; Reeves, J.A.; et al. Clinical activity and molecular correlates of response to atezolizumab alone or in combination with bevacizumab versus sunitinib in renal cell carcinoma. Nat. Med. 2018, 24, 749–757. [Google Scholar] [CrossRef]
- Dizman, N.; Lyou, Y.; Salgia, N.; Bergerot, P.G.; Hsu, J.; Enriquez, D.; Izatt, T.; Trent, J.M.; Byron, S.; Pal, S. Correlates of clinical benefit from immunotherapy and targeted therapy in metastatic renal cell carcinoma: Comprehensive genomic and transcriptomic analysis. J. Immunother. Cancer 2020, 8, e000953. [Google Scholar] [CrossRef]
- Voss, M.H.; Azad, A.A.; Hansen, A.R.; Gray, J.E.; Welsh, S.J.; Song, X.; Kuziora, M.; Meinecke, L.; Blando, J.; Achour, I.; et al. A Randomized Phase II Study of MEDI0680 in Combination with Durvalumab versus Nivolumab Monotherapy in Patients with Advanced or Metastatic Clear-Cell Renal Cell Carcinoma. Clin. Cancer Res. 2022, 28, 3032–3041. [Google Scholar] [CrossRef]
- Rini, B.I.; Powles, T.; Atkins, M.B.; Escudier, B.; McDermott, D.F.; Suarez, C.; Bracarda, S.; Stadler, W.M.; Donskov, F.; Lee, J.L.; et al. Atezolizumab plus bevacizumab versus sunitinib in patients with previously untreated metastatic renal cell carcinoma (IMmotion151): A multicentre, open-label, phase 3, randomised controlled trial. Lancet 2019, 393, 2404–2415. [Google Scholar] [CrossRef]
- Motzer, R.J.; Powles, T.; Atkins, M.B.; Escudier, B.; McDermott, D.F.; Alekseev, B.Y.; Lee, J.L.; Suarez, C.; Stroyakovskiy, D.; De Giorgi, U.; et al. Final Overall Survival and Molecular Analysis in IMmotion151, a Phase 3 Trial Comparing Atezolizumab Plus Bevacizumab vs. Sunitinib in Patients with Previously Untreated Metastatic Renal Cell Carcinoma. JAMA Oncol. 2022, 8, 275–280. [Google Scholar] [CrossRef]
- Motzer, R.J.; Penkov, K.; Haanen, J.; Rini, B.; Albiges, L.; Campbell, M.T.; Venugopal, B.; Kollmannsberger, C.; Negrier, S.; Uemura, M.; et al. Avelumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2019, 380, 1103–1115. [Google Scholar] [CrossRef]
- Haanen, J.; Larkin, J.; Choueiri, T.K.; Albiges, L.; Rini, B.I.; Atkins, M.B.; Schmidinger, M.; Penkov, K.; Michelon, E.; Wang, J.; et al. Extended follow-up from JAVELIN Renal 101: Subgroup analysis of avelumab plus axitinib versus sunitinib by the International Metastatic Renal Cell Carcinoma Database Consortium risk group in patients with advanced renal cell carcinoma. ESMO Open 2023, 8, 101210. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Rini, B.I.; McDermott, D.F.; Aren Frontera, O.; Hammers, H.J.; Carducci, M.A.; Salman, P.; Escudier, B.; Beuselinck, B.; Amin, A.; et al. Nivolumab plus ipilimumab versus sunitinib in first-line treatment for advanced renal cell carcinoma: Extended follow-up of efficacy and safety results from a randomised, controlled, phase 3 trial. Lancet Oncol. 2019, 20, 1370–1385. [Google Scholar] [CrossRef] [PubMed]
- Rini, B.I.; Signoretti, S.; Choueiri, T.K.; McDermott, D.F.; Motzer, R.J.; George, S.; Powles, T.; Donskov, F.; Tykodi, S.S.; Pal, S.K.; et al. Long-term outcomes with nivolumab plus ipilimumab versus sunitinib in first-line treatment of patients with advanced sarcomatoid renal cell carcinoma. J. Immunother. Cancer 2022, 10, e005445. [Google Scholar] [CrossRef] [PubMed]
- Curtis, D. Identification of specific genes involved in schizophrenia aetiology-what difference does it make? Br. J. Psychiatry 2022, 221, 437–439. [Google Scholar] [CrossRef]
- Rini, B.I.; Plimack, E.R.; Stus, V.; Gafanov, R.; Hawkins, R.; Nosov, D.; Pouliot, F.; Alekseev, B.; Soulieres, D.; Melichar, B.; et al. Pembrolizumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2019, 380, 1116–1127. [Google Scholar] [CrossRef]
- Motzer, R.J.; Escudier, B.; McDermott, D.F.; George, S.; Hammers, H.J.; Srinivas, S.; Tykodi, S.S.; Sosman, J.A.; Procopio, G.; Plimack, E.R.; et al. Nivolumab versus Everolimus in Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2015, 373, 1803–1813. [Google Scholar] [CrossRef]
- Choueiri, T.K.; Powles, T.; Burotto, M.; Escudier, B.; Bourlon, M.T.; Zurawski, B.; Oyervides Juarez, V.M.; Hsieh, J.J.; Basso, U.; Shah, A.Y.; et al. Nivolumab plus Cabozantinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2021, 384, 829–841. [Google Scholar] [CrossRef]
- Motzer, R.; Alekseev, B.; Rha, S.Y.; Porta, C.; Eto, M.; Powles, T.; Grunwald, V.; Hutson, T.E.; Kopyltsov, E.; Mendez-Vidal, M.J.; et al. Lenvatinib plus Pembrolizumab or Everolimus for Advanced Renal Cell Carcinoma. N. Engl. J. Med. 2021, 384, 1289–1300. [Google Scholar] [CrossRef]
