
Hope on the Horizon: New and Future Therapies for Sickle Cell Disease


Abstract
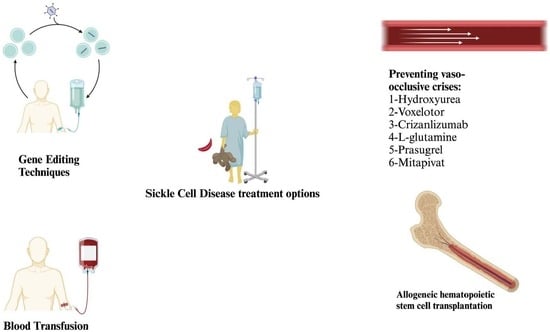
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Conventional Treatments
2.1. Hydroxyurea
2.2. Blood Transfusions
3. New Treatments
3.1. Voxelotor
3.2. Crizanlizumab
3.3. L-Glutamine
3.4. Stem Cell Transplantation
3.4.1. HCT with HLA-Matched Donors
3.4.2. HCT with Haploidentical Donors
4. Future Treatments
4.1. Prasugrel
4.2. Mitapivat
4.3. Gene Therapy
4.3.1. Advanced Gene Editing Techniques in SCD Therapy
Base Editing: Precision Redefined
Prime Editing: Direct Correction at the Molecular Level
Challenges and Future Directions
5. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brandow, A.M.; Liem, R.I. Advances in the diagnosis and treatment of sickle cell disease. J. Hematol. Oncol. 2022, 15, 20. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, R.K.; Patel, R.K.; Shah, V.; Nainiwal, L.; Trivedi, B. Hydroxyurea in sickle cell disease: Drug review. Indian J. Hematol. Blood Transfus. 2014, 30, 91–96. [Google Scholar] [CrossRef] [PubMed]
- Wong, T.E.; Brandow, A.M.; Lim, W.; Lottenberg, R. Update on the use of hydroxyurea therapy in sickle cell disease. Blood 2014, 124, 3850–3857. [Google Scholar] [CrossRef]
- Berthaut, I.; Bachir, D.; Kotti, S.; Chalas, C.; Stankovic, K.; Eustache, F.; Ravel, C.; Habibi, A.; Brailly-Tabard, S.; Lévy-Dutel, L.; et al. Adverse effect of hydroxyurea on spermatogenesis in patients with sickle cell anemia after 6 months of treatment. Blood 2017, 130, 2354–2356. [Google Scholar] [CrossRef]
- Virgous, C.; Lyons, L.; Sakwe, A.; Nayyar, T.; Goodwin, S.; Hildreth, J.; Osteen, K.; Bruner-Tran, K.; Alawode, O.; Bourne, P.; et al. Resumption of Spermatogenesis and Fertility Post Withdrawal of Hydroxyurea Treatment. Int. J. Mol. Sci. 2023, 24, 9374. [Google Scholar] [CrossRef] [PubMed]
- Joseph, L.; Jean, C.; Manceau, S.; Chalas, C.; Arnaud, C.; Kamdem, A.; Pondarre, C.; Habibi, A.; Bernaudin, F.; Allali, S.; et al. Effect of hydroxyurea exposure before puberty on sperm parameters in males with sickle cell disease. Blood 2021, 137, 826–829. [Google Scholar] [CrossRef] [PubMed]
- Piccin, A.; Murphy, C.; Eakins, E.; Rondinelli, M.B.; Daves, M.; Vecchiato, C.; Wolf, D.; Mc Mahon, C.; Smith, O.P. Insight into the complex pathophysiology of sickle cell anaemia and possible treatment. Eur. J. Haematol. 2019, 102, 319–330. [Google Scholar] [CrossRef]
- Steinberg, M.H.; Barton, F.; Castro, O.; Pegelow, C.H.; Ballas, S.K.; Kutlar, A.; Orringer, E.; Bellevue, R.; Olivieri, N.; Eckman, J.; et al. Effect of Hydroxyurea on Mortality and Morbidity in Adult Sickle Cell Anemia. JAMA 2003, 289, 1645–1651. [Google Scholar] [CrossRef]
- Zhou, B.; Mo, X.; Liu, X.; Qiu, W.; Yen, Y. Human ribonucleotide reductase M2 subunit gene amplification and transcriptional regulation in a homogeneous staining chromosome region responsible for the mechanism of drug resistance. Cytogenet. Genome Res. 2001, 95, 34–42. [Google Scholar] [CrossRef]
- Snyder, R.D. The role of deoxynucleoside triphosphate pools in the inhibition of DNA-excision repair and replication in human cells by hydroxyurea. Mutat. Res. Repair Rep. 1984, 131, 163–172. [Google Scholar] [CrossRef]
