
Store-Operated Ca2+ Entry as a Putative Target of Flecainide for the Treatment of Arrhythmogenic Cardiomyopathy
 , , ,
, , , 
Abstract
:1. Introduction
2. Pathological Background and Treatment of ACM

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Encoded Protein | Estimated Frequency (%) | Chromosomal Location |
|---|---|---|---|
| Desmosomes | |||
| PKP2 | Plakophilin-2 | 19–46 | 12p11.21 |
| DSP | Desmoplakin | 1–16 | 6p24.3 |
| DSG2 | Desmoglein-2 | 2.5–10 | 18q12.1 |
| DSC2 | Desmocollin-2 | 1–8 | 18q12.1 |
| JUP | Junction plakoglobin | Rare | 17q21.2 |
| Adherens junctions | |||
| CTNNA3 | Catenin-α3 | Rare | 10q21.3 |
| CDH2 | Cadherin 2 | Rare | 18q12.1 |
| Cytoskeleton | |||
| LMNA | Lamin A/C | Rare | 1q22 |
| DES | Desmin | Rare | 2q35 |
| FLNC | Filamin C | Rare | 7q32.1 |
| TTN | Titin | Rare | 2q31.2 |
| Ion transport | |||
| SCN5A | NaV1.5 | Rare | 3p22.2 |
| PLN | Phospholamban | Rare | 6q22.31 |
3. Anti-Arrhythmic Effects of Flecainide: From Nav1.5 to RyR2
4. The Rationale for Using Flecainide to Treat Ca2+-Dependent Ventricular Arrhythmias in ACM
5. Intracellular Ca2+ Oscillations Regulate MSC fate
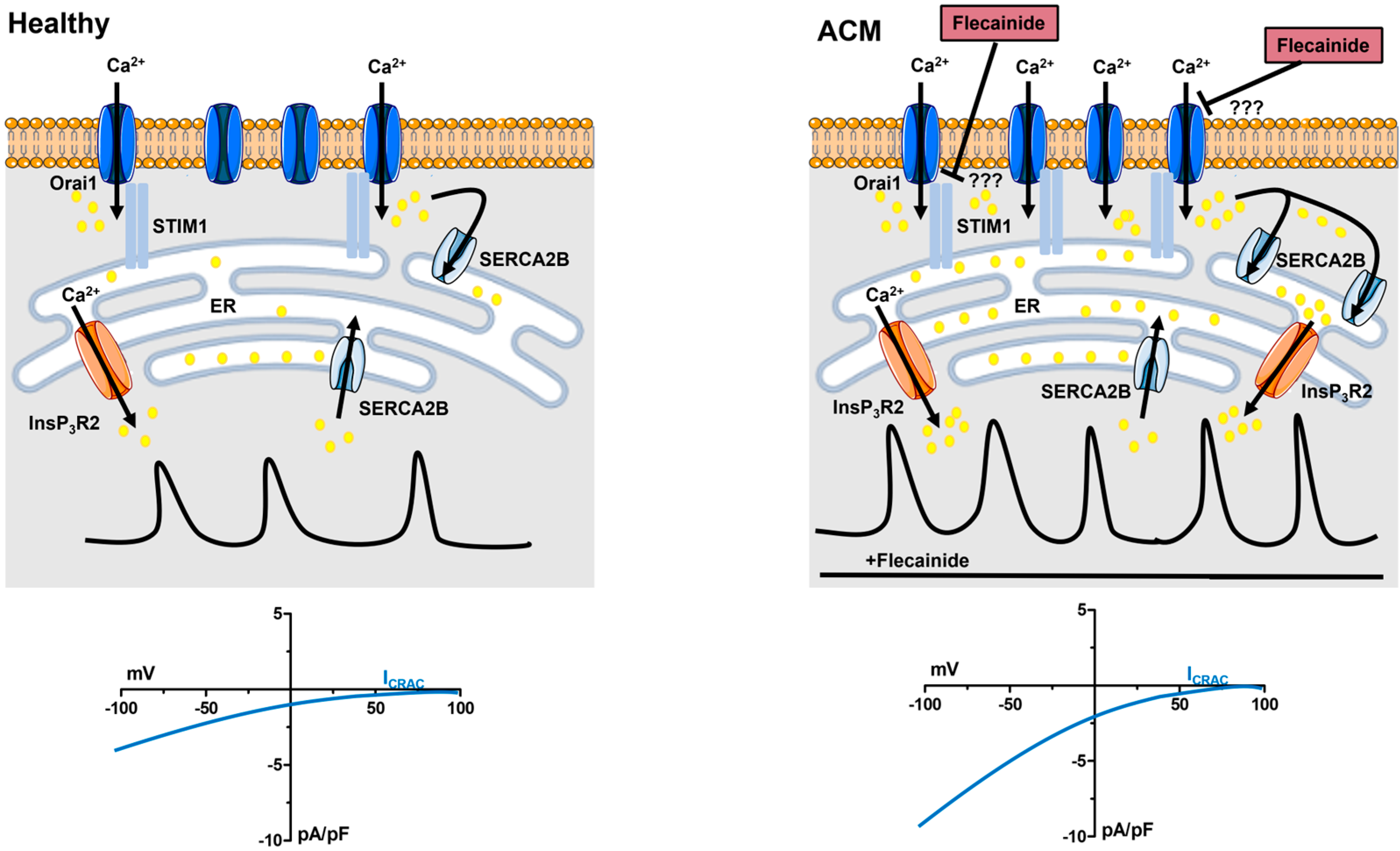
6. Flecainide Inhibits SOCE and Prevents Ca2+-Dependent Fibro-Adipogenic Differentiation in ACM C-hMSCs
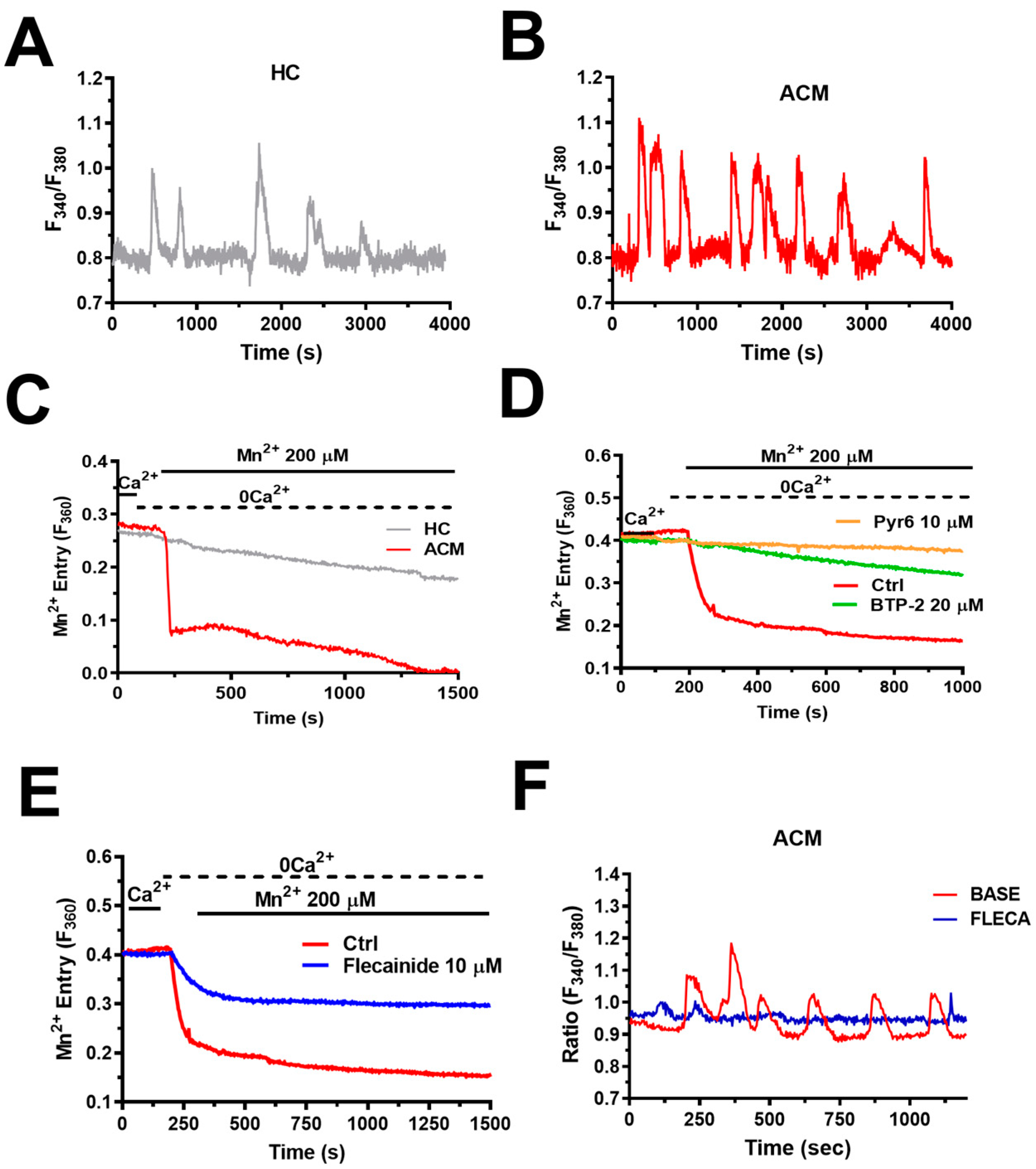
6.1. Intracellular Ca2+ Oscillations Are Up-Regulated in ACM C-hMSCs
6.2. Intracellular Ca2+ Oscillations in ACM C-hMSCs: Is There Any Role for Lysosomal Ca2+ Mobilization?
6.3. Intracellular Ca2+ Oscillations Drive Fibro-Adipogenic Differentiation in ACM C-hMSCs
6.4. Flecainide Abolishes Intracellular Ca2+ Oscillations and Fibro-Adipogenic Differentiation by Targeting SOCE
7. Future Directions
8. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Austin, K.M.; Trembley, M.A.; Chandler, S.F.; Sanders, S.P.; Saffitz, J.E.; Abrams, D.J.; Pu, W.T. Molecular mechanisms of arrhythmogenic cardiomyopathy. Nat. Rev. Cardiol. 2019, 16, 519–537. [Google Scholar] [CrossRef]
- Corrado, D.; Link, M.S.; Calkins, H. Arrhythmogenic Right Ventricular Cardiomyopathy. N. Engl. J. Med. 2017, 376, 61–72. [Google Scholar] [CrossRef]
- Ermakov, S.; Scheinman, M. Arrhythmogenic Right Ventricular Cardiomyopathy–Antiarrhythmic Therapy. Arrhythmia Electrophysiol. Rev. 2015, 4, 86–89. [Google Scholar] [CrossRef] [Green Version]
- Maione, A.S.; Faris, P.; Iengo, L.; Catto, V.; Bisonni, L.; Lodola, F.; Negri, S.; Casella, M.; Guarino, A.; Polvani, G.; et al. Ca2+ dysregulation in cardiac stromal cells sustains fibro-adipose remodeling in Arrhythmogenic Cardiomyopathy and can be modulated by flecainide. J. Transl. Med. 2022, 20, 522. [Google Scholar] [CrossRef]
- Emrich, S.M.; Yoast, R.E.; Trebak, M. Physiological Functions of CRAC Channels. Annu. Rev. Physiol. 2021, 84, 355–379. [Google Scholar] [CrossRef]
- Moccia, F.; Brunetti, V.; Perna, A.; Guerra, G.; Soda, T.; Berra-Romani, R. The Molecular Heterogeneity of Store-Operated Ca2+ Entry in Vascular Endothelial Cells: The Different roles of Orai1 and TRPC1/TRPC4 Channels in the Transition from Ca2+-Selective to Non-Selective Cation Currents. Int. J. Mol. Sci. 2023, 24, 3259. [Google Scholar] [CrossRef]
- Prakriya, M.; Lewis, R.S. Store-Operated Calcium Channels. Physiol. Rev. 2015, 95, 1383–1436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, L.H.; Mousawi, F.; Yang, X.; Roger, S. ATP-induced Ca2+-signalling mechanisms in the regulation of mesenchymal stem cell migration. Cell. Mol. Life Sci. 2017, 74, 3697–3710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Roger, S.; Yang, X.B.; Jiang, L.H. Role of the store-operated Ca2+ channel in ATP-induced Ca2+ signalling in mesenchymal stem cells and regulation of cell functions. Front. Biosci. (Landmark Ed.) 2021, 26, 1737–1745. [Google Scholar] [CrossRef] [PubMed]
- Ahamad, N.; Singh, B.B. Calcium channels and their role in regenerative medicine. World J. Stem Cells 2021, 13, 260–280. [Google Scholar] [CrossRef]
- James, C.A.; Jongbloed, J.D.H.; Hershberger, R.E.; Morales, A.; Judge, D.P.; Syrris, P.; Pilichou, K.; Domingo, A.M.; Murray, B.; Cadrin-Tourigny, J.; et al. International Evidence Based Reappraisal of Genes Associated With Arrhythmogenic Right Ventricular Cardiomyopathy Using the Clinical Genome Resource Framework. Circ. Genom. Precis. Med. 2021, 14, e003273. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Gras, E.; Lombardi, R.; Giocondo, M.J.; Willerson, J.T.; Schneider, M.D.; Khoury, D.S.; Marian, A.J. Suppression of canonical Wnt/beta-catenin signaling by nuclear plakoglobin recapitulates phenotype of arrhythmogenic right ventricular cardiomyopathy. J. Clin. Investig. 2006, 116, 2012–2021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sommariva, E.; Brambilla, S.; Carbucicchio, C.; Gambini, E.; Meraviglia, V.; Dello Russo, A.; Farina, F.M.; Casella, M.; Catto, V.; Pontone, G.; et al. Cardiac mesenchymal stromal cells are a source of adipocytes in arrhythmogenic cardiomyopathy. Eur. Heart J. 2016, 37, 1835–1846. [Google Scholar] [CrossRef] [Green Version]