- Hansen, U.K.; Ramskov, S.; Bjerregaard, A.M.; Borch, A.; Andersen, R.; Draghi, A.; Donia, M.; Bentzen, A.K.; Marquard, A.M.; Szallasi, Z.; et al. Tumor-Infiltrating T Cells From Clear Cell Renal Cell Carcinoma Patients Recognize Neoepitopes Derived From Point and Frameshift Mutations. Front. Immunol. 2020, 11, 373. [Google Scholar] [CrossRef]
- Braun, D.A.; Hou, Y.; Bakouny, Z.; Ficial, M.; Sant’ Angelo, M.; Forman, J.; Ross-Macdonald, P.; Berger, A.C.; Jegede, O.A.; Elagina, L.; et al. Interplay of somatic alterations and immune infiltration modulates response to PD-1 blockade in advanced clear cell renal cell carcinoma. Nat. Med. 2020, 26, 909–918. [Google Scholar] [CrossRef]
- Bruni, D.; Angell, H.K.; Galon, J. The immune contexture and Immunoscore in cancer prognosis and therapeutic efficacy. Nat. Rev. Cancer 2020, 20, 662–680. [Google Scholar] [CrossRef]
- Mlecnik, B.; Bifulco, C.; Bindea, G.; Marliot, F.; Lugli, A.; Lee, J.J.; Zlobec, I.; Rau, T.T.; Berger, M.D.; Nagtegaal, I.D.; et al. Multicenter International Society for Immunotherapy of Cancer Study of the Consensus Immunoscore for the Prediction of Survival and Response to Chemotherapy in Stage III Colon Cancer. J. Clin. Oncol. 2020, 38, 3638–3651. [Google Scholar] [CrossRef]
- Ascierto, P.A.; Marincola, F.M.; Fox, B.A.; Galon, J. No time to die: The consensus immunoscore for predicting survival and response to chemotherapy of locally advanced colon cancer patients in a multicenter international study. Oncoimmunology 2020, 9, 1826132. [Google Scholar] [CrossRef]
- Li, F.; Li, C.; Cai, X.; Xie, Z.; Zhou, L.; Cheng, B.; Zhong, R.; Xiong, S.; Li, J.; Chen, Z.; et al. The association between CD8+ tumor-infiltrating lymphocytes and the clinical outcome of cancer immunotherapy: A systematic review and meta-analysis. EClinicalMedicine 2021, 41, 101134. [Google Scholar] [CrossRef]
- Jin, Y.; Tan, A.; Feng, J.; Xu, Z.; Wang, P.; Ruan, P.; Luo, R.; Weng, Y.; Peng, M. Prognostic Impact of Memory CD8+ T Cells on Immunotherapy in Human Cancers: A Systematic Review and Meta-Analysis. Front. Oncol. 2021, 11, 698076. [Google Scholar] [CrossRef]
- Litchfield, K.; Reading, J.L.; Puttick, C.; Thakkar, K.; Abbosh, C.; Bentham, R.; Watkins, T.B.K.; Rosenthal, R.; Biswas, D.; Rowan, A.; et al. Meta-analysis of tumor- and T cell-intrinsic mechanisms of sensitization to checkpoint inhibition. Cell 2021, 184, 596–614.e514. [Google Scholar] [CrossRef]
- Fridman, W.H.; Zitvogel, L.; Sautes-Fridman, C.; Kroemer, G. The immune contexture in cancer prognosis and treatment. Nat. Rev. Clin. Oncol. 2017, 14, 717–734. [Google Scholar] [CrossRef]
- Rooney, M.S.; Shukla, S.A.; Wu, C.J.; Getz, G.; Hacohen, N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 2015, 160, 48–61. [Google Scholar] [CrossRef]
- Motzer, R.J.; Robbins, P.B.; Powles, T.; Albiges, L.; Haanen, J.B.; Larkin, J.; Mu, X.J.; Ching, K.A.; Uemura, M.; Pal, S.K.; et al. Avelumab plus axitinib versus sunitinib in advanced renal cell carcinoma: Biomarker analysis of the phase 3 JAVELIN Renal 101 trial. Nat. Med. 2020, 26, 1733–1741. [Google Scholar] [CrossRef]
- Kim, J.H.; Kim, G.H.; Ryu, Y.M.; Kim, S.Y.; Kim, H.D.; Yoon, S.K.; Cho, Y.M.; Lee, J.L. Clinical implications of the tumor microenvironment using multiplexed immunohistochemistry in patients with advanced or metastatic renal cell carcinoma treated with nivolumab plus ipilimumab. Front. Oncol. 2022, 12, 969569. [Google Scholar] [CrossRef]
- Wherry, E.J.; Kurachi, M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499. [Google Scholar] [CrossRef] [PubMed]
- Buchbinder, E.I.; Desai, A. CTLA-4 and PD-1 Pathways: Similarities, Differences, and Implications of Their Inhibition. Am. J. Clin. Oncol. 2016, 39, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Blackburn, S.D.; Shin, H.; Haining, W.N.; Zou, T.; Workman, C.J.; Polley, A.; Betts, M.R.; Freeman, G.J.; Vignali, D.A.; Wherry, E.J. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat. Immunol. 2009, 10, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Anderson, A.C.; Joller, N.; Kuchroo, V.K. Lag-3, Tim-3, and TIGIT: Co-inhibitory Receptors with Specialized Functions in Immune Regulation. Immunity 2016, 44, 989–1004. [Google Scholar] [CrossRef] [PubMed]
- Andrews, L.P.; Yano, H.; Vignali, D.A.A. Inhibitory receptors and ligands beyond PD-1, PD-L1 and CTLA-4: Breakthroughs or backups. Nat. Immunol. 2019, 20, 1425–1434. [Google Scholar] [CrossRef]
- Wei, S.C.; Duffy, C.R.; Allison, J.P. Fundamental Mechanisms of Immune Checkpoint Blockade Therapy. Cancer Discov. 2018, 8, 1069–1086. [Google Scholar] [CrossRef]
