
Harnessing Immune Response in Acute Myeloid Leukemia

 , , ,
, , ,  and
and
Abstract
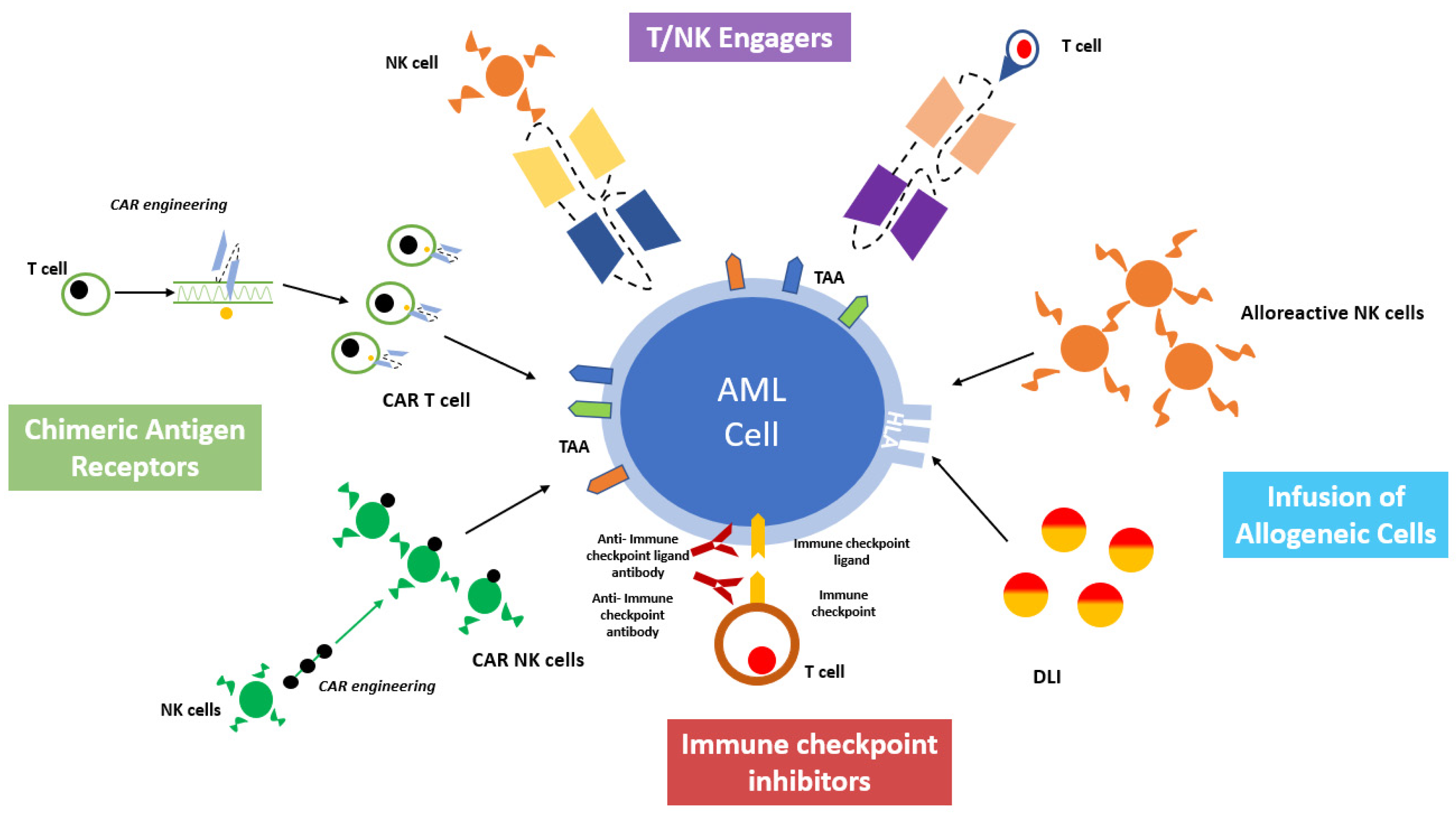
:1. Introduction
2. T and NK Cells
3. Principles of Adoptive Immunotherapy in Allogeneic Stem Cell Transplantation
3.1. Graft versus Leukemia Effect
3.2. Donor’s Lymphocytes Infusion
4. Immune Checkpoint Inhibitors (ICI) as Immunotherapeutic Strategies for AML
5. Target Antigens for Cell-Based Immunotherapy in AML
5.1. CD33
5.2. CD123
5.3. CLL-1
5.4. FLT3
5.5. NKG2D
6. Chimeric Antigen Receptor Cells
6.1. CAR-T
6.2. CAR-NK
7. Strategies for T and NK Cells Engagement
7.1. BiTEs and DARTs
Mechanism and Structure
7.2. BiKEs and TRiKEs
7.3. DARPins
7.4. ANKETsTM
8. New Toxicity Profile of Cell-Based Immunotherapy
9. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Kantarjian, H.; Kadia, T.; DiNardo, C.; Daver, N.; Borthakur, G.; Jabbour, E.; Garcia-Manero, G.; Konopleva, M.; Ravandi, F. Acute myeloid leukemia: Current progress and future directions. Blood Cancer J. 2021, 11, 2–25. [Google Scholar] [CrossRef] [PubMed]
- Döhner, H.; Wei, A.H.; Appelbaum, F.R.; Craddock, C.; DiNardo, C.D.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Godley, L.A.; Hasserjian, R.P.; et al. Diagnosis and management of AML in adults: 2022 recommendations from an international expert panel on behalf of the ELN. Blood 2022, 140, 1345–1377. [Google Scholar] [CrossRef]
- Kolb, H.J. Graft-versus-leukemia effects of transplantation and donor lymphocytes. Blood 2008, 112, 12–83. [Google Scholar] [CrossRef] [PubMed]
- June, C.H.; O’Connor, R.S.; Kawalekar, O.U.; Ghassemi, S.; Milone, M.C. CAR T cell immunotherapy for human cancer. Science 2018, 359, 1361–1365. [Google Scholar] [CrossRef]
- Tasian, S.K. Acute myeloid leukemia chimeric antigen receptor T-cell immunotherapy: How far up the road have we traveled? Ther. Adv. Hematol. 2018, 9, 135–148. [Google Scholar] [CrossRef] [PubMed]
- Marofi, F.; Al-Awad, A.S.; Sulaiman Rahman, H.; Markov, A.; Abdelbasset, W.K.; Ivanovna Enina, Y.; Mahmoodi, M.; Hassanzadeh, A.; Yazdanifar, M.; Stanley Chartrand, M.; et al. CAR-NK Cell: A New Paradigm in Tumor Immunotherapy. Front. Oncol. 2021, 11, 673276. [Google Scholar] [CrossRef]
- Isidori, A.; Daver, N.; Curti, A. Editorial: The Biological Landscape of Immunotherapy in AML. Front. Oncol. 2021, 11, 671252. [Google Scholar] [CrossRef]
- Vago, L.; Gojo, I. Immune escape and immunotherapy of acute myeloid leukemia. J. Clin. Investig. 2020, 130, 1552–1564. [Google Scholar] [CrossRef]
- van Luijn, M.M.; van den Ancker, W.; Chamuleau, M.E.; Ossenkoppele, G.J.; van Ham, S.M.; van de Loosdrecht, A.A. Impaired antigen presentation in neoplasia: Basic mechanisms and implications for acute myeloid leukemia. Immunotherapy 2010, 2, 85–97. [Google Scholar] [CrossRef]
- Williams, P.; Basu, S.; Garcia-Manero, G.; Hourigan, C.S.; Oetjen, K.A.; Cortes, J.E.; Ravandi, F.; Jabbour, E.J.; Al-Hamal, Z.; Konopleva, M.; et al. The distribution of T-cell subsets and the expression of immune checkpoint receptors and ligands in patients with newly diagnosed and relapsed acute myeloid leukemia. Cancer 2019, 125, 1470–1481. [Google Scholar] [CrossRef]
- Ciciarello, M.; Corradi, G.; Forte, D.; Cavo, M.; Curti, A. Emerging Bone Marrow Microenvironment-Driven Mechanisms of Drug Resistance in Acute Myeloid Leukemia: Tangle or Chance? Cancers 2021, 13, 5319. [Google Scholar] [CrossRef]
- Di Vito, C.; Mikulak, J.; Zaghi, E.; Pesce, S.; Marcenaro, E.; Mavilio, D. NK cells to cure cancer. Semin. Immunol. 2019, 41, 101272. [Google Scholar] [CrossRef]
- Kyrysyuk, O.; Wucherpfennig, K.W. Designing Cancer Immunotherapies That Engage T Cells and NK Cells. Annu. Rev. Immunol. 2023, 41, 17–38. [Google Scholar] [CrossRef]
- Sweeney, C.; Vyas, P. The Graft-Versus-Leukemia Effect in AML. Front. Oncol. 2019, 9, 1217. [Google Scholar] [CrossRef]
- Moretta, A.; Pende, D.; Locatelli, F.; Moretta, L. Activating and inhibitory killer immunoglobulin-like receptors (KIR) in haploidentical haemopoietic stem cell transplantation to cure high-risk leukaemias. Clin. Exp. Immunol. 2009, 157, 325–331. [Google Scholar] [CrossRef]
- Moretta, L.; Locatelli, F.; Pende, D.; Marcenaro, E.; Mingari, M.C.; Moretta, A. Killer Ig-like receptor mediated control of natural killer cell alloreactivity in haploidentical hematopoietic stem cell transplantation. Blood 2011, 117, 764–771. [Google Scholar] [CrossRef]
- Ruggeri, L.; Capanni, M.; Urbani, E.; Perruccio, K.; Shlomchik, W.D.; Tosti, A.; Posati, S.; Rogaia, D.; Frassoni, F.; Aversa, F.; et al. Effectiveness of donor natural killer cell alloreactivity in mismatched hematopoietic transplants. Science 2002, 295, 2097–2100. [Google Scholar] [CrossRef]
- Giebel, S.; Locatelli, F.; Lamparelli, T.; Velardi, A.; Davies, S.; Frumento, G.; Maccario, R.; Bonetti, F.; Wojnar, J.; Martinetti, M.; et al. Survival advantage with KIR ligand incompatibility in hematopoietic stem cell transplantation from unrelated donors. Blood 2003, 102, 814–819. [Google Scholar] [CrossRef]