- Ware, R.E. How I use hydroxyurea to treat young patients with sickle cell anemia. Blood 2010, 115, 5300–5311. [Google Scholar] [CrossRef] [PubMed]
- Elford, H.L. Effect of hydroxyurea on ribonucleotide reductase. Biochem. Biophys. Res. Commun. 1968, 33, 129–135. [Google Scholar] [CrossRef]
- Charache, S. Mechanism of action of hydroxyurea in the management of sickle cell anemia in adults. Semin. Hematol. 1997, 34, 15–21. [Google Scholar] [PubMed]
- Nahavandi, M.; Tavakkoli, F.; Wyche, M.Q.; Perlin, E.; Winter, W.P.; Castro, O. Nitric oxide and cyclic GMP levels in sickle cell patients receiving hydroxyurea. Br. J. Haematol. 2002, 119, 855–857. [Google Scholar] [CrossRef]
- Morris, C.R.; Vichinsky, E.P.; Van Warmerdam, J.; Machado, L.; Kepka-Lenhart, D.; Morris, S.M.; Kuypers, F.A. Hydroxyurea and Arginine Therapy: Impact on Nitric Oxide Production in Sickle Cell Disease. J. Pediatr. Hematol. Oncol. 2003, 25, 629–634. [Google Scholar] [CrossRef] [PubMed]
- Raththagala, M.; Karunarathne, W.; Kryziniak, M.; McCracken, J.; Spence, D.M. Hydroxyurea stimulates the release of ATP from rabbit erythrocytes through an increase in calcium and nitric oxide production. Eur. J. Pharmacol. 2010, 645, 32–38. [Google Scholar] [CrossRef][Green Version]
- Musiałek, M.W.; Rybaczek, D. Hydroxyurea—The Good, the Bad and the Ugly. Genes 2021, 12, 1096. [Google Scholar] [CrossRef]
- Li, Q.; Henry, E.R.; Hofrichter, J.; Smith, J.F.; Cellmer, T.; Dunkelberger, E.B.; Metaferia, B.B.; Jones-Straehle, S.; Boutom, S.; Christoph, G.W.; et al. Kinetic assay shows that increasing red cell volume could be a treatment for sickle cell disease. Proc. Natl. Acad. Sci. USA 2017, 114, E689–E696. [Google Scholar] [CrossRef]
- Garnier, Y.; Ferdinand, S.; Connes, P.; Garnier, M.; Etienne-Julan, M.; Lemonne, N.; Romana, M. Decrease of externalized phosphatidylserine density on red blood cell-derived microparticles in SCA patients treated with hydroxycarbamide. Br. J. Haematol. 2018, 182, 448–451. [Google Scholar] [CrossRef]
- Davies, S.C.; Gilmore, A. The role of hydroxyurea in the management of sickle cell disease. Blood Rev. 2003, 17, 99–109. [Google Scholar] [CrossRef]
- Chou, S.T. Transfusion therapy for sickle cell disease: A balancing act. Hematology 2013, 2013, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Kuriri, F.A.; Ahmed, A.; Alanazi, F.; Alhumud, F.; Hakami, M.A.; Ahmed, O.A.B. Red Blood Cell Alloimmunization and Autoimmunization in Blood Transfusion-Dependent Sickle Cell Disease and β-Thalassemia Patients in Al-Ahsa Region, Saudi Arabia. Anemia 2023, 2023, 3239960. [Google Scholar] [CrossRef] [PubMed]
- Vissa, M.; Vichinsky, E. Voxelotor for the treatment of sickle cell disease. Expert Rev. Hematol. 2021, 14, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Herity, L.B.; Vaughan, D.M.; Rodriguez, L.R.; Lowe, D.K. Voxelotor: A Novel Treatment for Sickle Cell Disease. Ann. Pharmacother. 2021, 55, 240–245. [Google Scholar] [CrossRef] [PubMed]