- Lasorsa, F.; di Meo, N.A.; Rutigliano, M.; Milella, M.; Ferro, M.; Pandolfo, S.D.; Crocetto, F.; Tataru, O.S.; Autorino, R.; Battaglia, M.; et al. Immune Checkpoint Inhibitors in Renal Cell Carcinoma: Molecular Basis and Rationale for Their Use in Clinical Practice. Biomedicines 2023, 11, 1071. [Google Scholar] [CrossRef]
- Gros, A.; Robbins, P.F.; Yao, X.; Li, Y.F.; Turcotte, S.; Tran, E.; Wunderlich, J.R.; Mixon, A.; Farid, S.; Dudley, M.E.; et al. PD-1 identifies the patient-specific CD8+ tumor-reactive repertoire infiltrating human tumors. J. Clin. Investig. 2014, 124, 2246–2259. [Google Scholar] [CrossRef]
- Zhou, G.; Sprengers, D.; Boor, P.P.C.; Doukas, M.; Schutz, H.; Mancham, S.; Pedroza-Gonzalez, A.; Polak, W.G.; de Jonge, J.; Gaspersz, M.; et al. Antibodies Against Immune Checkpoint Molecules Restore Functions of Tumor-Infiltrating T Cells in Hepatocellular Carcinomas. Gastroenterology 2017, 153, 1107–1119.e1110. [Google Scholar] [CrossRef]
- Simoni, Y.; Becht, E.; Fehlings, M.; Loh, C.Y.; Koo, S.L.; Teng, K.W.W.; Yeong, J.P.S.; Nahar, R.; Zhang, T.; Kared, H.; et al. Bystander CD8+ T cells are abundant and phenotypically distinct in human tumour infiltrates. Nature 2018, 557, 575–579. [Google Scholar] [CrossRef]
- Ohmura, H.; Yamaguchi, K.; Hanamura, F.; Ito, M.; Makiyama, A.; Uchino, K.; Shimokawa, H.; Tamura, S.; Esaki, T.; Mitsugi, K.; et al. OX40 and LAG3 are associated with better prognosis in advanced gastric cancer patients treated with anti-programmed death-1 antibody. Br. J. Cancer 2020, 122, 1507–1517. [Google Scholar] [CrossRef]
- Yarchoan, M.; Cope, L.; Ruggieri, A.N.; Anders, R.A.; Noonan, A.M.; Goff, L.W.; Goyal, L.; Lacy, J.; Li, D.; Patel, A.K.; et al. Multicenter randomized phase II trial of atezolizumab with or without cobimetinib in biliary tract cancers. J. Clin. Investig. 2021, 131, e152670. [Google Scholar] [CrossRef]
- Thommen, D.S.; Koelzer, V.H.; Herzig, P.; Roller, A.; Trefny, M.; Dimeloe, S.; Kiialainen, A.; Hanhart, J.; Schill, C.; Hess, C.; et al. A transcriptionally and functionally distinct PD-1+ CD8+ T cell pool with predictive potential in non-small-cell lung cancer treated with PD-1 blockade. Nat. Med. 2018, 24, 994–1004. [Google Scholar] [CrossRef]
- Tawbi, H.A.; Schadendorf, D.; Lipson, E.J.; Ascierto, P.A.; Matamala, L.; Castillo Gutierrez, E.; Rutkowski, P.; Gogas, H.J.; Lao, C.D.; De Menezes, J.J.; et al. Relatlimab and Nivolumab versus Nivolumab in Untreated Advanced Melanoma. N. Engl. J. Med. 2022, 386, 24–34. [Google Scholar] [CrossRef]
- Amaria, R.N.; Postow, M.; Burton, E.M.; Tetzlaff, M.T.; Ross, M.I.; Torres-Cabala, C.; Glitza, I.C.; Duan, F.; Milton, D.R.; Busam, K.; et al. Neoadjuvant relatlimab and nivolumab in resectable melanoma. Nature 2022, 611, 155–160. [Google Scholar] [CrossRef]
- Curigliano, G.; Gelderblom, H.; Mach, N.; Doi, T.; Tai, D.; Forde, P.M.; Sarantopoulos, J.; Bedard, P.L.; Lin, C.C.; Hodi, F.S.; et al. Phase I/Ib Clinical Trial of Sabatolimab, an Anti-TIM-3 Antibody, Alone and in Combination with Spartalizumab, an Anti-PD-1 Antibody, in Advanced Solid Tumors. Clin. Cancer Res. 2021, 27, 3620–3629. [Google Scholar] [CrossRef]
- Giraldo, N.A.; Becht, E.; Vano, Y.; Petitprez, F.; Lacroix, L.; Validire, P.; Sanchez-Salas, R.; Ingels, A.; Oudard, S.; Moatti, A.; et al. Tumor-Infiltrating and Peripheral Blood T-Cell Immunophenotypes Predict Early Relapse in Localized Clear Cell Renal Cell Carcinoma. Clin. Cancer Res. 2017, 23, 4416–4428. [Google Scholar] [CrossRef]
- Dai, S.; Zeng, H.; Liu, Z.; Jin, K.; Jiang, W.; Wang, Z.; Lin, Z.; Xiong, Y.; Wang, J.; Chang, Y.; et al. Intratumoral CXCL13+CD8+T cell infiltration determines poor clinical outcomes and immunoevasive contexture in patients with clear cell renal cell carcinoma. J. Immunother. Cancer 2021, 9, e001823. [Google Scholar] [CrossRef]
- Qi, Y.; Xia, Y.; Lin, Z.; Qu, Y.; Qi, Y.; Chen, Y.; Zhou, Q.; Zeng, H.; Wang, J.; Chang, Y.; et al. Tumor-infiltrating CD39+CD8+ T cells determine poor prognosis and immune evasion in clear cell renal cell carcinoma patients. Cancer Immunol. Immunother. 2020, 69, 1565–1576. [Google Scholar] [CrossRef]
- Cai, C.; Xu, Y.F.; Wu, Z.J.; Dong, Q.; Li, M.Y.; Olson, J.C.; Rabinowitz, Y.M.; Wang, L.H.; Sun, Y. Tim-3 expression represents dysfunctional tumor infiltrating T cells in renal cell carcinoma. World J. Urol. 2016, 34, 561–567. [Google Scholar] [CrossRef]
- Pignon, J.C.; Jegede, O.; Shukla, S.A.; Braun, D.A.; Horak, C.E.; Wind-Rotolo, M.; Ishii, Y.; Catalano, P.J.; Grosha, J.; Flaifel, A.; et al. irRECIST for the Evaluation of Candidate Biomarkers of Response to Nivolumab in Metastatic Clear Cell Renal Cell Carcinoma: Analysis of a Phase II Prospective Clinical Trial. Clin. Cancer Res. 2019, 25, 2174–2184. [Google Scholar] [CrossRef] [PubMed]