- Ruggeri, L.; Mancusi, A.; Burchielli, E.; Aversa, F.; Martelli, M.F.; Velardi, A. Natural killer cell alloreactivity and haplo-identical hematopoietic transplantation. Cytotherapy 2006, 8, 554–558. [Google Scholar] [CrossRef]
- Marcenaro, E.; Carlomagno, S.; Pesce, S.; Della Chiesa, M.; Moretta, A.; Sivori, S. Role of alloreactive KIR2DS1(+) NK cells in haploidentical hematopoietic stem cell transplantation. J. Leukoc. Biol. 2011, 90, 661–667. [Google Scholar] [CrossRef]
- Pende, D.; Marcenaro, S.; Falco, M.; Martini, S.; Bernardo, M.E.; Montagna, D.; Romeo, E.; Cognet, C.; Martinetti, M.; Maccario, R.; et al. Anti-leukemia activity of alloreactive NK cells in KIR ligand-mismatched haploidentical HSCT for pediatric patients: Evaluation of the functional role of activating KIR and redefinition of inhibitory KIR specificity. Blood 2009, 113, 3119–3129. [Google Scholar] [CrossRef]
- Minetto, P.; Guolo, F.; Pesce, S.; Greppi, M.; Obino, V.; Ferretti, E.; Sivori, S.; Genova, C.; Lemoli, R.M.; Marcenaro, E. Harnessing NK Cells for Cancer Treatment. Front. Immunol. 2019, 10, 2836. [Google Scholar] [CrossRef]
- Liu, L.; Chang, Y.J.; Xu, L.P.; Zhang, X.H.; Wang, Y.; Liu, K.Y.; Huang, X.J. Reversal of T Cell Exhaustion by the First Donor Lymphocyte Infusion Is Associated with the Persistently Effective Antileukemic Responses in Patients with Relapsed AML after Allo-HSCT. Biol. Blood Marrow Transplant. 2018, 24, 1350–1359. [Google Scholar] [CrossRef]
- Bachireddy, P.; Wu, C.J. Understanding anti-leukemia responses to donor lymphocyte infusion. Oncoimmunology 2014, 3, e28187. [Google Scholar] [CrossRef]
- Bachireddy, P.; Hainz, U.; Rooney, M.; Pozdnyakova, O.; Aldridge, J.; Zhang, W.; Liao, X.; Hodi, F.S.; O’Connell, K.; Haining, W.N.; et al. Reversal of in situ T-cell exhaustion during effective human antileukemia responses to donor lymphocyte infusion. Blood 2014, 123, 1412–1421. [Google Scholar] [CrossRef]
- Dazzi, F.; Szydlo, R.M.; Craddock, C.; Cross, N.C.; Kaeda, J.; Chase, A.; Olavarria, E.; van Rhee, F.; Kanfer, E.; Apperley, J.F.; et al. Comparison of single-dose and escalating-dose regimens of donor lymphocyte infusion for relapse after allografting for chronic myeloid leukemia. Blood 2000, 95, 67–71. [Google Scholar] [CrossRef]
- Kolb, H.J.; Mittermüller, J.; Clemm, C.; Holler, E.; Ledderose, G.; Brehm, G.; Heim, M.; Wilmanns, W. Donor leukocyte transfusions for treatment of recurrent chronic myelogenous leukemia in marrow transplant patients. Blood 1990, 76, 2462–2465. [Google Scholar] [CrossRef]
- Drobyski, W.R.; Keever, C.A.; Roth, M.S.; Koethe, S.; Hanson, G.; McFadden, P.; Gottschall, J.L.; Ash, R.C.; van Tuinen, P.; Horowitz, M.M. Salvage immunotherapy using donor leukocyte infusions as treatment for relapsed chronic myelogenous leukemia after allogeneic bone marrow transplantation: Efficacy and toxicity of a defined T-cell dose. Blood 1993, 82, 2310–2318. [Google Scholar] [CrossRef]
- Drobyski, W.R.; Hessner, M.J.; Klein, J.P.; Kabler-Babbitt, C.; Vesole, D.H.; Keever-Taylor, C.A. T-Cell Depletion Plus Salvage Immunotherapy With Donor Leukocyte Infusions as a Strategy to Treat Chronic-Phase Chronic Myelogenous Leukemia Patients Undergoing HLA-Identical Sibling Marrow Transplantation. Blood 1999, 94, 434–441. [Google Scholar] [CrossRef]
- Tsirigotis, P.; Byrne, M.; Schmid, C.; Baron, F.; Ciceri, F.; Esteve, J.; Gorin, N.C.; Giebel, S.; Mohty, M.; Savani, B.N.; et al. Relapse of AML after hematopoietic stem cell transplantation: Methods of monitoring and preventive strategies. A review from the ALWP of the EBMT. Bone Marrow Transplant. 2016, 51, 1431–1438. [Google Scholar] [CrossRef]
- Wang, Y.; Wu, D.P.; Liu, Q.F.; Qin, Y.Z.; Wang, J.B.; Xu, L.P.; Liu, Y.R.; Zhu, H.H.; Chen, J.; Dai, M.; et al. In adults with t(8;21)AML, posttransplant RUNX1/RUNX1T1-based MRD monitoring, rather than c-KIT mutations, allows further risk stratification. Blood 2014, 124, 1880–1886. [Google Scholar] [CrossRef]
- Shayegi, N.; Kramer, M.; Bornhäuser, M.; Schaich, M.; Schetelig, J.; Platzbecker, U.; Röllig, C.; Heiderich, C.; Landt, O.; Ehninger, G.; et al. The level of residual disease based on mutant NPM1 is an independent prognostic factor for relapse and survival in AML. Blood 2013, 122, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Buccisano, F.; Hourigan, C.S.; Walter, R.B. The Prognostic Significance of Measurable (“Minimal”) Residual Disease in Acute Myeloid Leukemia. Curr. Hematol. Malig. Rep. 2017, 12, 547–556. [Google Scholar] [CrossRef] [PubMed]