- Henry, E.R.; Metaferia, B.; Li, Q.; Harper, J.; Best, R.B.; Glass, K.E.; Cellmer, T.; Dunkelberger, E.B.; Conrey, A.; Thein, S.L.; et al. Treatment of sickle cell disease by increasing oxygen affinity of hemoglobin. Blood 2021, 138, 1172–1181. [Google Scholar] [CrossRef]
- Vichinsky, E.; Hoppe, C.C.; Ataga, K.I.; Ware, R.E.; Nduba, V.; El-Beshlawy, A.; Hassab, H.; Achebe, M.M.; Alkindi, S.; Brown, R.C.; et al. A Phase 3 Randomized Trial of Voxelotor in Sickle Cell Disease. N. Engl. J. Med. 2019, 381, 509–519. [Google Scholar] [CrossRef]
- Haroun, E.; Dutta, D.; Lim, S.H. Effects of GBT1118, a voxelotor analog, on intestinal pathophysiology in sickle cell disease. Br. J. Haematol. 2023, 202, 184–194. [Google Scholar] [CrossRef]
- Leibovitch, J.N.; Tambe, A.V.; Cimpeanu, E.; Poplawska, M.; Jafri, F.; Dutta, D.; Lim, S.H. l-glutamine, crizanlizumab, voxelotor, and cell-based therapy for adult sickle cell disease: Hype or hope? Blood Rev. 2022, 53, 100925. [Google Scholar] [CrossRef]
- Blair, H.A. Crizanlizumab: First Approval. Drugs 2020, 80, 79–84. [Google Scholar] [CrossRef]
- Gardner, R. Crizanlizumab in vaso-occlusive crisis caused by sickle cell disease. Drugs Today 2020, 56, 705–714. [Google Scholar] [CrossRef]
- Ataga, K.I.; Kutlar, A.; Kanter, J.; Liles, D.; Cancado, R.; Friedrisch, J.; Guthrie, T.H.; Knight-Madden, J.; Alvarez, O.A.; Gordeuk, V.R.; et al. Crizanlizumab for the Prevention of Pain Crises in Sickle Cell Disease. N. Engl. J. Med. 2017, 376, 429–439. [Google Scholar] [CrossRef] [PubMed]
- Kuriri, F.A.; O’Malley, C.J.; Jackson, D.E. Molecular mechanisms of immunoreceptors in platelets. Thromb. Res. 2019, 176, 108–114. [Google Scholar] [CrossRef]
- Karki, N.R.; Kutlar, A. P-Selectin Blockade in the Treatment of Painful Vaso-Occlusive Crises in Sickle Cell Disease: A Spotlight on Crizanlizumab. J. Pain Res. 2021, 14, 849–856. [Google Scholar] [CrossRef] [PubMed]
- Kutlar, A.; Kanter, J.; Liles, D.K.; Alvarez, O.A.; Cançado, R.D.; Friedrisch, J.R.; Knight-Madden, J.M.; Bruederle, A.; Shi, M.; Zhu, Z.; et al. Effect of crizanlizumab on pain crises in subgroups of patients with sickle cell disease: A SUSTAIN study analysis. Am. J. Hematol. 2019, 94, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Riley, T.R.; Riley, T.T. Profile of crizanlizumab and its potential in the prevention of pain crises in sickle cell disease: Evidence to date. J. Blood Med. 2019, 10, 307–311. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency’s (EMA) Committee for Medicinal Products for Human Use (CHMP). Revocation of Authorisation for Sickle Cell Disease Medicine Adakveo; EMA: Amsterdam, The Netherlands, 2023. [Google Scholar]
- Niihara, Y.; Miller, S.T.; Kanter, J.; Lanzkron, S.; Smith, W.R.; Hsu, L.L.; Gordeuk, V.R.; Viswanathan, K.; Sarnaik, S.; Osunkwo, I.; et al. A Phase 3 Trial of l-Glutamine in Sickle Cell Disease. N. Engl. J. Med. 2018, 379, 226–235. [Google Scholar] [CrossRef]
- Johnson, F.L.; Look, A.T.; Gockerman, J.; Ruggiero, M.R.; Dalla-Pozza, L.; Billings, F.T. Bone-Marrow Transplantation in a Patient with Sickle-Cell Anemia. N. Engl. J. Med. 1984, 311, 780–783. [Google Scholar] [CrossRef]