- Maione, A.S.; Stadiotti, I.; Pilato, C.A.; Perrucci, G.L.; Saverio, V.; Catto, V.; Vettor, G.; Casella, M.; Guarino, A.; Polvani, G.; et al. Excess TGF-beta1 Drives Cardiac Mesenchymal Stromal Cells to a Pro-Fibrotic Commitment in Arrhythmogenic Cardiomyopathy. Int. J. Mol. Sci. 2021, 22, 2673. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.D.; Ambler, S.K.; Mitchell, M.D.; Long, C.S. The cardiac fibroblast: Therapeutic target in myocardial remodeling and failure. Annu. Rev. Pharmacol. Toxicol. 2005, 45, 657–687. [Google Scholar] [CrossRef] [PubMed]
- Camelliti, P.; Borg, T.K.; Kohl, P. Structural and functional characterisation of cardiac fibroblasts. Cardiovasc. Res. 2005, 65, 40–51. [Google Scholar] [CrossRef] [Green Version]
- Corrado, D.; Basso, C.; Judge, D.P. Arrhythmogenic Cardiomyopathy. Circ. Res. 2017, 121, 784–802. [Google Scholar] [CrossRef]
- Gaine, S.P.; Calkins, H. Antiarrhythmic Drug Therapy in Arrhythmogenic Right Ventricular Cardiomyopathy. Biomedicines 2023, 11, 1213. [Google Scholar] [CrossRef]
- Rolland, T.; Badenco, N.; Maupain, C.; Duthoit, G.; Waintraub, X.; Laredo, M.; Himbert, C.; Frank, R.; Hidden-Lucet, F.; Gandjbakhch, E. Safety and efficacy of flecainide associated with beta-blockers in arrhythmogenic right ventricular cardiomyopathy. EP Eur. 2022, 24, 278–284. [Google Scholar] [CrossRef]
- Lavalle, C.; Trivigno, S.; Vetta, G.; Magnocavallo, M.; Mariani, M.V.; Santini, L.; Forleo, G.B.; Grimaldi, M.; Badagliacca, R.; Lanata, L.; et al. Flecainide in Ventricular Arrhythmias: From Old Myths to New Perspectives. J. Clin. Med. 2021, 10, 3696. [Google Scholar] [CrossRef]
- Stevens, T.L.; Wallace, M.J.; Refaey, M.E.; Roberts, J.D.; Koenig, S.N.; Mohler, P.J. Arrhythmogenic Cardiomyopathy: Molecular Insights for Improved Therapeutic Design. J. Cardiovasc. Dev. Dis. 2020, 7, 21. [Google Scholar] [CrossRef]
- Salvage, S.C.; Chandrasekharan, K.H.; Jeevaratnam, K.; Dulhunty, A.F.; Thompson, A.J.; Jackson, A.P.; Huang, C.L. Multiple targets for flecainide action: Implications for cardiac arrhythmogenesis. Br. J. Pharmacol. 2018, 175, 1260–1278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salvage, S.C.; Huang, C.L.; Fraser, J.A.; Dulhunty, A.F. How does flecainide impact RyR2 channel function? J. Gen. Physiol. 2022, 154. [Google Scholar] [CrossRef]
- Andrikopoulos, G.K.; Pastromas, S.; Tzeis, S. Flecainide: Current status and perspectives in arrhythmia management. World J. Cardiol. 2015, 7, 76–85. [Google Scholar] [CrossRef]
- Benitah, J.P.; Gomez, A.M. Is the Debate on the Flecainide Action on the RYR2 in CPVT Closed? Circ. Res. 2021, 128, 332–334. [Google Scholar] [CrossRef] [PubMed]
- Aliot, E.; Capucci, A.; Crijns, H.J.; Goette, A.; Tamargo, J. Twenty-five years in the making: Flecainide is safe and effective for the management of atrial fibrillation. Europace 2011, 13, 161–173. [Google Scholar] [CrossRef]
- Nitta, J.; Sunami, A.; Marumo, F.; Hiraoka, M. States and sites of actions of flecainide on guinea-pig cardiac sodium channels. Eur. J. Pharmacol. 1992, 214, 191–197. [Google Scholar] [CrossRef]
- Horvath, B.; Hezso, T.; Kiss, D.; Kistamas, K.; Magyar, J.; Nanasi, P.P.; Banyasz, T. Late Sodium Current Inhibitors as Potential Antiarrhythmic Agents. Front. Pharmacol. 2020, 11, 413. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.K.; Russell, C.; Wang, S.Y. State-dependent block of wild-type and inactivation-deficient Na+ channels by flecainide. J. Gen. Physiol. 2003, 122, 365–374. [Google Scholar] [CrossRef]
- Belardinelli, L.; Liu, G.; Smith-Maxwell, C.; Wang, W.Q.; El-Bizri, N.; Hirakawa, R.; Karpinski, S.; Li, C.H.; Hu, L.; Li, X.J.; et al. A novel, potent, and selective inhibitor of cardiac late sodium current suppresses experimental arrhythmias. J. Pharmacol. Exp. Ther. 2013, 344, 23–32. [Google Scholar] [CrossRef]
- Melgari, D.; Zhang, Y.; El Harchi, A.; Dempsey, C.E.; Hancox, J.C. Molecular basis of hERG potassium channel blockade by the class Ic antiarrhythmic flecainide. J. Mol. Cell. Cardiol. 2015, 86, 42–53. [Google Scholar] [CrossRef] [Green Version]
- Kryshtal, D.O.; Blackwell, D.J.; Egly, C.L.; Smith, A.N.; Batiste, S.M.; Johnston, J.N.; Laver, D.R.; Knollmann, B.C. RYR2 Channel Inhibition Is the Principal Mechanism of Flecainide Action in CPVT. Circ. Res. 2021, 128, 321–331. [Google Scholar] [CrossRef]
- Bannister, M.L.; MacLeod, K.T.; George, C.H. Moving in the right direction: Elucidating the mechanisms of interaction between flecainide and the cardiac ryanodine receptor. Br. J. Pharmacol. 2022, 179, 2558–2563. [Google Scholar] [CrossRef]
- Hwang, H.S.; Hasdemir, C.; Laver, D.; Mehra, D.; Turhan, K.; Faggioni, M.; Yin, H.; Knollmann, B.C. Inhibition of cardiac Ca2+ release channels (RyR2) determines efficacy of class I antiarrhythmic drugs in catecholaminergic polymorphic ventricular tachycardia. Circ. Arrhythmia Electrophysiol. 2011, 4, 128–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Echt, D.S.; Liebson, P.R.; Mitchell, L.B.; Peters, R.W.; Obias-Manno, D.; Barker, A.H.; Arensberg, D.; Baker, A.; Friedman, L.; Greene, H.L.; et al. Mortality and morbidity in patients receiving encainide, flecainide, or placebo. The Cardiac Arrhythmia Suppression Trial. N. Engl. J. Med. 1991, 324, 781–788. [Google Scholar] [CrossRef] [PubMed]