- Bastos-Oreiro, M.; Perez-Corral, A.; Martínez-Laperche, C.; Bento, L.; Pascual, C.; Kwon, M.; Balsalobre, P.; Muñoz, C.; Buces, E.; Serrano, D.; et al. Prognostic impact of minimal residual disease analysis by flow cytometry in patients with acute myeloid leukemia before and after allogeneic hemopoietic stem cell transplantation. Eur. J. Haematol. 2014, 93, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Du, K.; Luo, Y.; Shi, J.; Cao, L.; Zheng, Y.; Zheng, G.; Zhao, Y.; Ye, X.; Cai, Z.; et al. Superiority of preemptive donor lymphocyte infusion based on minimal residual disease in acute leukemia patients after allogeneic hematopoietic stem cell transplantation. Transfusion 2014, 54, 1493–1500. [Google Scholar] [CrossRef]
- Yan, C.H.; Liu, D.H.; Liu, K.Y.; Xu, L.P.; Liu, Y.R.; Chen, H.; Han, W.; Wang, Y.; Qin, Y.Z.; Huang, X.J. Risk stratification–directed donor lymphocyte infusion could reduce relapse of standard-risk acute leukemia patients after allogeneic hematopoietic stem cell transplantation. Blood 2012, 119, 3256–3262. [Google Scholar] [CrossRef]
- Dominietto, A.; Pozzi, S.; Miglino, M.; Albarracin, F.; Piaggio, G.; Bertolotti, F.; Grasso, R.; Zupo, S.; Raiola, A.M.; Gobbi, M.; et al. Donor lymphocyte infusions for the treatment of minimal residual disease in acute leukemia. Blood 2007, 109, 5063–5064. [Google Scholar] [CrossRef]
- Georgi, J.A.; Stasik, S.; Bornhäuser, M.; Platzbecker, U.; Thiede, C. Analysis of Subset Chimerism for MRD-Detection and Pre-Emptive Treatment in AML. Front. Oncol. 2022, 12, 841608. [Google Scholar] [CrossRef]
- Sairafi, D.; Remberger, M.; Uhlin, M.; Ljungman, P.; Ringdén, O.; Mattsson, J. Leukemia Lineage-Specific Chimerism Analysis and Molecular Monitoring Improve Outcome of Donor Lymphocyte Infusions. Biol. Blood Marrow Transplant. 2010, 16, 1728–1737. [Google Scholar] [CrossRef]
- Schmid, C.; Labopin, M.; Schaap, N.; Veelken, H.; Schleuning, M.; Stadler, M.; Finke, J.; Hurst, E.; Baron, F.; Ringden, O.; et al. Prophylactic donor lymphocyte infusion after allogeneic stem cell transplantation in acute leukaemia—A matched pair analysis by the Acute Leukaemia Working Party of EBMT. Br. J. Haematol. 2019, 184, 782–787. [Google Scholar] [CrossRef]
- Poiré, X.; Graux, C.; Ory, A.; Herman, J.; Baron, F.; Schoemans, H.; Lewalle, P.; De Becker, A.; Deeren, D.; Berneman, Z.; et al. Sequential administration of low dose 5-azacytidine (AZA) and donor lymphocyte infusion (DLI) for patients with acute myeloid leukemia (AML) or myelodysplastic syndrome (MDS) in relapse after allogeneic stem cell transplantation (SCT): A prospective study from the Belgian Hematology Society (BHS). Bone Marrow Transplant. 2022, 57, 116–118. [Google Scholar] [CrossRef] [PubMed]
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.; Hwu, W.J.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; Kauh, J.; Odunsi, K.; et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med. 2012, 366, 2455–2465. [Google Scholar] [CrossRef] [PubMed]
- Baues, C.; Semrau, R.; Gaipl, U.S.; Bröckelmann, P.J.; Rosenbrock, J.; Engert, A.; Marnitz, S. Checkpoint inhibitors and radiation treatment in Hodgkin’s lymphoma: New study concepts of the German Hodgkin Study Group. Strahlenther. Onkol. 2017, 193, 95–99. [Google Scholar] [CrossRef] [PubMed]
- Maurer, S.; Zhong, X.; Prada, B.D.; Mascarenhas, J.; de Andrade, L.F. The Latest Breakthroughs in Immunotherapy for Acute Myeloid Leukemia, with a Special Focus on NKG2D Ligands. Int. J. Mol. Sci. 2022, 23, 15907. [Google Scholar] [CrossRef]
- Daver, N.; Garcia-Manero, G.; Basu, S.; Boddu, P.C.; Alfayez, M.; Cortes, J.E.; Konopleva, M.; Ravandi-Kashani, F.; Jabbour, E.; Kadia, T.; et al. Efficacy, safety, and biomarkers of response to azacitidine and nivolumab in relapsed/refractory acute myeloid leukemia: A nonrandomized, open-label, phase II study. Canc. Discov. 2019, 9, 3–83. [Google Scholar] [CrossRef]
- Gojo, I.; Stuart, R.K.; Webster, J.; Blackford, A.; Varela, J.C.; Morrow, J.; DeZern, A.E.; Foster, M.C.; Levis, M.J.; Coombs, C.C.; et al. Multi-center phase 2 study of pembroluzimab (pembro) and azacitidine (AZA) in patients with relapsed/refractory acute myeloid leukemia (AML) and in newly diagnosed (≥65 Years) AML patients. Blood 2019, 134 (Suppl. S1), 832. [Google Scholar] [CrossRef]
- Ruggeri, L.; Urbani, E.; André, P.; Mancusi, A.; Tosti, A.; Topini, F.; Bléry, M.; Animobono, L.; Romagné, F.; Wagtmann, N.; et al. Effects of anti-NKG2A antibody administration on leukemia and normal hematopoietic cells. Haematologica 2016, 101, 5–633. [Google Scholar] [CrossRef]
- Roberto, A.; Di Vito, C.; Zaghi, E.; Mazza, E.M.C.; Capucetti, A.; Calvi, M.; Tentorio, P.; Zanon, V.; Sarina, B.; Mariotti, J.; et al. The early expansion of anergic NKG2Apos/CD56dim/CD16neg natural killer represents a therapeutic target in haploidentical hematopoietic stem cell transplantation. Haematologica 2018, 103, 1390–1402. [Google Scholar] [CrossRef]
- André, P.; Denis, C.; Soulas, C.; Bourbon-Caillet, C.; Lopez, J.; Arnoux, T.; Bléry, M.; Bonnafous, C.; Gauthier, L.; Morel, A. Anti-NKG2A mAb is a checkpoint inhibitor that promotes anti-tumor immunity by unleashing both T and NK cells. Cell 2018, 175, 1731–1743.e13. [Google Scholar] [CrossRef]