- Ficial, M.; Jegede, O.A.; Sant’Angelo, M.; Hou, Y.; Flaifel, A.; Pignon, J.C.; Braun, D.A.; Wind-Rotolo, M.; Sticco-Ivins, M.A.; Catalano, P.J.; et al. Expression of T-Cell Exhaustion Molecules and Human Endogenous Retroviruses as Predictive Biomarkers for Response to Nivolumab in Metastatic Clear Cell Renal Cell Carcinoma. Clin. Cancer Res. 2021, 27, 1371–1380. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Shi, L.Z.; Zhao, H.; Chen, J.; Xiong, L.; He, Q.; Chen, T.; Roszik, J.; Bernatchez, C.; Woodman, S.E.; et al. Loss of IFN-gamma Pathway Genes in Tumor Cells as a Mechanism of Resistance to Anti-CTLA-4 Therapy. Cell 2016, 167, 397–404.e399. [Google Scholar] [CrossRef] [PubMed]
- Negrier, S.; Escudier, B.; Lasset, C.; Douillard, J.Y.; Savary, J.; Chevreau, C.; Ravaud, A.; Mercatello, A.; Peny, J.; Mousseau, M.; et al. Recombinant human interleukin-2, recombinant human interferon alfa-2a, or both in metastatic renal-cell carcinoma. Groupe Francais d’Immunotherapie. N. Engl. J. Med. 1998, 338, 1272–1278. [Google Scholar] [CrossRef]
- Klapper, J.A.; Downey, S.G.; Smith, F.O.; Yang, J.C.; Hughes, M.S.; Kammula, U.S.; Sherry, R.M.; Royal, R.E.; Steinberg, S.M.; Rosenberg, S. High-dose interleukin-2 for the treatment of metastatic renal cell carcinoma: A retrospective analysis of response and survival in patients treated in the surgery branch at the National Cancer Institute between 1986 and 2006. Cancer 2008, 113, 293–301. [Google Scholar] [CrossRef]
- Allard, C.B.; Gelpi-Hammerschmidt, F.; Harshman, L.C.; Choueiri, T.K.; Faiena, I.; Modi, P.; Chung, B.I.; Tinay, I.; Singer, E.A.; Chang, S.L. Contemporary trends in high-dose interleukin-2 use for metastatic renal cell carcinoma in the United States. Urol. Oncol. 2015, 33, 496.e11-6. [Google Scholar] [CrossRef][Green Version]
- Ayers, M.; Lunceford, J.; Nebozhyn, M.; Murphy, E.; Loboda, A.; Kaufman, D.R.; Albright, A.; Cheng, J.D.; Kang, S.P.; Shankaran, V.; et al. IFN-gamma-related mRNA profile predicts clinical response to PD-1 blockade. J. Clin. Investig. 2017, 127, 2930–2940. [Google Scholar] [CrossRef]
- Alburquerque-Bejar, J.J.; Navajas-Chocarro, P.; Saigi, M.; Ferrero-Andres, A.; Morillas, J.M.; Vilarrubi, A.; Gomez, A.; Mate, J.L.; Munoz-Marmol, A.M.; Romero, O.A.; et al. MYC activation impairs cell-intrinsic IFNgamma signaling and confers resistance to anti-PD1/PD-L1 therapy in lung cancer. Cell Rep. Med. 2023, 4, 101006. [Google Scholar] [CrossRef]
- Ozga, A.J.; Chow, M.T.; Luster, A.D. Chemokines and the immune response to cancer. Immunity 2021, 54, 859–874. [Google Scholar] [CrossRef]
- Gudowska-Sawczuk, M.; Kudelski, J.; Mroczko, B. The Role of Chemokine Receptor CXCR3 and Its Ligands in Renal Cell Carcinoma. Int. J. Mol. Sci. 2020, 21, 8582. [Google Scholar] [CrossRef]
- Choueiri, T.K.; Fishman, M.N.; Escudier, B.; McDermott, D.F.; Drake, C.G.; Kluger, H.; Stadler, W.M.; Perez-Gracia, J.L.; McNeel, D.G.; Curti, B.; et al. Immunomodulatory Activity of Nivolumab in Metastatic Renal Cell Carcinoma. Clin. Cancer Res. 2016, 22, 5461–5471. [Google Scholar] [CrossRef] [PubMed]
- Wardell, C.M.; MacDonald, K.N.; Levings, M.K.; Cook, L. Cross talk between human regulatory T cells and antigen-presenting cells: Lessons for clinical applications. Eur. J. Immunol. 2021, 51, 27–38. [Google Scholar] [CrossRef]
- Alfaro, C.; Teijeira, A.; Onate, C.; Perez, G.; Sanmamed, M.F.; Andueza, M.P.; Alignani, D.; Labiano, S.; Azpilikueta, A.; Rodriguez-Paulete, A.; et al. Tumor-Produced Interleukin-8 Attracts Human Myeloid-Derived Suppressor Cells and Elicits Extrusion of Neutrophil Extracellular Traps (NETs). Clin. Cancer Res. 2016, 22, 3924–3936. [Google Scholar] [CrossRef] [PubMed]
- Berlato, C.; Khan, M.N.; Schioppa, T.; Thompson, R.; Maniati, E.; Montfort, A.; Jangani, M.; Canosa, M.; Kulbe, H.; Hagemann, U.B.; et al. A CCR4 antagonist reverses the tumor-promoting microenvironment of renal cancer. J. Clin. Investig. 2017, 127, 801–813. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Li, Q.J.; Feng, Y.; Zhang, Y.; Markowitz, G.J.; Ning, S.; Deng, Y.; Zhao, J.; Jiang, S.; Yuan, Y.; et al. TGF-beta-miR-34a-CCL22 signaling-induced Treg cell recruitment promotes venous metastases of HBV-positive hepatocellular carcinoma. Cancer Cell 2012, 22, 291–303. [Google Scholar] [CrossRef]