- Pantlin, P.G.; Bober, R.M.; Bernard, M.L.; Khatib, S.; Polin, G.M.; Rogers, P.A.; Morin, D.P. Class 1C antiarrhythmic drugs in atrial fibrillation and coronary artery disease. J. Cardiovasc. Electrophysiol. 2020, 31, 607–611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashraf, H.; Ko, N.K.; Ladia, V.; Agasthi, P.; Prendiville, T.; O’Herlihy, F.; Pujari, S.H.; Mulpuru, S.K.; Scott, L.; Sorajja, D. Use of Flecainide in Stable Coronary Artery Disease: An Analysis of Its Safety in Both Nonobstructive and Obstructive Coronary Artery Disease. Am. J. Cardiovasc. Drugs 2021, 21, 563–572. [Google Scholar] [CrossRef]
- Turturiello, D.; Cappato, R. The many NOs to the use of Class IC antiarrhythmics: Weren’t the guidelines too strict? Eur. Heart J. Suppl. 2022, 24, I47–I53. [Google Scholar] [CrossRef]
- Ermakov, S.; Gerstenfeld, E.P.; Svetlichnaya, Y.; Scheinman, M.M. Use of flecainide in combination antiarrhythmic therapy in patients with arrhythmogenic right ventricular cardiomyopathy. Heart Rhythm. 2017, 14, 564–569. [Google Scholar] [CrossRef]
- Towbin, J.A.; McKenna, W.J.; Abrams, D.J.; Ackerman, M.J.; Calkins, H.; Darrieux, F.C.C.; Daubert, J.P.; de Chillou, C.; DePasquale, E.C.; Desai, M.Y.; et al. 2019 HRS expert consensus statement on evaluation, risk stratification, and management of arrhythmogenic cardiomyopathy: Executive summary. Heart Rhythm. 2019, 16, e373–e407. [Google Scholar] [CrossRef] [Green Version]
- Moccia, F.; Lodola, F.; Stadiotti, I.; Pilato, C.A.; Bellin, M.; Carugo, S.; Pompilio, G.; Sommariva, E.; Maione, A.S. Calcium as a Key Player in Arrhythmogenic Cardiomyopathy: Adhesion Disorder or Intracellular Alteration? Int. J. Mol. Sci. 2019, 20, 3986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerrone, M.; Montnach, J.; Lin, X.; Zhao, Y.T.; Zhang, M.; Agullo-Pascual, E.; Leo-Macias, A.; Alvarado, F.J.; Dolgalev, I.; Karathanos, T.V.; et al. Plakophilin-2 is required for transcription of genes that control calcium cycling and cardiac rhythm. Nat. Commun. 2017, 8, 106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eisner, D.A.; Caldwell, J.L.; Kistamas, K.; Trafford, A.W. Calcium and Excitation-Contraction Coupling in the Heart. Circ. Res. 2017, 121, 181–195. [Google Scholar] [CrossRef]
- Kim, J.C.; Perez-Hernandez, M.; Alvarado, F.J.; Maurya, S.R.; Montnach, J.; Yin, Y.; Zhang, M.; Lin, X.; Vasquez, C.; Heguy, A.; et al. Disruption of Ca2+i Homeostasis and Connexin 43 Hemichannel Function in the Right Ventricle Precedes Overt Arrhythmogenic Cardiomyopathy in Plakophilin-2-Deficient Mice. Circulation 2019, 140, 1015–1030. [Google Scholar] [CrossRef]
- van Opbergen, C.J.M.; Bagwan, N.; Maurya, S.R.; Kim, J.C.; Smith, A.N.; Blackwell, D.J.; Johnston, J.N.; Knollmann, B.C.; Cerrone, M.; Lundby, A.; et al. Exercise Causes Arrhythmogenic Remodeling of Intracellular Calcium Dynamics in Plakophilin-2-Deficient Hearts. Circulation 2022, 145, 1480–1496. [Google Scholar] [CrossRef]
- Chelko, S.P.; Keceli, G.; Carpi, A.; Doti, N.; Agrimi, J.; Asimaki, A.; Beti, C.B.; Miyamoto, M.; Amat-Codina, N.; Bedja, D.; et al. Exercise triggers CAPN1-mediated AIF truncation, inducing myocyte cell death in arrhythmogenic cardiomyopathy. Sci. Transl. Med. 2021, 13, eabf0891. [Google Scholar] [CrossRef] [PubMed]
- Priori, S.G.; Chen, S.R. Inherited dysfunction of sarcoplasmic reticulum Ca2+ handling and arrhythmogenesis. Circ. Res. 2011, 108, 871–883. [Google Scholar] [CrossRef]
- Venetucci, L.; Denegri, M.; Napolitano, C.; Priori, S.G. Inherited calcium channelopathies in the pathophysiology of arrhythmias. Nat. Rev. Cardiol. 2012, 9, 561–575. [Google Scholar] [CrossRef]
- Moreau, A.; Reisqs, J.B.; Delanoe-Ayari, H.; Pierre, M.; Janin, A.; Deliniere, A.; Bessiere, F.; Meli, A.C.; Charrabi, A.; Lafont, E.; et al. Deciphering DSC2 arrhythmogenic cardiomyopathy electrical instability: From ion channels to ECG and tailored drug therapy. Clin. Transl. Med. 2021, 11, e319. [Google Scholar] [CrossRef]
- Forostyak, O.; Forostyak, S.; Kortus, S.; Sykova, E.; Verkhratsky, A.; Dayanithi, G. Physiology of Ca2+ signalling in stem cells of different origins and differentiation stages. Cell Calcium 2016, 59, 57–66. [Google Scholar] [CrossRef]
- Torre, E.C.; Bicer, M.; Cottrell, G.S.; Widera, D.; Tamagnini, F. Time-Dependent Reduction of Calcium Oscillations in Adipose-Derived Stem Cells Differentiating towards Adipogenic and Osteogenic Lineage. Biomolecules 2021, 11, 1400. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F.; Ruffinatti, F.A.; Zuccolo, E. Intracellular Ca2+ Signals to Reconstruct A Broken Heart: Still A Theoretical Approach? Curr. Drug Targets 2015, 16, 793–815. [Google Scholar] [CrossRef] [PubMed]
- Faris, P.; Casali, C.; Negri, S.; Iengo, L.; Biggiogera, M.; Maione, A.S.; Moccia, F. Nicotinic Acid Adenine Dinucleotide Phosphate Induces Intracellular Ca2+ Signalling and Stimulates Proliferation in Human Cardiac Mesenchymal Stromal Cells. Front. Cell Dev. Biol. 2022, 10, 874043. [Google Scholar] [CrossRef] [PubMed]
- Kawano, S.; Shoji, S.; Ichinose, S.; Yamagata, K.; Tagami, M.; Hiraoka, M. Characterization of Ca2+ signaling pathways in human mesenchymal stem cells. Cell Calcium 2002, 32, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Kawano, S.; Otsu, K.; Shoji, S.; Yamagata, K.; Hiraoka, M. Ca2+ oscillations regulated by Na+-Ca2+ exchanger and plasma membrane Ca2+ pump induce fluctuations of membrane currents and potentials in human mesenchymal stem cells. Cell Calcium 2003, 34, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Kawano, S.; Otsu, K.; Kuruma, A.; Shoji, S.; Yanagida, E.; Muto, Y.; Yoshikawa, F.; Hirayama, Y.; Mikoshiba, K.; Furuichi, T. ATP autocrine/paracrine signaling induces calcium oscillations and NFAT activation in human mesenchymal stem cells. Cell Calcium 2006, 39, 313–324. [Google Scholar] [CrossRef]