- Devillier, R.; Furst, S.; Chammard, A.B.; Pagliardini, T.; Harbi, S.; Maisano, V.; Granata, A.; Legrand, F.; Pakradouni, J.; Boher, J.M.; et al. Safety of Anti-NKG2A Blocking Antibody MonalizumabAs Maintenance Therapy after Allogeneic Hematopoietic Stem Cell Transplantation: A Phase I Study. Blood 2021, 138, 1817. [Google Scholar] [CrossRef]
- Pabst, T.; Vey, N.; Adès, L.; Bacher, U.; Bargetzi, M.; Fung, S.; Gaidano, G.; Gandini, D.; Hultberg, A.; Johnson, A.; et al. Results from a phase I/II trial of cusatuzumab combined with azacitidine in patients with newly diagnosed acute myeloid leukemia who are ineligible for intensive chemotherapy. Haematologica 2023, 108, 1793. [Google Scholar] [CrossRef] [PubMed]
- Daver, N.; Alotaibi, A.S.; Bücklein, V.; Subklewe, M. T-cell-based immunotherapy of acute myeloid leukemia: Current concepts and future developments. Leukemia 2021, 35, 1843–1863. [Google Scholar] [CrossRef] [PubMed]
- Paul, S.P.; Taylor, L.S.; Stansbury, E.K.; McVicar, D.W. Myeloid specific human CD33 is an inhibitory receptor with differential ITIM function in recruiting the phosphatases SHP-1 and SHP-2. Blood 2000, 96, 483–490. [Google Scholar] [CrossRef] [PubMed]
- Burnett, A.K.; Hills, R.K.; Milligan, D.; Kjeldsen, L.; Kell, J.; Russell, N.H.; Yin, J.A.; Hunter, A.; Goldstone, A.H.; Wheatley, K. Identification of patients with acute myeloblastic leukemia who benefit from the addition of gemtuzumab ozogamicin: Results of the MRC AML15 trial. J. Clin. Oncol. 2011, 29, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Burnett, A.K.; Russell, N.H.; Hills, R.K.; Kell, J.; Freeman, S.; Kjeldsen, L.; Hunter, A.E.; Yin, J.; Craddock, C.F.; Dufva, I.H.; et al. Addition of gemtuzumab ozogamicin to induction chemotherapy improves survival in older patients with acute myeloid leukemia. J. Clin. Oncol. 2012, 30, 3924–3931. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.; Pautas, C.; Terré, C.; Raffoux, E.; Turlure, P.; Caillot, D.; Legrand, O.; Thomas, X.; Gardin, C.; Gogat-Marchant, K.; et al. Gemtuzumab ozogamicin for de novo acute myeloid leukemia: Final efficacy and safety updates from the open-label, phase III ALFA-0701 trial. Haematologica 2019, 104, 113–119. [Google Scholar] [CrossRef]
- Hills, R.K.; Castaigne, S.; Appelbaum, F.R.; Delaunay, J.; Petersdorf, S.; Othus, M.; Estey, E.H.; Dombret, H.; Chevret, S.; Ifrah, N.; et al. Addition of gemtuzumab ozogamicin to induction chemotherapy in adult patients with acute myeloid leukaemia: A meta-analysis of individual patient data from randomised controlled trials. Lancet Oncol. 2014, 15, 986–996. [Google Scholar] [CrossRef]
- Reusch, U.; Harrington, K.H.; Gudgeon, C.J.; Fucek, I.; Ellwanger, K.; Weichel, M.; Knackmuss, S.H.; Zhukovsky, E.A.; Fox, J.A.; Kunkel, L.A.; et al. Characterization of CD33/CD3 Tetravalent Bispecific Tandem Diabodies (TandAbs) for the Treatment of Acute Myeloid Leukemia. Clin. Cancer Res. 2016, 22, 5829–5838. [Google Scholar] [CrossRef]
- Ravandi, F.; Walter, R.B.; Subklewe, M.; Buecklein, V.; Jongen-Lavrencic, M.; Paschka, P.; Ossenkoppele, G.J.; Kantarjian, H.M.; Hindoyan, A.; Agarwal, S.K.; et al. Updated results from phase I dose-escalation study of AMG 330, a bispecific T-cell engager molecule, in patients with relapsed/refractory acute myeloid leukemia (R/R AML). J. Clin. Oncol. 2020, 38, 7508. [Google Scholar] [CrossRef]
- Subklewe, M.; Stein, A.; Walter, R.B.; Bhatia, R.; Wei, A.H.; Ritchie, D.; Bücklein, V.; Vachhani, P.; Dai, T.; Hindoyan, A.; et al. Preliminary Results from a Phase 1 First-in-Human Study of AMG 673, a Novel Half-Life Extended (HLE) Anti-CD33/CD3 BiTE® (Bispecific T-Cell Engager) in Patients with Relapsed/Refractory (R/R) Acute Myeloid Leukemia (AML). Blood 2019, 134 (Suppl. S1), 833. [Google Scholar] [CrossRef]
- Felices, M.; Warlick, E.; Juckett, M.; Weisdorf, D.; Vallera, D.; Miller, S.; Wangen, R.; Lewis, D.; Knox, J.; Schroeder, M.; et al. 444 GTB-3550 tri-specific killer engager TriKE™ drives NK cells expansion and cytotoxicity in acute myeloid leukemia (AML) and myelodysplastic syndromes (MDS) patients. J. Immunother. Cancer 2021, 9, A473. [Google Scholar] [CrossRef]
- El Achi, H.; Dupont, E.; Paul, S.; Khoury, J.D. CD123 as a Biomarker in Hematolymphoid Malignancies: Principles of Detection and Targeted Therapies. Cancers 2020, 12, 3087. [Google Scholar] [CrossRef] [PubMed]
- Bras, A.E.; de Haas, V.; van Stigt, A.; Jongen-Lavrencic, M.; Beverloo, H.B.; TeMarvelde, J.G.; Zwaan, C.M.; van Dongen, J.J.M.; Leusen, J.H.W.; van der Velden, V.H.J. CD123 expression levels in 846 acute leukemia patients based on standardized immunophenotyping. Cytom. B Clin. Cytom. 2019, 96, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Testa, U.; Riccioni, R.; Militi, S.; Coccia, E.; Stellacci, E.; Samoggia, P.; Latagliata, R.; Mariani, G.; Rossini, A.; Battistini, A.; et al. Elevatedexpression of IL-3Ralpha in acute myelogenousleukemiaisassociated with enhancedblastproliferation, increasedcellularity, and poorprognosis. Blood 2002, 100, 2980–2988. [Google Scholar] [CrossRef] [PubMed]