- Schalper, K.A.; Carleton, M.; Zhou, M.; Chen, T.; Feng, Y.; Huang, S.P.; Walsh, A.M.; Baxi, V.; Pandya, D.; Baradet, T.; et al. Elevated serum interleukin-8 is associated with enhanced intratumor neutrophils and reduced clinical benefit of immune-checkpoint inhibitors. Nat. Med. 2020, 26, 688–692. [Google Scholar] [CrossRef]
- Yuen, K.C.; Liu, L.F.; Gupta, V.; Madireddi, S.; Keerthivasan, S.; Li, C.; Rishipathak, D.; Williams, P.; Kadel, E.E., III; Koeppen, H.; et al. High systemic and tumor-associated IL-8 correlates with reduced clinical benefit of PD-L1 blockade. Nat. Med. 2020, 26, 693–698. [Google Scholar] [CrossRef]
- Li, J.Y.; Ou, Z.L.; Yu, S.J.; Gu, X.L.; Yang, C.; Chen, A.X.; Di, G.H.; Shen, Z.Z.; Shao, Z.M. The chemokine receptor CCR4 promotes tumor growth and lung metastasis in breast cancer. Breast Cancer Res. Treat. 2012, 131, 837–848. [Google Scholar] [CrossRef]
- Lee, J.H.; Cho, Y.S.; Lee, J.Y.; Kook, M.C.; Park, J.W.; Nam, B.H.; Bae, J.M. The chemokine receptor CCR4 is expressed and associated with a poor prognosis in patients with gastric cancer. Ann. Surg. 2009, 249, 933–941. [Google Scholar] [CrossRef]
- Liu, Q.; Rexiati, M.; Yang, Y.; Wang, W.G.; Azhati, B.; Saimaiti, W.; Wang, Y.J. Expression of chemokine receptor 4 was associated with poor survival in renal cell carcinoma. Med. Oncol. 2014, 31, 882. [Google Scholar] [CrossRef]
- Bui, T.O.; Dao, V.T.; Nguyen, V.T.; Feugeas, J.P.; Pamoukdjian, F.; Bousquet, G. Genomics of Clear-cell Renal Cell Carcinoma: A Systematic Review and Meta-analysis. Eur. Urol. 2022, 81, 349–361. [Google Scholar] [CrossRef]
- Haase, V.H. The VHL/HIF oxygen-sensing pathway and its relevance to kidney disease. Kidney Int. 2006, 69, 1302–1307. [Google Scholar] [CrossRef]
- Masoud, G.N.; Li, W. HIF-1alpha pathway: Role, regulation and intervention for cancer therapy. Acta Pharm. Sin. B 2015, 5, 378–389. [Google Scholar] [CrossRef]
- McGettrick, A.F.; O’Neill, L.A.J. The Role of HIF in Immunity and Inflammation. Cell Metab. 2020, 32, 524–536. [Google Scholar] [CrossRef]
- Clark, P.E. The role of VHL in clear-cell renal cell carcinoma and its relation to targeted therapy. Kidney Int. 2009, 76, 939–945. [Google Scholar] [CrossRef]
- Bourhis, M.; Palle, J.; Galy-Fauroux, I.; Terme, M. Direct and Indirect Modulation of T Cells by VEGF-A Counteracted by Anti-Angiogenic Treatment. Front. Immunol. 2021, 12, 616837. [Google Scholar] [CrossRef] [PubMed]
- Horikawa, N.; Abiko, K.; Matsumura, N.; Hamanishi, J.; Baba, T.; Yamaguchi, K.; Yoshioka, Y.; Koshiyama, M.; Konishi, I. Expression of Vascular Endothelial Growth Factor in Ovarian Cancer Inhibits Tumor Immunity through the Accumulation of Myeloid-Derived Suppressor Cells. Clin. Cancer Res. 2017, 23, 587–599. [Google Scholar] [CrossRef]
- Courau, T.; Nehar-Belaid, D.; Florez, L.; Levacher, B.; Vazquez, T.; Brimaud, F.; Bellier, B.; Klatzmann, D. TGF-beta and VEGF cooperatively control the immunotolerant tumor environment and the efficacy of cancer immunotherapies. JCI Insight 2016, 1, e85974. [Google Scholar] [CrossRef]
- Wheeler, K.C.; Jena, M.K.; Pradhan, B.S.; Nayak, N.; Das, S.; Hsu, C.D.; Wheeler, D.S.; Chen, K.; Nayak, N.R. VEGF may contribute to macrophage recruitment and M2 polarization in the decidua. PLoS ONE 2018, 13, e0191040. [Google Scholar] [CrossRef] [PubMed]
- Motz, G.T.; Santoro, S.P.; Wang, L.P.; Garrabrant, T.; Lastra, R.R.; Hagemann, I.S.; Lal, P.; Feldman, M.D.; Benencia, F.; Coukos, G. Tumor endothelium FasL establishes a selective immune barrier promoting tolerance in tumors. Nat. Med. 2014, 20, 607–615. [Google Scholar] [CrossRef] [PubMed]
- Geindreau, M.; Ghiringhelli, F.; Bruchard, M. Vascular Endothelial Growth Factor, a Key Modulator of the Anti-Tumor Immune Response. Int. J. Mol. Sci. 2021, 22, 4871. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Langenkamp, E.; Georganaki, M.; Loskog, A.; Fuchs, P.F.; Dieterich, L.C.; Kreuger, J.; Dimberg, A. VEGF suppresses T-lymphocyte infiltration in the tumor microenvironment through inhibition of NF-kappaB-induced endothelial activation. FASEB J. 2015, 29, 227–238. [Google Scholar] [CrossRef]
- Dirkx, A.E.; Oude Egbrink, M.G.; Kuijpers, M.J.; van der Niet, S.T.; Heijnen, V.V.; Bouma-ter Steege, J.C.; Wagstaff, J.; Griffioen, A.W. Tumor angiogenesis modulates leukocyte-vessel wall interactions in vivo by reducing endothelial adhesion molecule expression. Cancer Res. 2003, 63, 2322–2329. [Google Scholar]
- Meder, L.; Schuldt, P.; Thelen, M.; Schmitt, A.; Dietlein, F.; Klein, S.; Borchmann, S.; Wennhold, K.; Vlasic, I.; Oberbeck, S.; et al. Combined VEGF and PD-L1 Blockade Displays Synergistic Treatment Effects in an Autochthonous Mouse Model of Small Cell Lung Cancer. Cancer Res. 2018, 78, 4270–4281. [Google Scholar] [CrossRef]