- Berridge, M.J. Inositol trisphosphate and calcium oscillations. Biochem. Soc. Symp. 2007, 74, 1–7. [Google Scholar] [CrossRef]
- Berridge, M.J. Inositol trisphosphate and calcium signalling mechanisms. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2009, 1793, 933–940. [Google Scholar] [CrossRef] [Green Version]
- Miyakawa, T.; Maeda, A.; Yamazawa, T.; Hirose, K.; Kurosaki, T.; Iino, M. Encoding of Ca2+ signals by differential expression of IP3 receptor subtypes. EMBO J. 1999, 18, 1303–1308. [Google Scholar] [CrossRef]
- Dai, J.M.; Kuo, K.H.; Leo, J.M.; van Breemen, C.; Lee, C.H. Mechanism of ACh-induced asynchronous calcium waves and tonic contraction in porcine tracheal muscle bundle. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 290, L459–L469. [Google Scholar] [CrossRef] [Green Version]
- Di Capite, J.; Ng, S.W.; Parekh, A.B. Decoding of cytoplasmic Ca2+ oscillations through the spatial signature drives gene expression. Curr. Biol. 2009, 19, 853–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuccolo, E.; Kheder, D.A.; Lim, D.; Perna, A.; Nezza, F.D.; Botta, L.; Scarpellino, G.; Negri, S.; Martinotti, S.; Soda, T.; et al. Glutamate triggers intracellular Ca2+ oscillations and nitric oxide release by inducing NAADP- and InsP3 -dependent Ca2+ release in mouse brain endothelial cells. J. Cell. Physiol. 2019, 234, 3538–3554. [Google Scholar] [CrossRef] [PubMed]
- Balducci, V.; Faris, P.; Balbi, C.; Costa, A.; Negri, S.; Rosti, V.; Bollini, S.; Moccia, F. The human amniotic fluid stem cell secretome triggers intracellular Ca2+ oscillations, NF-kappaB nuclear translocation and tube formation in human endothelial colony-forming cells. J. Cell. Mol. Med. 2021, 25, 8074–8086. [Google Scholar] [CrossRef] [PubMed]
- Berra-Romani, R.; Faris, P.; Pellavio, G.; Orgiu, M.; Negri, S.; Forcaia, G.; Var-Gaz-Guadarrama, V.; Garcia-Carrasco, M.; Botta, L.; Sancini, G.; et al. Histamine induces intracellular Ca2+ oscillations and nitric oxide release in endothelial cells from brain microvascular circulation. J. Cell. Physiol. 2020, 235, 1515–1530. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F.; Dragoni, S.; Lodola, F.; Bonetti, E.; Bottino, C.; Guerra, G.; Laforenza, U.; Rosti, V.; Tanzi, F. Store-dependent Ca2+ entry in endothelial progenitor cells as a perspective tool to enhance cell-based therapy and adverse tumour vascularization. Curr. Med. Chem. 2012, 19, 5802–5818. [Google Scholar] [CrossRef] [PubMed]
- Thillaiappan, N.B.; Chavda, A.P.; Tovey, S.C.; Prole, D.L.; Taylor, C.W. Ca2+ signals initiate at immobile IP3 receptors adjacent to ER-plasma membrane junctions. Nat. Commun. 2017, 8, 1505. [Google Scholar] [CrossRef] [Green Version]
- Taylor, C.W.; Machaca, K. IP3 receptors and store-operated Ca2+ entry: A license to fill. Curr. Opin. Cell Biol. 2018, 57, 1–7. [Google Scholar] [CrossRef]
- Emrich, S.M.; Yoast, R.E.; Xin, P.; Arige, V.; Wagner, L.E.; Hempel, N.; Gill, D.L.; Sneyd, J.; Yule, D.I.; Trebak, M. Omnitemporal choreographies of all five STIM/Orai and IP3Rs underlie the complexity of mammalian Ca2+ signaling. Cell Rep. 2021, 34, 108760. [Google Scholar] [CrossRef]
- Lewis, R.S. Store-Operated Calcium Channels: From Function to Structure and Back Again. Cold Spring Harb. Perspect. Biol. 2020, 12, a035055. [Google Scholar] [CrossRef]
- Brandman, O.; Liou, J.; Park, W.S.; Meyer, T. STIM2 is a feedback regulator that stabilizes basal cytosolic and endoplasmic reticulum Ca2+ levels. Cell 2007, 131, 1327–1339. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, M.; Ong, H.L.; Saadi, H.; Son, G.Y.; Shokatian, Z.; Terry, L.E.; Trebak, M.; Yule, D.I.; Ambudkar, I. Functional communication between IP3R and STIM2 at subthreshold stimuli is a critical checkpoint for initiation of SOCE. Proc. Natl. Acad. Sci. USA 2022, 119, e2114928118. [Google Scholar] [CrossRef]
- Subedi, K.P.; Ong, H.L.; Son, G.Y.; Liu, X.; Ambudkar, I.S. STIM2 Induces Activated Conformation of STIM1 to Control Orai1 Function in ER-PM Junctions. Cell Rep. 2018, 23, 522–534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mignen, O.; Constantin, B.; Potier-Cartereau, M.; Penna, A.; Gautier, M.; Gueguinou, M.; Renaudineau, Y.; Shoji, K.F.; Felix, R.; Bayet, E.; et al. Constitutive calcium entry and cancer: Updated views and insights. Eur. Biophys. J. 2017, 46, 395–413. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F.; Zuccolo, E.; Soda, T.; Tanzi, F.; Guerra, G.; Mapelli, L.; Lodola, F.; D’Angelo, E. Stim and Orai proteins in neuronal Ca2+ signaling and excitability. Front. Cell. Neurosci. 2015, 9, 153. [Google Scholar] [CrossRef] [Green Version]
- Zuccolo, E.; Laforenza, U.; Negri, S.; Botta, L.; Berra-Romani, R.; Faris, P.; Scarpellino, G.; Forcaia, G.; Pellavio, G.; Sancini, G.; et al. Muscarinic M5 receptors trigger acetylcholine-induced Ca2+ signals and nitric oxide release in human brain microvascular endothelial cells. J. Cell. Physiol. 2019, 234, 4540–4562. [Google Scholar] [CrossRef]