- Al-Mawali, A.; Pinto, A.D.; Al-Zadjali, S. CD34+CD38-CD123+ Cells Are Present in Virtually All Acute Myeloid Leukaemia Blasts: A Promising Single Unique Phenotype for Minimal Residual Disease Detection. Acta Haematol. 2017, 138, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Jordan, C.T.; Upchurch, D.; Szilvassy, S.J.; Guzman, M.L.; Howard, D.S.; Pettigrew, A.L.; Meyerrose, T.; Rossi, R.; Grimes, B.; Rizzieri, D.A.; et al. The interleukin-3 receptor alpha chain is a unique marker for human acute myelogenous leukemia stem cells. Leukemia 2000, 14, 1777–1784. [Google Scholar] [CrossRef] [PubMed]
- Pollyea, D.A.; Gutman, J.A.; Gore, L.; Smith, C.A.; Jordan, C.T. Targeting acute myeloid leukemia stem cells: A review and principles for the development of clinical trials. Haematologica 2014, 99, 1277–1284. [Google Scholar] [CrossRef]
- Uckun, F.M.; Lin, T.L.; Mims, A.S.; Patel, P.; Lee, C.; Shahidzadeh, A.; Shami, P.J.; Cull, E.; Cogle, C.R.; Watts, J.A. Clinical Phase 1B Study of the CD3xCD123 Bispecific Antibody APVO436 in Patients with Relapsed/Refractory Acute Myeloid Leukemia or Myelodysplastic Syndrome. Cancers 2021, 13, 4113. [Google Scholar] [CrossRef]
- Ravandi, F.; Bashey, A.; Stock, W.; Foran, M.J.; Mawad, R.; Egan, D.; Blu, W.; Yang, A.; Pastore, A.; Johnson, C.; et al. Complete Responses in Relapsed/Refractory Acute Myeloid Leukemia (AML) Patients on a Weekly Dosing Schedule of Vibecotamab (XmAb14045), a CD123 x CD3 T Cell-Engaging Bispecific Antibody; Initial Results of a Phase 1 Study. Blood 2020, 136 (Suppl. S1), 4–5. [Google Scholar] [CrossRef]
- Aldoss, I.; Uy, G.L.; Vey, N.; Emadi, A.; Sayre, P.H.; Walter, R.B.; Foster, M.C.; Arellano, M.L.; Godwin, J.E.; Wieduwilt, M.J.; et al. Flotetuzumab As Salvage Therapy for Primary Induction Failure and Early Relapse Acute Myeloid Leukemia. Blood 2020, 136 (Suppl. S1), 16–18. [Google Scholar] [CrossRef]
- Boyiadzis, M.; Desai, P.; Daskalakis, N.; Donnellan, W.; Ferrante, L.; Goldberg, J.D.; Grunwald, M.R.; Guttke, C.; Li, X.; Perez-Simon, J.A.; et al. First-in-human study of JNJ-63709178, a CD123/CD3 targeting antibody, in relapsed/refractory acute myeloid leukemia. Clin. Transl. Sci. 2023, 16, 3–435. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Padmanabhan, I.S.; Parmar, S.; Gong, Y. Targeting CLL-1 for acute myeloid leukemia therapy. J. Hematol. Oncol. 2019, 12, 41. [Google Scholar] [CrossRef] [PubMed]
- Chiffoleau, E. C-Type Lectin-Like Receptors as Emerging Orchestrators of Sterile Inflammation Represent Potential Therapeutic Targets. Front. Immunol. 2018, 9, 227. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Zhang, M.; Sun, R.; Lyu, H.; Xiao, X.; Zhang, X.; Li, F.; Xie, D.; Xiong, X.; Wang, J.; et al. First-in-human phase I study of CLL-1 CAR-T cells in adults with relapsed/refractory acute myeloid leukemia. J. Hematol. Oncol. 2022, 15, 1. [Google Scholar] [CrossRef] [PubMed]
- Kazi, J.U.; Rönnstrand, L. FMS-like Tyrosine Kinase 3/FLT3: From Basic Science to Clinical Implications. Physiol. Rev. 2019, 99, 1433–1466. [Google Scholar] [CrossRef] [PubMed]
- Gebru, M.T.; Wang, H.G. Therapeutic targeting of FLT3 and associated drug resistance in acute myeloid leukemia. J. Hematol. Oncol. 2020, 13, 155. [Google Scholar] [CrossRef]
- Sallman, D.A.; Brayer, J.B.; Poire, X.; Havelange, V.; Awada, A.; Lewalle, P.; Odunsi, K.; Wang, E.S.; Lonez, C.; Lequertier, T.; et al. Results from the Completed Dose-Escalation of the Hematological Arm of the Phase I Think Study Evaluating Multiple Infusions of NKG2D-Based CAR T-Cells as Standalone Therapy in Relapse/Refractory Acute Myeloid Leukemia and Myelodysplastic Syndrome Patients. Blood 2019, 134 (Suppl. S1), 3826. [Google Scholar] [CrossRef]
- Luo, Q.; Luo, W.; Zhu, Q.; Huang, H.; Peng, H.; Liu, R.; Xie, M.; Li, S.; Li, M.; Hu, X.; et al. Tumor-Derived Soluble MICA Obstructs the NKG2D Pathway to Restrain NK Cytotoxicity. Aging Dis. 2020, 11, 118–128. [Google Scholar] [CrossRef]
- Larson, R.C.; Maus, M.V. Recent advances and discoveries in the mechanisms and functions of CAR T cells. Nat. Rev. Cancer 2021, 21, 145–161. [Google Scholar] [CrossRef]
- Vishwasrao, P.; Li, G.; Boucher, J.C.; Smith, D.L.; Hui, S.K. Emerging CAR T Cell Strategies for the Treatment of AML. Cancers 2022, 14, 1241. [Google Scholar] [CrossRef]
- Hofmann, S.; Schubert, M.L.; Wang, L.; He, B.; Neuber, B.; Dreger, P.; Müller-Tidow, C.; Schmitt, M. Chimeric Antigen Receptor (CAR) T Cell Therapy in Acute Myeloid Leukemia (AML). J. Clin. Med. 2019, 8, 200. [Google Scholar] [CrossRef]