- Voron, T.; Colussi, O.; Marcheteau, E.; Pernot, S.; Nizard, M.; Pointet, A.L.; Latreche, S.; Bergaya, S.; Benhamouda, N.; Tanchot, C.; et al. VEGF-A modulates expression of inhibitory checkpoints on CD8+ T cells in tumors. J. Exp. Med. 2015, 212, 139–148. [Google Scholar] [CrossRef]
- Yang, J.C.; Haworth, L.; Sherry, R.M.; Hwu, P.; Schwartzentruber, D.J.; Topalian, S.L.; Steinberg, S.M.; Chen, H.X.; Rosenberg, S.A. A randomized trial of bevacizumab, an anti-vascular endothelial growth factor antibody, for metastatic renal cancer. N. Engl. J. Med. 2003, 349, 427–434. [Google Scholar] [CrossRef]
- Escudier, B.; Pluzanska, A.; Koralewski, P.; Ravaud, A.; Bracarda, S.; Szczylik, C.; Chevreau, C.; Filipek, M.; Melichar, B.; Bajetta, E.; et al. Bevacizumab plus interferon alfa-2a for treatment of metastatic renal cell carcinoma: A randomised, double-blind phase III trial. Lancet 2007, 370, 2103–2111. [Google Scholar] [CrossRef]
- Motzer, R.J.; Hutson, T.E.; Tomczak, P.; Michaelson, M.D.; Bukowski, R.M.; Oudard, S.; Negrier, S.; Szczylik, C.; Pili, R.; Bjarnason, G.A.; et al. Overall survival and updated results for sunitinib compared with interferon alfa in patients with metastatic renal cell carcinoma. J. Clin. Oncol. 2009, 27, 3584–3590. [Google Scholar] [CrossRef]
- Motzer, R.J.; Hutson, T.E.; Tomczak, P.; Michaelson, M.D.; Bukowski, R.M.; Rixe, O.; Oudard, S.; Negrier, S.; Szczylik, C.; Kim, S.T.; et al. Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N. Engl. J. Med. 2007, 356, 115–124. [Google Scholar] [CrossRef]
- Sternberg, C.N.; Davis, I.D.; Mardiak, J.; Szczylik, C.; Lee, E.; Wagstaff, J.; Barrios, C.H.; Salman, P.; Gladkov, O.A.; Kavina, A.; et al. Pazopanib in locally advanced or metastatic renal cell carcinoma: Results of a randomized phase III trial. J. Clin. Oncol. 2010, 28, 1061–1068. [Google Scholar] [CrossRef]
- Rini, B.I.; Escudier, B.; Tomczak, P.; Kaprin, A.; Szczylik, C.; Hutson, T.E.; Michaelson, M.D.; Gorbunova, V.A.; Gore, M.E.; Rusakov, I.G.; et al. Comparative effectiveness of axitinib versus sorafenib in advanced renal cell carcinoma (AXIS): A randomised phase 3 trial. Lancet 2011, 378, 1931–1939. [Google Scholar] [CrossRef] [PubMed]
- Ravaud, A.; Motzer, R.J.; Pandha, H.S.; George, D.J.; Pantuck, A.J.; Patel, A.; Chang, Y.H.; Escudier, B.; Donskov, F.; Magheli, A.; et al. Adjuvant Sunitinib in High-Risk Renal-Cell Carcinoma after Nephrectomy. N. Engl. J. Med. 2016, 375, 2246–2254. [Google Scholar] [CrossRef] [PubMed]
- Miao, D.; Margolis, C.A.; Gao, W.; Voss, M.H.; Li, W.; Martini, D.J.; Norton, C.; Bosse, D.; Wankowicz, S.M.; Cullen, D.; et al. Genomic correlates of response to immune checkpoint therapies in clear cell renal cell carcinoma. Science 2018, 359, 801–806. [Google Scholar] [CrossRef] [PubMed]
- Guo, T.; Wang, T.; Zhang, J.; Chen, S.; Wang, X. HIF1A predicts the efficacy of anti-PD-1 therapy in advanced clear cell renal cell carcinoma. Transl. Oncol. 2022, 26, 101554. [Google Scholar] [CrossRef] [PubMed]
- Khattak, M.A.; Abed, A.; Reid, A.L.; McEvoy, A.C.; Millward, M.; Ziman, M.; Gray, E.S. Role of Serum Vascular Endothelial Growth Factor (VEGF) as a Potential Biomarker of Response to Immune Checkpoint Inhibitor Therapy in Advanced Melanoma: Results of a Pilot Study. Front. Oncol. 2020, 10, 1041. [Google Scholar] [CrossRef]
- Brown, L.C.; Zhu, J.; Desai, K.; Kinsey, E.; Kao, C.; Lee, Y.H.; Pabla, S.; Labriola, M.K.; Tran, J.; Dragnev, K.H.; et al. Evaluation of tumor microenvironment and biomarkers of immune checkpoint inhibitor response in metastatic renal cell carcinoma. J. Immunother. Cancer 2022, 10, e005249. [Google Scholar] [CrossRef]
- Raimondi, A.; Sepe, P.; Zattarin, E.; Mennitto, A.; Stellato, M.; Claps, M.; Guadalupi, V.; Verzoni, E.; de Braud, F.; Procopio, G. Predictive Biomarkers of Response to Immunotherapy in Metastatic Renal Cell Cancer. Front. Oncol. 2020, 10, 1644. [Google Scholar] [CrossRef]
- Liu, Y.; Zugazagoitia, J.; Ahmed, F.S.; Henick, B.S.; Gettinger, S.N.; Herbst, R.S.; Schalper, K.A.; Rimm, D.L. Immune Cell PD-L1 Colocalizes with Macrophages and Is Associated with Outcome in PD-1 Pathway Blockade Therapy. Clin. Cancer Res. 2020, 26, 970–977. [Google Scholar] [CrossRef]
- Sharpe, A.H.; Pauken, K.E. The diverse functions of the PD1 inhibitory pathway. Nat. Rev. Immunol. 2018, 18, 153–167. [Google Scholar] [CrossRef]