- Hsieh, M.M.; Fitzhugh, C.D.; Tisdale, J.F. Allogeneic hematopoietic stem cell transplantation for sickle cell disease: The time is now. Blood 2011, 118, 1197–1207. [Google Scholar] [CrossRef]
- Cappelli, B.; Volt, F.; Tozatto-Maio, K.; Scigliuolo, G.M.; Ferster, A.; Dupont, S.; Simões, B.P.; Al-Seraihy, A.; Aljurf, M.D.; Almohareb, F.; et al. Risk factors and outcomes according to age at transplantation with an HLA-identical sibling for sickle cell disease. Haematologica 2019, 104, e543–e546. [Google Scholar] [CrossRef]
- Sheth, S.; Bhatia, M. Hematopoietic stem cell transplantation in sickle cell disease: Patient selection and special considerations. J. Blood Med. 2015, 6, 229–238. [Google Scholar] [CrossRef]
- Walters, M.C.; Hardy, K.; Edwards, S.; Adamkiewicz, T.; Barkovich, J.; Bernaudin, F.; Buchanan, G.R.; Bunin, N.; Dickerhoff, R.; Giller, R.; et al. Pulmonary, Gonadal, and Central Nervous System Status after Bone Marrow Transplantation for Sickle Cell Disease. Biol. Blood Marrow Transplant. 2010, 16, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Gluckman, E.; Cappelli, B.; Bernaudin, F.; Labopin, M.; Volt, F.; Carreras, J.; Simões, B.P.; Ferster, A.; Dupont, S.; de la Fuente, J.; et al. Sickle cell disease: An international survey of results of HLA-identical sibling hematopoietic stem cell transplantation. Blood 2017, 129, 1548–1556. [Google Scholar] [CrossRef] [PubMed]
- Tozatto-Maio, K.; Torres, M.A.; Degaide, N.H.S.; Cardoso, J.F.; Volt, F.; Pinto, A.C.S.; Oliveira, D.; Elayoubi, H.; Kashima, S.; Loiseau, P.; et al. HLA-Matched Unrelated Donors for Patients with Sickle Cell Disease: Results of International Donor Searches. Biol. Blood Marrow Transplant. 2020, 26, 2034–2039. [Google Scholar] [CrossRef] [PubMed]
- Gluckman, E.; Fuente, J.D.; Cappelli, B.; Scigliuolo, G.M.; Volt, F.; Tozatto-Maio, K.; Rocha, V.; Tommaso, M.; O’Boyle, F.; Smiers, F.; et al. The role of HLA matching in unrelated donor hematopoietic stem cell transplantation for sickle cell disease in Europe. Bone Marrow Transplant. 2020, 55, 1946–1954. [Google Scholar] [CrossRef]
- Bolaños-Meade, J.; Fuchs, E.J.; Luznik, L.; Lanzkron, S.M.; Gamper, C.J.; Jones, R.J.; Brodsky, R.A. HLA-haploidentical bone marrow transplantation with posttransplant cyclophosphamide expands the donor pool for patients with sickle cell disease. Blood 2012, 120, 4285–4291. [Google Scholar] [CrossRef]
- Walters, M.C.; De Castro, L.M.; Sullivan, K.M.; Krishnamurti, L.; Kamani, N.; Bredeson, C.; Neuberg, D.; Hassell, K.L.; Farnia, S.; Campbell, A.; et al. Indications and Results of HLA-Identical Sibling Hematopoietic Cell Transplantation for Sickle Cell Disease. Biol. Blood Marrow Transplant. 2016, 22, 207–211. [Google Scholar] [CrossRef]
- Jiang, H.; Fu, D.; Bidgoli, A.; Paczesny, S. T Cell Subsets in Graft Versus Host Disease and Graft Versus Tumor. Front. Immunol. 2021, 12, 761448. [Google Scholar] [CrossRef]
- Aydin, M.; Dovern, E.; Leeflang, M.M.; de la Fuente, J.; Kassim, A.A.; Biemond, B.J.; Nur, E. Haploidentical Allogeneic Stem Cell Transplantation in Sickle Cell Disease: A Systematic Review and Meta-Analysis. Transplant. Cell. Ther. 2021, 27, 1004.e1–1004.e8. [Google Scholar] [CrossRef]