- Yoast, R.E.; Emrich, S.M.; Zhang, X.; Xin, P.; Johnson, M.T.; Fike, A.J.; Walter, V.; Hempel, N.; Yule, D.I.; Sneyd, J.; et al. The native ORAI channel trio underlies the diversity of Ca2+ signaling events. Nat. Commun. 2020, 11, 2444. [Google Scholar] [CrossRef] [PubMed]
- Vaeth, M.; Yang, J.; Yamashita, M.; Zee, I.; Eckstein, M.; Knosp, C.; Kaufmann, U.; Karoly Jani, P.; Lacruz, R.S.; Flockerzi, V.; et al. ORAI2 modulates store-operated calcium entry and T cell-mediated immunity. Nat. Commun. 2017, 8, 14714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eckstein, M.; Vaeth, M.; Aulestia, F.J.; Costiniti, V.; Kassam, S.N.; Bromage, T.G.; Pedersen, P.; Issekutz, T.; Idaghdour, Y.; Moursi, A.M.; et al. Differential regulation of Ca2+ influx by ORAI channels mediates enamel mineralization. Sci. Signal. 2019, 12, eaav4663. [Google Scholar] [CrossRef]
- Lee, S.H.; Park, Y.; Song, M.; Srikanth, S.; Kim, S.; Kang, M.K.; Gwack, Y.; Park, N.H.; Kim, R.H.; Shin, K.H. Orai1 mediates osteogenic differentiation via BMP signaling pathway in bone marrow mesenchymal stem cells. Biochem. Biophys. Res. Commun. 2016, 473, 1309–1314. [Google Scholar] [CrossRef] [Green Version]
- Ahamad, N.; Sun, Y.; Nascimento Da Conceicao, V.; Xavier Paul Ezhilan, C.R.D.; Natarajan, M.; Singh, B.B. Differential activation of Ca2+ influx channels modulate stem cell potency, their proliferation/viability and tissue regeneration. NPJ Regen. Med. 2021, 6, 67. [Google Scholar] [CrossRef]
- Ahamad, N.; Sun, Y.; Singh, B.B. Increasing cytosolic Ca2+ levels restore cell proliferation and stem cell potency in aged MSCs. Stem Cell Res. 2021, 56, 102560. [Google Scholar] [CrossRef]
- Peng, H.; Hao, Y.; Mousawi, F.; Roger, S.; Li, J.; Sim, J.A.; Ponnambalam, S.; Yang, X.; Jiang, L.H. Purinergic and Store-Operated Ca2+ Signaling Mechanisms in Mesenchymal Stem Cells and Their Roles in ATP-Induced Stimulation of Cell Migration. Stem Cells 2016, 34, 2102–2114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, T.J.; Seong, J.; Ouyang, M.; Sun, J.; Lu, S.; Hong, J.P.; Wang, N.; Wang, Y. Substrate rigidity regulates Ca2+ oscillation via RhoA pathway in stem cells. J. Cell. Physiol. 2009, 218, 285–293. [Google Scholar] [CrossRef] [Green Version]
- Tao, R.; Sun, H.Y.; Lau, C.P.; Tse, H.F.; Lee, H.C.; Li, G.R. Cyclic ADP ribose is a novel regulator of intracellular Ca2+ oscillations in human bone marrow mesenchymal stem cells. J. Cell. Mol. Med. 2011, 15, 2684–2696. [Google Scholar] [CrossRef] [Green Version]
- Sauer, H.; Sharifpanah, F.; Hatry, M.; Steffen, P.; Bartsch, C.; Heller, R.; Padmasekar, M.; Howaldt, H.P.; Bein, G.; Wartenberg, M. NOS inhibition synchronizes calcium oscillations in human adipose tissue-derived mesenchymal stem cells by increasing gap-junctional coupling. J. Cell. Physiol. 2011, 226, 1642–1650. [Google Scholar] [CrossRef]
- Mestril, S.; Kim, R.; Hinman, S.S.; Gomez, S.M.; Allbritton, N.L. Stem/Proliferative and Differentiated Cells within Primary Murine Colonic Epithelium Display Distinct Intracellular Free Ca2+ Signal Codes. Adv. Healthc. Mater. 2021, 10, e2101318. [Google Scholar] [CrossRef]
- Okada, H.; Okabe, K.; Tanaka, S. Finely-Tuned Calcium Oscillations in Osteoclast Differentiation and Bone Resorption. Int. J. Mol. Sci. 2020, 22, 180. [Google Scholar] [CrossRef]
- Tyser, R.C.; Miranda, A.M.; Chen, C.M.; Davidson, S.M.; Srinivas, S.; Riley, P.R. Calcium handling precedes cardiac differentiation to initiate the first heartbeat. eLife 2016, 5, e17113. [Google Scholar] [CrossRef] [PubMed]
- Pchelintseva, E.; Djamgoz, M.B.A. Mesenchymal stem cell differentiation: Control by calcium-activated potassium channels. J. Cell. Physiol. 2018, 233, 3755–3768. [Google Scholar] [CrossRef] [PubMed]
- Titushkin, I.; Sun, S.; Shin, J.; Cho, M. Physicochemical control of adult stem cell differentiation: Shedding light on potential molecular mechanisms. J. Biomed. Biotechnol. 2010, 2010, 743476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, S.; Liu, Y.; Lipsky, S.; Cho, M. Physical manipulation of calcium oscillations facilitates osteodifferentiation of human mesenchymal stem cells. FASEB J. 2007, 21, 1472–1480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Y.L.; Chou, R.H.; Chen, L.T.; Shyu, W.C.; Hsieh, S.C.; Wu, C.S.; Zeng, H.J.; Yeh, S.P.; Yang, D.M.; Hung, S.C.; et al. EZH2 regulates neuronal differentiation of mesenchymal stem cells through PIP5K1C-dependent calcium signaling. J. Biol. Chem. 2011, 286, 9657–9667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lepski, G.; Jannes, C.E.; Maciaczyk, J.; Papazoglou, A.; Mehlhorn, A.T.; Kaiser, S.; Teixeira, M.J.; Marie, S.K.; Bischofberger, J.; Nikkhah, G. Limited Ca2+ and PKA-pathway dependent neurogenic differentiation of human adult mesenchymal stem cells as compared to fetal neuronal stem cells. Exp. Cell Res. 2010, 316, 216–231. [Google Scholar] [CrossRef] [PubMed]
- Mozolewski, P.; Jeziorek, M.; Schuster, C.M.; Bading, H.; Frost, B.; Dobrowolski, R. The role of nuclear Ca2+ in maintaining neuronal homeostasis and brain health. J. Cell Sci. 2021, 134. [Google Scholar] [CrossRef]
- Hanna, H.; Andre, F.M.; Mir, L.M. Electrical control of calcium oscillations in mesenchymal stem cells using microsecond pulsed electric fields. Stem Cell Res. Ther. 2017, 8, 91. [Google Scholar] [CrossRef] [Green Version]