- Miller, J.S.; Soignier, Y.; Panoskaltsis-Mortari, A.; McNearney, S.A.; Yun, G.H.; Fautsch, S.K.; McKenna, D.; Le, C.; Defor, T.E.; Burns, L.J.; et al. Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood 2005, 105, 3051–3057. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Tricot, G.; Szmania, S.; Rosen, N.; Garg, T.K.; Malaviarachchi, P.A.; Moreno, A.; DuPont, B.; Hsu, K.C.; Baxter-Lowe, L.A.; et al. Infusion of haplo-identical killer immunoglobulin-like receptor ligand mismatched NK cells for relapsed myeloma in the setting of autologous stem cell transplantation. Br. J. Haematol. 2008, 143, 641–653. [Google Scholar] [CrossRef]
- Rubnitz, J.E.; Inaba, H.; Ribeiro, R.C.; Pounds, S.; Rooney, B.; Bell, T.; Pui, C.H.; Leung, W. NKAML: A pilot study to determine the safety and feasibility of haploidentical natural killer cell transplantation in childhood acute myeloid leukemia. J. Clin. Oncol. 2010, 28, 955–959. [Google Scholar] [CrossRef] [PubMed]
- Curti, A.; Ruggeri, L.; D’Addio, A.; Bontadini, A.; Dan, E.; Motta, M.R.; Trabanelli, S.; Giudice, V.; Urbani, E.; Martinelli, G.; et al. Successful transfer of alloreactive haploidentical KIR ligand-mismatched natural killer cells after infusion in elderly high risk acute myeloid leukemia patients. Blood 2011, 118, 3273–3279. [Google Scholar] [CrossRef] [PubMed]
- Curti, A.; Ruggeri, L.; Parisi, S.; Bontadini, A.; Dan, E.; Motta, M.R.; Rizzi, S.; Trabanelli, S.; Ocadlikova, D.; Lecciso, M.; et al. Larger Size of Donor Alloreactive NK Cell Repertoire Correlates with Better Response to NK Cell Immunotherapy in Elderly Acute Myeloid Leukemia Patients. Clin. Cancer Res. 2016, 22, 1914–1921. [Google Scholar] [CrossRef]
- Daher, M.; Melo Garcia, L.; Li, Y.; Rezvani, K. CAR-NK cells: The next wave of cellular therapy for cancer. Clin. Transl. Immunol. 2021, 10, e1274. [Google Scholar] [CrossRef]
- Zhang, L.; Meng, Y.; Feng, X.; Han, Z. CAR-NK cells for cancer immunotherapy: From bench to bedside. Biomark. Res. 2022, 10, 12. [Google Scholar] [CrossRef]
- Albinger, N.; Pfeifer, R.; Nitsche, M.; Mertlitz, S.; Campe, J.; Stein, K.; Kreyenberg, H.; Schubert, R.; Quadflieg, M.; Schneider, D.; et al. Primary CD33-targeting CAR-NK cells for the treatment of acute myeloid leukemia. Blood Cancer J. 2022, 12, 61. [Google Scholar] [CrossRef]
- Staerz, U.D.; Kanagawa, O.; Bevan, M.J. Hybrid antibodies can target sites for attack by T cells. Nature 1985, 314, 628–631. [Google Scholar] [CrossRef]
- Perez, P.; Hoffman, R.W.; Shaw, S.; Bluestone, J.A.; Segal, D.M. Specific targeting of cytotoxic T cells by anti-T3 linked to anti-target cell antibody. Nature 1985, 316, 354–356. [Google Scholar] [CrossRef] [PubMed]
- Offner, S.; Hofmeister, R.; Romaniuk, A.; Kufer, P.; Baeuerle, P.A. Induction of regular cytolytic T cell synapses by bispecific single-chain antibody constructs on MHC class I-negative tumor cells. Mol. Immunol. 2006, 43, 763–771. [Google Scholar] [CrossRef]
- Sun, Z.J.; Kim, K.S.; Wagner, G.; Reinherz, E.L. Mechanisms contributing to T cell receptor signaling and assembly revealed by the solution structure of an ectodomain fragment of the CD3 epsilon gamma heterodimer. Cell 2001, 105, 913–923. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Wu, B.; Brandl, C.; Johnson, J.; Wolf, A.; Chow, A.; Doshi, S. Blinatumomab, a Bispecific T-cell Engager (BiTE(®)) for CD-19 Targeted Cancer Immunotherapy: Clinical Pharmacology and Its Implications. Clin. Pharmacokinet. 2016, 55, 1271–1288. [Google Scholar] [CrossRef] [PubMed]
- Labrijn, A.F.; Janmaat, M.L.; Reichert, J.M.; Parren, P.W.H.I. Bispecific antibodies: A mechanistic review of the pipeline. Nat. Rev. Drug Discov. 2019, 18, 585–608. [Google Scholar] [CrossRef] [PubMed]
- Brischwein, K.; Parr, L.; Pflanz, S.; Volkland, J.; Lumsden, J.; Klinger, M.; Locher, M.; Hammond, S.A.; Kiener, P.; Kufer, P.; et al. Strictly target cell-dependent activation of T cells by bispecific single-chain antibody constructs of the BiTE class. J. Immunother. 2007, 30, 798–807. [Google Scholar] [CrossRef]
- Dreier, T.; Lorenczewski, G.; Brandl, C.; Hoffmann, P.; Syring, U.; Hanakam, F.; Kufer, P.; Riethmuller, G.; Bargou, R.; Baeuerle, P.A. Extremely potent, rapid and costimulation-independent cytotoxic T-cell response against lymphoma cells catalyzed by a single-chain bispecific antibody. Int. J. Cancer 2002, 100, 690–697. [Google Scholar] [CrossRef]
- Rader, C. DARTs take aim at BiTEs. Blood 2011, 117, 4403–4404. [Google Scholar] [CrossRef]
- Anegón, I.; Cuturi, M.C.; Trinchieri, G.; Perussia, B. Interaction of Fc receptor (CD16) ligands induces transcription of interleukin 2 receptor (CD25) and lymphokine genes and expression of their products in human natural killer cells. J. Exp. Med. 1988, 167, 452–472. [Google Scholar] [CrossRef]