- Torres, L.; Conran, N. Emerging pharmacotherapeutic approaches for the management of sickle cell disease. Expert Opin. Pharmacother. 2019, 20, 173–186. [Google Scholar] [CrossRef]
- Jakubowski, J.A.; Zhou, C.; Jurcevic, S.; Winters, K.J.; Lachno, D.R.; Frelinger, A.L.; Gupta, N.; Howard, J.; Payne, C.D.; Mant, T.G. A phase 1 study of prasugrel in patients with sickle cell disease: Effects on biomarkers of platelet activation and coagulation. Thromb. Res. 2014, 133, 190–195. [Google Scholar] [CrossRef]
- Wun, T.; Soulieres, D.; Frelinger, A.L.; Krishnamurti, L.; Novelli, E.M.; Kutlar, A.; Ataga, K.I.; Knupp, C.L.; McMahon, L.E.; Strouse, J.J.; et al. A double-blind, randomized, multicenter phase 2 study of prasugrel versus placebo in adult patients with sickle cell disease. J. Hematol. Oncol. 2013, 6, 17. [Google Scholar] [CrossRef] [PubMed]
- Heeney, M.M.; Hoppe, C.C.; Abboud, M.R.; Inusa, B.; Kanter, J.; Ogutu, B.; Brown, P.B.; Heath, L.E.; Jakubowski, J.A.; Zhou, C.; et al. A Multinational Trial of Prasugrel for Sickle Cell Vaso-Occlusive Events. N. Engl. J. Med. 2016, 374, 625–635. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.Z.; Conrey, A.; Frey, I.; Nichols, J.; Menapace, L.A.; Tumburu, L.; Lequang, T.; Li, Q.; Dunkelberger, E.; Henry, E.; et al. Phase 1 Multiple Ascending Dose Study of Safety, Tolerability, and Pharmacokinetics/Pharmacodynamics of Mitapivat (AG-348) in Subjects with Sickle Cell Disease. Blood 2020, 136, 21–22. [Google Scholar] [CrossRef]
- Rab, M.A.E.; Bos, J.; van Oirschot, B.A.; van Straaten, S.; Kosinski, P.A.; Chubukov, V.; Kim, H.; Mangus, H.; Schutgens, R.E.G.; Pasterkamp, G.; et al. Decreased activity and stability of pyruvate kinase in sickle cell disease: A novel target for mitapivat therapy. Blood 2021, 137, 2997–3001. [Google Scholar] [CrossRef] [PubMed]
- Howard, J.; Kuo, K.H.; Oluyadi, A.; Shao, H.; Morris, S.; Zaidi, A.U.; Van Beers, E.J.; Thien, S.L. A Phase 2/3, Randomized, Double-Blind, Placebo-Controlled Study of Mitapivat in Patients with Sickle Cell Disease. Blood 2021, 138, 3109. [Google Scholar] [CrossRef]
- van Dijk, M.J.; Rab, M.A.; Rijneveld, A.W.; Nur, E.; Bartels, M.; Jans, J.J.; van Solinge, W.W.; Schutgens, R.E.; van Wijk, R.; Van Beers, E.J. Safety and Efficacy of Mitapivat (AG-348), an Oral Activator of Pyruvate Kinase R, in Subjects with Sickle Cell Disease: A Phase 2, Open-Label Study (ESTIMATE). Blood 2021, 138, 2047. [Google Scholar] [CrossRef]
- Xu, J.Z.; Conrey, A.K.; Frey, I.C.; Gwaabe, E.; Menapace, L.A.; Tumburu, L.; Lundt, M.; Lequang, T.; Li, Q.; Glass, K.; et al. A phase 1 dose escalation study of the pyruvate kinase activator mitapivat (AG-348) in sickle cell disease. Blood 2022, 140, 2053–2062. [Google Scholar] [CrossRef]
- Esrick, E.B.; Lehmann, L.E.; Biffi, A.; Achebe, M.; Brendel, C.; Ciuculescu, M.F.; Daley, H.; MacKinnon, B.; Morris, E.; Federico, A.; et al. Post-Transcriptional Genetic Silencing of BCL11A to Treat Sickle Cell Disease. N. Engl. J. Med. 2021, 384, 205–215. [Google Scholar] [CrossRef]