- Thrivikraman, G.; Madras, G.; Basu, B. Electrically driven intracellular and extracellular nanomanipulators evoke neurogenic/cardiomyogenic differentiation in human mesenchymal stem cells. Biomaterials 2016, 77, 26–43. [Google Scholar] [CrossRef]
- Masoumi, N.; Ghollasi, M.; Raheleh, H.; Eftekhari, E.; Ghiasi, M. Carbachol, along with calcium, indicates new strategy in neural differentiation of human adipose tissue-derived mesenchymal stem cells in vitro. Regen. Ther. 2023, 23, 60–66. [Google Scholar] [CrossRef]
- Samanta, K.; Mirams, G.R.; Parekh, A.B. Sequential forward and reverse transport of the Na+ Ca2+ exchanger generates Ca2+ oscillations within mitochondria. Nat. Commun. 2018, 9, 156. [Google Scholar] [CrossRef] [Green Version]
- Berra-Romani, R.; Raqeeb, A.; Torres-Jácome, J.; Guzman-Silva, A.; Guerra, G.; Tanzi, F.; Moccia, F. The mechanism of injury-induced intracellular calcium concentration oscillations in the endothelium of excised rat aorta. J. Vasc. Res. 2012, 49, 65–76. [Google Scholar] [CrossRef] [Green Version]
- Scorza, S.I.; Milano, S.; Saponara, I.; Certini, M.; De Zio, R.; Mola, M.G.; Procino, G.; Carmosino, M.; Moccia, F.; Svelto, M.; et al. TRPML1-Induced Lysosomal Ca2+ Signals Activate AQP2 Translocation and Water Flux in Renal Collecting Duct Cells. Int. J. Mol. Sci. 2023, 24, 1647. [Google Scholar] [CrossRef]
- Zuccolo, E.; Laforenza, U.; Ferulli, F.; Pellavio, G.; Scarpellino, G.; Tanzi, M.; Turin, I.; Faris, P.; Lucariello, A.; Maestri, M.; et al. Stim and Orai mediate constitutive Ca2+ entry and control endoplasmic reticulum Ca2+ refilling in primary cultures of colorectal carcinoma cells. Oncotarget 2018, 9, 31098–31119. [Google Scholar] [CrossRef] [Green Version]
- Faris, P.; Rumolo, A.; Tapella, L.; Tanzi, M.; Metallo, A.; Conca, F.; Negri, S.; Lefkimmiatis, K.; Pedrazzoli, P.; Lim, D.; et al. Store-Operated Ca2+ Entry Is Up-Regulated in Tumour-Infiltrating Lymphocytes from Metastatic Colorectal Cancer Patients. Cancers 2022, 14, 3312. [Google Scholar] [CrossRef]
- Moccia, F.; Negri, S.; Faris, P.; Perna, A.; De Luca, A.; Soda, T.; Romani, R.B.; Guerra, G. Targeting Endolysosomal Two-Pore Channels to Treat Cardiovascular Disorders in the Novel COronaVIrus Disease 2019. Front. Physiol. 2021, 12, 629119. [Google Scholar] [CrossRef]
- Negri, S.; Faris, P.; Moccia, F. Endolysosomal Ca2+ signaling in cardiovascular health and disease. Int. Rev. Cell Mol. Biol. 2021, 363, 203–269. [Google Scholar] [CrossRef]
- Kilpatrick, B.S.; Eden, E.R.; Schapira, A.H.; Futter, C.E.; Patel, S. Direct mobilisation of lysosomal Ca2+ triggers complex Ca2+ signals. J. Cell Sci. 2013, 126, 60–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moccia, F.; Zuccolo, E.; Di Nezza, F.; Pellavio, G.; Faris, P.S.; Negri, S.; De Luca, A.; Laforenza, U.; Ambrosone, L.; Rosti, V.; et al. Nicotinic acid adenine dinucleotide phosphate activates two-pore channel TPC1 to mediate lysosomal Ca2+ release in endothelial colony-forming cells. J. Cell. Physiol. 2021, 236, 688–705. [Google Scholar] [CrossRef]
- Gul, R.; Park, D.R.; Shawl, A.I.; Im, S.Y.; Nam, T.S.; Lee, S.H.; Ko, J.K.; Jang, K.Y.; Kim, D.; Kim, U.H. Nicotinic Acid Adenine Dinucleotide Phosphate (NAADP) and Cyclic ADP-Ribose (cADPR) Mediate Ca2+ Signaling in Cardiac Hypertrophy Induced by beta-Adrenergic Stimulation. PLoS ONE 2016, 11, e0149125. [Google Scholar] [CrossRef]
- Capel, R.A.; Bolton, E.L.; Lin, W.K.; Aston, D.; Wang, Y.; Liu, W.; Wang, X.; Burton, R.A.; Bloor-Young, D.; Shade, K.T.; et al. Two-pore Channels (TPC2s) and Nicotinic Acid Adenine Dinucleotide Phosphate (NAADP) at Lysosomal-Sarcoplasmic Reticular Junctions Contribute to Acute and Chronic beta-Adrenoceptor Signaling in the Heart. J. Biol. Chem. 2015, 290, 30087–30098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davidson, S.M.; Foote, K.; Kunuthur, S.; Gosain, R.; Tan, N.; Tyser, R.; Zhao, Y.J.; Graeff, R.; Ganesan, A.; Duchen, M.R.; et al. Inhibition of NAADP signalling on reperfusion protects the heart by preventing lethal calcium oscillations via two-pore channel 1 and opening of the mitochondrial permeability transition pore. Cardiovasc. Res. 2015, 108, 357–366. [Google Scholar] [CrossRef] [Green Version]
- Sbano, L.; Bonora, M.; Marchi, S.; Baldassari, F.; Medina, D.L.; Ballabio, A.; Giorgi, C.; Pinton, P. TFEB-mediated increase in peripheral lysosomes regulates store-operated calcium entry. Sci. Rep. 2017, 7, 40797. [Google Scholar] [CrossRef]
- Reyes Gaido, O.E.; Nkashama, L.J.; Schole, K.L.; Wang, Q.; Umapathi, P.; Mesubi, O.O.; Konstantinidis, K.; Luczak, E.D.; Anderson, M.E. CaMKII as a Therapeutic Target in Cardiovascular Disease. Annu. Rev. Pharmacol. Toxicol. 2023, 63, 249–272. [Google Scholar] [CrossRef] [PubMed]
- Smedler, E.; Uhlen, P. Frequency decoding of calcium oscillations. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2014, 1840, 964–969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parekh, A.B. Decoding cytosolic Ca2+ oscillations. Trends Biochem. Sci. 2011, 36, 78–87. [Google Scholar] [CrossRef]
- Rostas, J.A.P.; Skelding, K.A. Calcium/Calmodulin-Stimulated Protein Kinase II (CaMKII): Different Functional Outcomes from Activation, Depending on the Cellular Microenvironment. Cells 2023, 12, 401. [Google Scholar] [CrossRef]
- De Koninck, P.; Schulman, H. Sensitivity of CaM kinase II to the frequency of Ca2+ oscillations. Science 1998, 279, 227–230. [Google Scholar] [CrossRef] [Green Version]