- de Ruiter, E.J.; Mulder, F.J.; Koomen, B.M.; Speel, E.J.; van den Hout, M.; de Roest, R.H.; Bloemena, E.; Devriese, L.A.; Willems, S.M. Comparison of three PD-L1 immunohistochemical assays in head and neck squamous cell carcinoma (HNSCC). Mod. Pathol. 2021, 34, 1125–1132. [Google Scholar] [CrossRef]
- Wang, M.; Wang, S.; Trapani, J.A.; Neeson, P.J. Challenges of PD-L1 testing in non-small cell lung cancer and beyond. J. Thorac. Dis. 2020, 12, 4541–4548. [Google Scholar] [CrossRef]
- Chen, S.; Crabill, G.A.; Pritchard, T.S.; McMiller, T.L.; Wei, P.; Pardoll, D.M.; Pan, F.; Topalian, S.L. Mechanisms regulating PD-L1 expression on tumor and immune cells. J. Immunother. Cancer 2019, 7, 305. [Google Scholar] [CrossRef]
- Cha, J.H.; Chan, L.C.; Li, C.W.; Hsu, J.L.; Hung, M.C. Mechanisms Controlling PD-L1 Expression in Cancer. Mol. Cell 2019, 76, 359–370. [Google Scholar] [CrossRef]
- Vidotto, T.; Melo, C.M.; Castelli, E.; Koti, M.; Dos Reis, R.B.; Squire, J.A. Emerging role of PTEN loss in evasion of the immune response to tumours. Br. J. Cancer 2020, 122, 1732–1743. [Google Scholar] [CrossRef]
- Barsoum, I.B.; Smallwood, C.A.; Siemens, D.R.; Graham, C.H. A mechanism of hypoxia-mediated escape from adaptive immunity in cancer cells. Cancer Res. 2014, 74, 665–674. [Google Scholar] [CrossRef]
- Paver, E.C.; Cooper, W.A.; Colebatch, A.J.; Ferguson, P.M.; Hill, S.K.; Lum, T.; Shin, J.S.; O’Toole, S.; Anderson, L.; Scolyer, R.A.; et al. Programmed death ligand-1 (PD-L1) as a predictive marker for immunotherapy in solid tumours: A guide to immunohistochemistry implementation and interpretation. Pathology 2021, 53, 141–156. [Google Scholar] [CrossRef]
- Lantuejoul, S.; Sound-Tsao, M.; Cooper, W.A.; Girard, N.; Hirsch, F.R.; Roden, A.C.; Lopez-Rios, F.; Jain, D.; Chou, T.Y.; Motoi, N.; et al. PD-L1 Testing for Lung Cancer in 2019: Perspective From the IASLC Pathology Committee. J. Thorac. Oncol. 2020, 15, 499–519. [Google Scholar] [CrossRef]
- Motzer, R.J.; Tannir, N.M.; McDermott, D.F.; Aren Frontera, O.; Melichar, B.; Choueiri, T.K.; Plimack, E.R.; Barthelemy, P.; Porta, C.; George, S.; et al. Nivolumab plus Ipilimumab versus Sunitinib in Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2018, 378, 1277–1290. [Google Scholar] [CrossRef]
- Powles, T.; Plimack, E.R.; Soulieres, D.; Waddell, T.; Stus, V.; Gafanov, R.; Nosov, D.; Pouliot, F.; Melichar, B.; Vynnychenko, I.; et al. Pembrolizumab plus axitinib versus sunitinib monotherapy as first-line treatment of advanced renal cell carcinoma (KEYNOTE-426): Extended follow-up from a randomised, open-label, phase 3 trial. Lancet Oncol. 2020, 21, 1563–1573. [Google Scholar] [CrossRef]
- Carretero-Gonzalez, A.; Lora, D.; Martin Sobrino, I.; Saez Sanz, I.; Bourlon, M.T.; Anido Herranz, U.; Martinez Chanza, N.; Castellano, D.; de Velasco, G. The Value of PD-L1 Expression as Predictive Biomarker in Metastatic Renal Cell Carcinoma Patients: A Meta-Analysis of Randomized Clinical Trials. Cancers 2020, 12, 1945. [Google Scholar] [CrossRef]
- Xiao, X.; Wu, Y.; Shen, F.; MuLaTiAize, Y.; Xinhua, N. Osimertinib Improves the Immune Microenvironment of Lung Cancer by Downregulating PD-L1 Expression of Vascular Endothelial Cells and Enhances the Antitumor Effect of Bevacizumab. J. Oncol. 2022, 2022, 1531353. [Google Scholar] [CrossRef] [PubMed]
- Piao, W.; Li, L.; Saxena, V.; Iyyathurai, J.; Lakhan, R.; Zhang, Y.; Lape, I.T.; Paluskievicz, C.; Hippen, K.L.; Lee, Y.; et al. PD-L1 signaling selectively regulates T cell lymphatic transendothelial migration. Nat. Commun. 2022, 13, 2176. [Google Scholar] [CrossRef] [PubMed]
- Lane, R.S.; Femel, J.; Breazeale, A.P.; Loo, C.P.; Thibault, G.; Kaempf, A.; Mori, M.; Tsujikawa, T.; Chang, Y.H.; Lund, A.W. IFNgamma-activated dermal lymphatic vessels inhibit cytotoxic T cells in melanoma and inflamed skin. J. Exp. Med. 2018, 215, 3057–3074. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.; Fan, W.; Liu, T.; Zhu, B.; Liu, Y.; Wang, S.; Wu, J.; Liu, J.; Zou, F.; Wei, J.; et al. TREM2+ macrophages suppress CD8+ T-cell infiltration after transarterial chemoembolisation in hepatocellular carcinoma. J. Hepatol. 2023, 79, 126–140. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, B.; Porter, E.G.; Stewart, J.C.; Ferreira, C.R.; Schipma, M.J.; Dykhuizen, E.C. PBRM1 Regulates the Expression of Genes Involved in Metabolism and Cell Adhesion in Renal Clear Cell Carcinoma. PLoS ONE 2016, 11, e0153718. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research, N. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature 2013, 499, 43–49. [Google Scholar] [CrossRef]