- Komor, A.C.; Kim, Y.B.; Packer, M.S.; Zuris, J.A.; Liu, D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 2016, 533, 420–424. [Google Scholar] [CrossRef]
- Gaudelli, N.M.; Komor, A.C.; Rees, H.A.; Packer, M.S.; Badran, A.H.; Bryson, D.I.; Liu, D.R. Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature 2017, 551, 464–471. [Google Scholar] [CrossRef]
- Zeng, J.; Wu, Y.; Ren, C.; Bonanno, J.; Shen, A.H.; Shea, D.; Gehrke, J.M.; Clement, K.; Luk, K.; Yao, Q.; et al. Therapeutic base editing of human hematopoietic stem cells. Nat. Med. 2020, 26, 535–541. [Google Scholar] [CrossRef]
- Park, S.H.; Bao, G. CRISPR/Cas9 gene editing for curing sickle cell disease. Transfus. Apher. Sci. 2021, 60, 103060. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Georgakopoulou, A.; Newby, G.A.; Everette, K.A.; Nizamis, E.; Paschoudi, K.; Vlachaki, E.; Gil, S.; Anderson, A.K.; Koob, T.; et al. In vivo base editing by a single i.v. vector injection for treatment of hemoglobinopathies. J. Clin. Investig. 2022, 7, 162939. [Google Scholar] [CrossRef] [PubMed]
- Mayuranathan, T.; Newby, G.A.; Feng, R.; Yao, Y.; Mayberry, K.D.; Lazzarotto, C.R.; Li, Y.; Levine, R.M.; Nimmagadda, N.; Dempsey, E.; et al. Potent and uniform fetal hemoglobin induction via base editing. Nat. Genet. 2023, 55, 1210–1220. [Google Scholar] [CrossRef]
- Leibowitz, M.L.; Papathanasiou, S.; Doerfler, P.A.; Blaine, L.J.; Sun, L.; Yao, Y.; Zhang, C.-Z.; Weiss, M.J.; Pellman, D. Chromothripsis as an on-target consequence of CRISPR–Cas9 genome editing. Nat. Genet. 2021, 53, 895–905. [Google Scholar] [CrossRef] [PubMed]
- Kosicki, M.; Tomberg, K.; Bradley, A. Repair of double-strand breaks induced by CRISPR–Cas9 leads to large deletions and complex rearrangements. Nat. Biotechnol. 2018, 36, 765–771. [Google Scholar] [CrossRef]
- Li, C.; Georgakopoulou, A.; Newby, G.A.; Chen, P.J.; Everette, K.A.; Paschoudi, K.; Vlachaki, E.; Gil, S.; Anderson, A.K.; Koob, T.; et al. In vivo HSC prime editing rescues Sickle Cell Disease in a mouse model. Blood 2023, 141, 2085–2099. [Google Scholar] [CrossRef]
- Sweeney, C.L.; De Ravin, S.S. The promise of in vivo HSC prime editing. Blood 2023, 141, 2039–2040. [Google Scholar] [CrossRef]
- Anzalone, A.V.; Randolph, P.B.; Davis, J.R.; Sousa, A.A.; Koblan, L.W.; Levy, J.M.; Chen, P.J.; Wilson, C.; Newby, G.A.; Raguram, A.; et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 2019, 576, 149–157. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuriri, F.A. Hope on the Horizon: New and Future Therapies for Sickle Cell Disease. J. Clin. Med. 2023, 12, 5692. https://doi.org/10.3390/jcm12175692
Kuriri FA. Hope on the Horizon: New and Future Therapies for Sickle Cell Disease. Journal of Clinical Medicine. 2023; 12(17):5692. https://doi.org/10.3390/jcm12175692
Chicago/Turabian StyleKuriri, Fahd A. 2023. "Hope on the Horizon: New and Future Therapies for Sickle Cell Disease" Journal of Clinical Medicine 12, no. 17: 5692. https://doi.org/10.3390/jcm12175692
APA StyleKuriri, F. A. (2023). Hope on the Horizon: New and Future Therapies for Sickle Cell Disease. Journal of Clinical Medicine, 12(17), 5692. https://doi.org/10.3390/jcm12175692