- Dupont, G.; Houart, G.; De Koninck, P. Sensitivity of CaM kinase II to the frequency of Ca2+ oscillations: A simple model. Cell Calcium 2003, 34, 485–497. [Google Scholar] [CrossRef]
- Markoulaki, S.; Matson, S.; Ducibella, T. Fertilization stimulates long-lasting oscillations of CaMKII activity in mouse eggs. Dev. Biol. 2004, 272, 15–25. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Ye, J.; Meng, Y.; Ai, W.; Su, H.; Zheng, J.; Liu, F.; Zhu, X.; Wang, L.; Gao, P.; et al. Calcium Supplementation Enhanced Adipogenesis and Improved Glucose Homeostasis Through Activation of Camkii and PI3K/Akt Signaling Pathway in Porcine Bone Marrow Mesenchymal Stem Cells (pBMSCs) and Mice Fed High Fat Diet (HFD). Cell. Physiol. Biochem. 2018, 51, 154–172. [Google Scholar] [CrossRef]
- Trebak, M.; Kinet, J.P. Calcium signalling in T cells. Nat. Rev. Immunol. 2019, 19, 154–169. [Google Scholar] [CrossRef]
- Abjorsbraten, K.S.; Skaaraas, G.; Cunen, C.; Bjornstad, D.M.; Binder, K.M.G.; Bojarskaite, L.; Jensen, V.; Nilsson, L.N.G.; Rao, S.B.; Tang, W.; et al. Impaired astrocytic Ca2+ signaling in awake-behaving Alzheimer’s disease transgenic mice. eLife 2022, 11, e75055. [Google Scholar] [CrossRef]
- Protasi, F.; Girolami, B.; Roccabianca, S.; Rossi, D. Store-operated calcium entry: From physiology to tubular aggregate myopathy. Curr. Opin. Pharmacol. 2023, 68, 102347. [Google Scholar] [CrossRef]
- Masson, B.; Le Ribeuz, H.; Sabourin, J.; Laubry, L.; Woodhouse, E.; Foster, R.; Ruchon, Y.; Dutheil, M.; Boet, A.; Ghigna, M.R.; et al. Orai1 Inhibitors as Potential Treatments for Pulmonary Arterial Hypertension. Circ. Res. 2022, 131, e102–e119. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F.; Zuccolo, E.; Poletto, V.; Turin, I.; Guerra, G.; Pedrazzoli, P.; Rosti, V.; Porta, C.; Montagna, D. Targeting Stim and Orai Proteins as an Alternative Approach in Anticancer Therapy. Curr. Med. Chem. 2016, 23, 3450–3480. [Google Scholar] [CrossRef] [PubMed]
- Sabourin, J.; Beauvais, A.; Luo, R.; Montani, D.; Benitah, J.P.; Masson, B.; Antigny, F. The SOCE Machinery: An Unbalanced Knowledge between Left and Right Ventricular Pathophysiology. Cells 2022, 11, 3282. [Google Scholar] [CrossRef]
- Sabourin, J.; Bartoli, F.; Antigny, F.; Gomez, A.M.; Benitah, J.P. Transient Receptor Potential Canonical (TRPC)/Orai1-dependent Store-operated Ca2+ Channels: NEW TARGETS OF ALDOSTERONE IN CARDIOMYOCYTES. J. Biol. Chem. 2016, 291, 13394–13409. [Google Scholar] [CrossRef] [Green Version]
- Riva, B.; Pessolano, E.; Quaglia, E.; Cordero-Sanchez, C.; Bhela, I.P.; Topf, A.; Serafini, M.; Cox, D.; Harris, E.; Garibaldi, M.; et al. STIM1 and ORAI1 mutations leading to tubular aggregate myopathies are sensitive to the Store-operated Ca2+-entry modulators CIC-37 and CIC-39. Cell Calcium 2022, 105, 102605. [Google Scholar] [CrossRef]
- Luo, R.; Gomez, A.M.; Benitah, J.P.; Sabourin, J. Targeting Orai1-Mediated Store-Operated Ca2+ Entry in Heart Failure. Front. Cell Dev. Biol. 2020, 8, 586109. [Google Scholar] [CrossRef]
- Ma, G.; Wen, S.; Huang, Y.; Zhou, Y. The STIM-Orai Pathway: Light-Operated Ca2+ Entry Through Engineered CRAC Channels. Adv. Exp. Med. Biol. 2017, 993, 117–138. [Google Scholar] [CrossRef]
- Azimi, I.; Stevenson, R.J.; Zhang, X.; Meizoso-Huesca, A.; Xin, P.; Johnson, M.; Flanagan, J.U.; Chalmers, S.B.; Yoast, R.E.; Kapure, J.S.; et al. A new selective pharmacological enhancer of the Orai1 Ca2+ channel reveals roles for Orai1 in smooth and skeletal muscle functions. ACS Pharmacol. Transl. Sci. 2020, 3, 135–147. [Google Scholar] [CrossRef] [Green Version]
- Moccia, F.; Dragoni, S.; Poletto, V.; Rosti, V.; Tanzi, F.; Ganini, C.; Porta, C. Orai1 and Transient Receptor Potential Channels as novel molecular targets to impair tumor neovascularisation in renal cell carcinoma and other malignancies. Anti-Cancer Agents Med. Chem. 2014, 14, 296–312. [Google Scholar]
- Moccia, F.; Bonetti, E.; Dragoni, S.; Fontana, J.; Lodola, F.; Romani, R.B.; Laforenza, U.; Rosti, V.; Tanzi, F. Hematopoietic progenitor and stem cells circulate by surfing on intracellular Ca2+ waves: A novel target for cell-based therapy and anti-cancer treatment? Curr. Signal Transduct. Ther. 2012, 7, 161–176. [Google Scholar]
- Andrikopoulos, P.; Baba, A.; Matsuda, T.; Djamgoz, M.B.; Yaqoob, M.M.; Eccles, S.A. Ca2+ influx through reverse mode Na+/ Ca2+ exchange is critical for vascular endothelial growth factor-mediated extracellular signal-regulated kinase (ERK) 1/2 activation and angiogenic functions of human endothelial cells. J. Biol. Chem. 2011, 286, 37919–37931. [Google Scholar] [CrossRef] [Green Version]
- Andrikopoulos, P.; Fraser, S.P.; Patterson, L.; Ahmad, Z.; Burcu, H.; Ottaviani, D.; Diss, J.K.; Box, C.; Eccles, S.A.; Djamgoz, M.B. Angiogenic functions of voltage-gated Na+ Channels in human endothelial cells: Modulation of vascular endothelial growth factor (VEGF) signaling. J. Biol. Chem. 2011, 286, 16846–16860. [Google Scholar] [CrossRef] [Green Version]
- Stauderman, K.A. CRAC channels as targets for drug discovery and development. Cell Calcium 2018, 74, 147–159. [Google Scholar] [CrossRef]