- Varela, I.; Tarpey, P.; Raine, K.; Huang, D.; Ong, C.K.; Stephens, P.; Davies, H.; Jones, D.; Lin, M.L.; Teague, J.; et al. Exome sequencing identifies frequent mutation of the SWI/SNF complex gene PBRM1 in renal carcinoma. Nature 2011, 469, 539–542. [Google Scholar] [CrossRef]
- Zhou, H.; Liu, J.; Zhang, Y.; Huang, Y.; Shen, J.; Yang, Y.; Fang, W.; Zhang, L. PBRM1 mutation and preliminary response to immune checkpoint blockade treatment in non-small cell lung cancer. NPJ Precis. Oncol. 2020, 4, 6. [Google Scholar] [CrossRef]
- Braun, D.A.; Ishii, Y.; Walsh, A.M.; Van Allen, E.M.; Wu, C.J.; Shukla, S.A.; Choueiri, T.K. Clinical Validation of PBRM1 Alterations as a Marker of Immune Checkpoint Inhibitor Response in Renal Cell Carcinoma. JAMA Oncol. 2019, 5, 1631–1633. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, B.; Zhao, G.; Lee, Y.; Buzdin, A.; Mu, X.; Zhao, J.; Chen, H.; Li, X. Spatial transcriptomics: Technologies, applications and experimental considerations. Genomics 2023, 115, 110671. [Google Scholar] [CrossRef]
- Hu, B.; Sajid, M.; Lv, R.; Liu, L.; Sun, C. A review of spatial profiling technologies for characterizing the tumor microenvironment in immuno-oncology. Front. Immunol. 2022, 13, 996721. [Google Scholar] [CrossRef]
- Kumar, T.; Nee, K.; Wei, R.; He, S.; Nguyen, Q.H.; Bai, S.; Blake, K.; Pein, M.; Gong, Y.; Sei, E.; et al. A spatially resolved single-cell genomic atlas of the adult human breast. Nature 2023. [Google Scholar] [CrossRef]
- Phillips, D.; Matusiak, M.; Gutierrez, B.R.; Bhate, S.S.; Barlow, G.L.; Jiang, S.; Demeter, J.; Smythe, K.S.; Pierce, R.H.; Fling, S.P.; et al. Immune cell topography predicts response to PD-1 blockade in cutaneous T cell lymphoma. Nat. Commun. 2021, 12, 6726. [Google Scholar] [CrossRef]
- Locke, D.; Hoyt, C.C. Companion diagnostic requirements for spatial biology using multiplex immunofluorescence and multispectral imaging. Front. Mol. Biosci. 2023, 10, 1051491. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Trial Name | Trial Features | Treatment Arm (s) | Control Arm | Significant Findings (Treatment vs. Control) |
|---|---|---|---|---|
| Immotion 151 [58,59] | Phase III, multicenter RCT on previously untreated metastatic RCC | atezolizumab + bevacizumab | sunitinib | OS PD-L1+: 38.7 mo vs. 31.6 mo PFS: PD-L1+: 11.2 mo vs. 7.7 mo (HR = 0.74) |
| JAVELIN Renal 101 [60,61] | Phase III clinical trial of previously untreated advanced-RCC patients | avelumab + axitinb | sunitinib | OS PD-L1+: NR vs. 36.2 mo (HR = 0.81) PFS PD-L1+: 13.9 mo vs. 8.2 mo (HR = 0.58) |
| CheckMate 214 [62,63,64] | Phase III clinical trial of previously untreated advanced-ccRCC patients | nivolumab + ipilumumab | sunitinib | OS PD-L1+: 66.8 mo vs. 23.9 mo (HR = 0.57) OS PD-L1−: 59.2 mo vs. 41.9 mo (HR = 0.77) PFS PD-L1+: NR vs. 5.6 mo PFS PD-L1−: 9 mo vs. 5.4 mo |
| KEYNOTE-426 [65] | Phase III clinical trial on previously untreated advanced-ccRCC patients | pembrolizumab + axitinib | sunitinib | OS PD-L1+: HR = 0.54 OS PD-L1−: HR = 0.59, NS PFS PD-L+: 15.3 mo vs. 8.9 mo (HR = 0.62) PFS PD-L1−: 15 mo vs. 12.5 mo (HR = 0.87, NS) |
| CheckMate 025 [66] | Phase III clinical trial on advanced ccRCC patients previously on antiangiogenic therapy | nivolumab | everolimus | OS PD-L1+: 21.8 mo vs. 18.8 mo (HR = 0.79)OS PD-L1−: 27.4 mo vs. 21.2 mo (HR = 0.77)PFS PD-L1+: 4.6 mo vs. 4.4 mo (HR = 0.88) |
| CheckMate 9ER [67] | Phase III clinical trial on previously untreated advanced-ccRCC patients | nivolumab + cabazantinib | sunitinib | OS PD-L1+: HR = 0.80 PFS PD-L1+: HR = 0.49 |
| CLEAR [68] | Phase III clinical trial on systemic therapy naïve patients with advanced RCC | pembrolizumab + lenvatinib; lenvatinib + everoliums | sunitinib | PFS PD-L1+; P+L vs. S: HR = 0.40 PFS PD-L1−, P+L vs. S: HR = 0.39 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martin, S.D.; Bhuiyan, I.; Soleimani, M.; Wang, G. Biomarkers for Immune Checkpoint Inhibitors in Renal Cell Carcinoma. J. Clin. Med. 2023, 12, 4987. https://doi.org/10.3390/jcm12154987
Martin SD, Bhuiyan I, Soleimani M, Wang G. Biomarkers for Immune Checkpoint Inhibitors in Renal Cell Carcinoma. Journal of Clinical Medicine. 2023; 12(15):4987. https://doi.org/10.3390/jcm12154987
Chicago/Turabian StyleMartin, Spencer D., Ishmam Bhuiyan, Maryam Soleimani, and Gang Wang. 2023. "Biomarkers for Immune Checkpoint Inhibitors in Renal Cell Carcinoma" Journal of Clinical Medicine 12, no. 15: 4987. https://doi.org/10.3390/jcm12154987
APA StyleMartin, S. D., Bhuiyan, I., Soleimani, M., & Wang, G. (2023). Biomarkers for Immune Checkpoint Inhibitors in Renal Cell Carcinoma. Journal of Clinical Medicine, 12(15), 4987. https://doi.org/10.3390/jcm12154987