- Gleason, M.K.; Verneris, M.R.; Todhunter, D.A.; Zhang, B.; McCullar, V.; Zhou, S.X.; Panoskaltsis-Mortari, A.; Weiner, L.M.; Vallera, D.A.; Miller, J.S. Bispecific and trispecific killer cell engagers directly activate human NK cells through CD16 signaling and induce cytotoxicity and cytokine production. Mol. Cancer Ther. 2012, 11, 2674–2684. [Google Scholar] [CrossRef]
- Hato, S.V.; Khong, A.; de Vries, I.J.M.; Lesterhuis, W.J. Targeting natural killer cells to acute myeloid leukemia in vitro with a CD16 x 33 bispecific killer cell engager and ADAM17 inhibition. Clin. Cancer Res. 2013, 19, 3844–3855. [Google Scholar] [CrossRef]
- Stumpp, M.T.; Binz, H.K.; Amstutz, P. DARPins: A new generation of protein therapeutics. Drug Discov. Today 2008, 13, 695–701. [Google Scholar] [CrossRef] [PubMed]
- Plückthun, A. Designed ankyrin repeat proteins (DARPins): Binding proteins for research, diagnostics, and therapy. Annu. Rev. Pharmacol. Toxicol. 2015, 55, 489–511. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, M.; Reichen, C.; Fischer, S.; Grübler, Y.; Eggenschwiler, A.; Marpakwar, R.; Looser, T.; Spitzli, P.; Herzog, C.; Villemagne, D.; et al. MP0533: A MultispecificDarpin CD3 Engager Targeting CD33, CD123, and CD70 for the Treatment of AML and MDS Designed to Selectively Target Leukemic Stem Cells. Blood 2022, 140 (Suppl. S1), 2251–2252. [Google Scholar] [CrossRef]
- Gauthier, L.; Virone-Oddos, A.; Beninga, J.; Rossi, B.; Nicolazzi, C.; Amara, C.; Blanchard-Alvarez, A.; Gourdin, N.; Courta, J.; Basset, A.; et al. Control of acute myeloid leukemia by a trifunctional NKp46-CD16a-NK cell engager targeting CD123. Nat. Biotechnol. 2023, 41, 1296–1306. [Google Scholar] [CrossRef]
- Stein, A.S.; Bajel, A.; Fleming, S.; Jongen-Lavrencic, M.; Garciaz, S.; Maiti, A.; Boissel, N.; De Botton, S.; Huls, G.A.; de Leeuw, D.C.; et al. An Open-Label, First-in-Human, Dose-Escalation Study of SAR443579 Administered as Single Agent By Intravenous Infusion in Patients with Relapsed or Refractory Acute Myeloid Leukemia (R/R AML), B-Cell Acute Lymphoblastic Leukemia (B-ALL) or High-Risk Myelodysplasia (HR-MDS). Blood 2022, 140 (Suppl. S1), 7476–7477. [Google Scholar] [CrossRef]
- Lee, D.W.; Gardner, R.; Porter, D.L.; Louis, C.U.; Ahmed, N.; Jensen, M.; Grupp, S.A.; Mackall, C.L. Current concepts in the diagnosis and management of cytokine release syndrome. Blood 2014, 124, 188–195, Erratum in Blood 2016, 128, 1533. [Google Scholar] [CrossRef]
- Brudno, J.N.; Kochenderfer, J.N. Toxicities of chimeric antigen receptor T cells: Recognition and management. Blood 2016, 127, 3321–3330. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Target Antigen | Type—Function | % of Expression | Potentialtarget Therapies |
|---|---|---|---|
| CD33 | Lineage-specific Sialic acid-binding receptor—role as negative regulator of cell activation | Up to 90% | CAR-T, CAR-NK, BiTEs, BiKEs, TRiKEs, DARPin |
| CD123 | Lineage-specific Alpha chain of the interleukin 3 receptor | Variable Higher in NPM1-mutated and FLT3-ITD AML | CAR-T, CAR-NK, DARTs, BiTEs, ANKET, DARPin |
| CLL-1 | Leukemia-associated C-type lectin-like receptor—role in regulation of innate and adaptive immunity | 90–95% | CAR-T, CAR-NK, BiTEs, TRiKEs, |
| (FLT3/CD135) | Leukemia-associated Class III receptor tyrosine kinases | >90% | CAR-T, BiTEs |
| NKG2DL | Not tumor-specific Ligand of C-type lectin-like receptor expressed on immune-effector cells-stimulates the cytotoxic activity | Upregulated in neoplastic cells | CAR-T, CAR-NK |
| Product | Target | Patient Population | Phase | NCT |
|---|---|---|---|---|
| TAA05 | FLT3 (CD135) | Relapsed/Refractory Acute Myeloid Leukemia | Early Phase I | NCT05445011 |
| CI-135 | FLT3 (CD135) | Relapsed/Refractory Acute Myeloid Leukemia | Early Phase I | NCT05266950 |
| Anti-FLT3 CAR T-cells | FLT3 (CD135) | Relapsed/Refractory Acute Myeloid Leukemia | Early Phase I | NCT05023707 |
| TAA-001 | FLT3 (CD135) | Relapsed/Refractory Acute Myeloid Leukemia | Early Phase I | NCT05432401 |
| Dual CD33/CLL1 CAR T Cells | CD33/CLL1 | Relapsed/Refractory Acute Myeloid Leukemia | Phase I | NCT05248685 |
| SC-DARIC33 | CD33 | Relapsed/Refractory Acute Myeloid Leukemia | Phase I | NCT05105152 |
| CD33-CAR T cells | CD33 | Relapsed/Refractory Acute Myeloid Leukemia | Phase I/II | NCT04835519 |
| NKG2D-CAR-T cells | NKG2D | Acute Myeloid Leukemia; Myelodysplastic Syndrome; Multiple Myeloma | Phase I | NCT02203825 |
| CD123-CAR T cells | CD123 | Relapsed/Refractory Acute Myeloid Leukemia | Phase I | NCT04318678 |
| UCART123 | CD123 | Relapsed/Refractory Acute Myeloid Leukemia | Phase I | NCT03190278 |
| CD123-CAR T cells | CD123 | Relapsed/Refractory Acute Myeloid Leukemia | Phase I | NCT04265963 |
| Product | Target | Patient Population | Phase | NCT |
|---|---|---|---|---|
| CD33 CAR NK | CD33/CLL1 | Relapsed/Refractory Acute Myeloid Leukemia | Early Phase I | NCT0521501 |
| CD33/CLL1 CAR-NK | Anti-CD33 | Relapsed/Refractory Acute Myeloid Leukemia | Phase I | NCT05008575 |
| JD023 | CD123 | Relapsed/Refractory Acute Myeloid Leukemia | Early Phase I | NCT05574608 |