- Tian, C.; Du, L.; Zhou, Y.; Li, M. Store-operated CRAC channel inhibitors: Opportunities and challenges. Future Med. Chem. 2016, 8, 817–832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schleifer, H.; Doleschal, B.; Lichtenegger, M.; Oppenrieder, R.; Derler, I.; Frischauf, I.; Glasnov, T.N.; Kappe, C.O.; Romanin, C.; Groschner, K. Novel pyrazole compounds for pharmacological discrimination between receptor-operated and store-operated Ca2+ entry pathways. Br. J. Pharmacol. 2012, 167, 1712–1722. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Xin, P.; Yoast, R.E.; Emrich, S.M.; Johnson, M.T.; Pathak, T.; Benson, J.C.; Azimi, I.; Gill, D.L.; Monteith, G.R.; et al. Distinct pharmacological profiles of ORAI1, ORAI2, and ORAI3 channels. Cell Calcium 2020, 91, 102281. [Google Scholar] [CrossRef]
- Chauvet, S.; Jarvis, L.; Chevallet, M.; Shrestha, N.; Groschner, K.; Bouron, A. Pharmacological Characterization of the Native Store-Operated Calcium Channels of Cortical Neurons from Embryonic Mouse Brain. Front. Pharmacol. 2016, 7, 486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuccolo, E.; Lim, D.; Kheder, D.A.; Perna, A.; Catarsi, P.; Botta, L.; Rosti, V.; Riboni, L.; Sancini, G.; Tanzi, F.; et al. Acetylcholine induces intracellular Ca2+ oscillations and nitric oxide release in mouse brain endothelial cells. Cell Calcium 2017, 66, 33–47. [Google Scholar] [CrossRef] [PubMed]
- Takezawa, R.; Cheng, H.; Beck, A.; Ishikawa, J.; Launay, P.; Kubota, H.; Kinet, J.P.; Fleig, A.; Yamada, T.; Penner, R. A pyrazole derivative potently inhibits lymphocyte Ca2+ influx and cytokine production by facilitating transient receptor potential melastatin 4 channel activity. Mol. Pharmacol. 2006, 69, 1413–1420. [Google Scholar] [CrossRef]
- Zitt, C.; Strauss, B.; Schwarz, E.C.; Spaeth, N.; Rast, G.; Hatzelmann, A.; Hoth, M. Potent inhibition of Ca2+ release-activated Ca2+ channels and T-lymphocyte activation by the pyrazole derivative BTP2. J. Biol. Chem. 2004, 279, 12427–12437. [Google Scholar] [CrossRef] [Green Version]
- Jairaman, A.; Prakriya, M. Molecular pharmacology of store-operated CRAC channels. Channels 2013, 7, 402–414. [Google Scholar] [CrossRef] [Green Version]
- DeHaven, W.I.; Smyth, J.T.; Boyles, R.R.; Bird, G.S.; Putney, J.W., Jr. Complex actions of 2-aminoethyldiphenyl borate on store-operated calcium entry. J. Biol. Chem. 2008, 283, 19265–19273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuccolo, E.; Bottino, C.; Diofano, F.; Poletto, V.; Codazzi, A.C.; Mannarino, S.; Campanelli, R.; Fois, G.; Marseglia, G.L.; Guerra, G.; et al. Constitutive Store-Operated Ca2+ Entry Leads to Enhanced Nitric Oxide Production and Proliferation in Infantile Hemangioma-Derived Endothelial Colony-Forming Cells. Stem Cells Dev. 2016, 25, 301–319. [Google Scholar] [CrossRef] [PubMed]
- Campanelli, R.; Codazzi, A.C.; Poletto, V.; Abba, C.; Catarsi, P.; Fois, G.; Avanzini, M.A.; Brazzelli, V.; Tzialla, C.; De Silvestri, A.; et al. Kinetic and Angiogenic Activity of Circulating Endothelial Colony Forming Cells in Patients with Infantile Haemangioma Receiving Propranolol. Thromb. Haemost. 2019, 119, 274–284. [Google Scholar] [CrossRef] [PubMed]
- Rahman, S.; Rahman, T. Unveiling some FDA-approved drugs as inhibitors of the store-operated Ca2+ entry pathway. Sci. Rep. 2017, 7, 12881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strack, S.; Barban, M.A.; Wadzinski, B.E.; Colbran, R.J. Differential inactivation of postsynaptic density-associated and soluble Ca2+/calmodulin-dependent protein kinase II by protein phosphatases 1 and 2A. J. Neurochem. 1997, 68, 2119–2128. [Google Scholar] [CrossRef] [Green Version]
- Shioda, N.; Fukunaga, K. Physiological and Pathological Roles of CaMKII-PP1 Signaling in the Brain. Int. J. Mol. Sci. 2017, 19, 20. [Google Scholar] [CrossRef] [Green Version]
- Aromolaran, A.A.; Blatter, L.A. Modulation of intracellular Ca2+ release and capacitative Ca2+ entry by CaMKII inhibitors in bovine vascular endothelial cells. Am. J. Physiol. Cell Physiol. 2005, 289, C1426–C1436. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Xue, J.; Sun, Z.; Liu, T.; Zhang, L.; Wang, L.; You, H.; Fan, Z.; Zheng, Y.; Luo, D. CaMKII Potentiates Store-Operated Ca2+ Entry Through Enhancing STIM1 Aggregation and Interaction with Orai1. Cell. Physiol. Biochem. 2018, 46, 1042–1054. [Google Scholar] [CrossRef]
- Ji, Y.; Guo, X.; Zhang, Z.; Huang, Z.; Zhu, J.; Chen, Q.H.; Gui, L. CaMKIIdelta meditates phenylephrine induced cardiomyocyte hypertrophy through store-operated Ca2+ entry. Cardiovasc. Pathol. 2017, 27, 9–17. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moccia, F.; Brunetti, V.; Soda, T.; Faris, P.; Scarpellino, G.; Berra-Romani, R. Store-Operated Ca2+ Entry as a Putative Target of Flecainide for the Treatment of Arrhythmogenic Cardiomyopathy. J. Clin. Med. 2023, 12, 5295. https://doi.org/10.3390/jcm12165295
Moccia F, Brunetti V, Soda T, Faris P, Scarpellino G, Berra-Romani R. Store-Operated Ca2+ Entry as a Putative Target of Flecainide for the Treatment of Arrhythmogenic Cardiomyopathy. Journal of Clinical Medicine. 2023; 12(16):5295. https://doi.org/10.3390/jcm12165295
Chicago/Turabian StyleMoccia, Francesco, Valentina Brunetti, Teresa Soda, Pawan Faris, Giorgia Scarpellino, and Roberto Berra-Romani. 2023. "Store-Operated Ca2+ Entry as a Putative Target of Flecainide for the Treatment of Arrhythmogenic Cardiomyopathy" Journal of Clinical Medicine 12, no. 16: 5295. https://doi.org/10.3390/jcm12165295