| NKG2D CAR-NK | NKG2D | Relapsed/Refractory Acute Myeloid Leukemia | Phase I | NCT05247957 |
| NKG2D CAR-NK | NKG2D | Relapsed/Refractory Acute Myeloid Leukemia | Phase I | NCT05734898 |
| Product | NCT | Target | Patient Population | Phase | Outcome |
|---|---|---|---|---|---|
| AMV 564 | NCT03144245 | CD3 × CD33 bispecific antibody (BiTE) | Relapsed/Refractory Acute Myeloid Leukemia | Phase I | 36 patients enrolled, median age 71 years Efficacy: Bone marrow blast reduction in 17 (49%) patients; 1 CR, 1 CRi, and 1 PR; 3 patients had hematologic improvement in neutrophil counts Safety: CRS ≥ grade 3: 0%, the most common Grade ≥ 3 AE has been anemia (11%) [58] |
| AMG 330 | NCT02520427 | CD3 × CD33 bispecific antibody (BiTE) | Relapsed/Refractory Acute Myeloid Leukemia | Phase I | 55 patients enrolled, median age 58 years Efficacy: 3 CR (1 MRD negative), 4 Cri, 1 MLFS Safety: treatment-related AEs in 89%. CRS 60%; grade ≥ 3 in 7% [59]. |
| AMG 673 | NCT03224819 | CD3 × CD33 bispecific antibody (BiTE-HLE) | Relapsed/Refractory Acute Myeloid Leukemia | Phase I | 30 patients enrolled, median age 67.5 years Efficacy:Bone marrow blast reduction in 12/27 (44%) evaluable patients. 1 CRi. Safety: CRS in 50% (grade ≥ 3 in 4/15, 13%). Treatment-related serious AEs in 37% (grade ≥ 3 in 15/30, 50%; the most common were abnormal hepatic enzymes (17%), CRS (13%), leukopenia (13%), thrombocytopenia (7%), and febrile neutropenia (7%) [60]. |
| APVO 436 | NCT03647800 | CD3 × CD123 bispecific antibody (BiTE) | Relapsed/Refractory Acute Myeloid Leukemia; Relapsed/Refractory Myelodysplastic Syndrome | Phase I/Ib | 39 AML patients enrolled, median age 69 years. Efficacy: SD in 22/34 (64.7%) evaluable AML patients. Safety: treatment-related serious adverse events (SAEs) in 13/46 (28.3%) patients, the most common being CRS (7/13). Treatment-related transient neurotoxicity in 5/46 (10.9%) patients. CRS grade ≥ 3 in 4/46 (8.7%) patients, anemia grade ≥ 3 in 2/46 (4.3%) patients, infusion related reaction ≥ 3(IRR) in 2/46 (4.3%) patients [68]. |
| Vibecotamab (XmAb14045) | NCT02730312 | CD3 × CD123 bispecific antibody (BiTE) | Relapsed/Refractory Acute Myeloid Leukemia; B-cell Acute Lymphoblastic Leukemia; Blast Phase Chronic Myeloid Leukemia, Blastic Plasmacytoid Dendritic Cell Neoplasm | Phase I | 104 AML patients enrolled, median age 63 years. Efficacy: ORR of 14% (2 CR, 3 Cri, 2 MLFS). SD in 36 (71%) patients. Safety: CRS in 62/106 (58.5%) patients, CRS grade ≥ 3 in 9/62 (15%) patients. IRR in 24% of patients [69]. |
| Flotetuzumab (MGD006) | NCT02152956 | CD3 × CD123 dual affinity retargeting antibody (DART) | Primary Induction Failure (PIF) and Early Relapse (ER) Acute Myeloid Leukemia | Phase I /II | 38 PIF/ER AML patients enrolled, median age 63 yrs. Efficay: CRR for PIF pts was 45.8% (11/24; 5 CR, 3 CRh, and 3 CRi); CRR for ER pts was 35.7% (5/14; 2 CR, 1 CRh, 1 CRi and 1 MLFS). Safety: CRS was the most frequent AE, (grade ≤ 2). 9/38 (23.7%) pts experienced grade 1–2 headache; two pts experienced grade 3 confusion of short duration and fully reversable [70]. |
| GEM333 | NCT03516760 | CD3 × CD33 bispecific antibody (BiTE) | Relapsed/Refractory Acute Myeloid Leukemia | Phase I | N/A |
| JNJ-67571244 | NCT03915379 | CD3 × CD33 bispecific antibody (BiTE) | Relapsed/Refractory Acute Myeloid Leukemia; high risk Myelodysplastic Syndrome | Phase I | N/A |
| JNJ-63709178 | NCT02715011 | CD3 × CD123 bispecific antibody (BiTE) | Relapsed/Refractory Acute Myeloid Leukemia | Phase I | 62 patients enrolled, median age 67 years. Efficacy: no blast reduction observed. Safety: 63% total drug-related AEs (grade ≥ 3 in 84% of patients). CRS in 43.5% of patients. IRR in 11.3% of patients [71]. |
| SAR440234 | NCT03594955 | CD3 × CD123 bispecific antibody (BiTE) | Relapsed/Refractory Acute Myeloid Leukemia; high risk Myelodysplastic Syndrome; B-cell Acute Lymphoblastic Leukemia | Phase I/II | 7 patients enrolled, none enrolled in the Dose Expansion Part because of early termination of the study Efficacy: data not collected or analyzed due to the early termination of the study. Safety: 100% drug-related AEs (85.7% serious AEs). 28.6% CRS. |
| GTB-3550 | NCT03214666 | CD16/IL-15/CD33 Tri-Specific Killer Engager (TriKE®) | Relapsed/Refractory Acute Myeloid Leukemia; high risk Myelodysplastic Syndrome; Advanced Systemic Mastocytosis | Phase I/II | 12 patients enrolled, 9 AML patients, 3 MDS patients. None enrolled in the Dose Expansion Part because of early termination of the study Efficacy:data not collected or analyzed due to the early termination of the study. Safety: 100% drug-related AEs (25% serious AEs). 16.7% CRS [61]. |
| AMG427 | NCT03541369 | CD3 × FLT3 bispecific antibody (BiTE) | Relapsed/Refractory Acute Myeloid Leukemia | Phase I | N/A |
| CLN-049 | NCT05143996 | CD3 × FLT3 bispecific antibody (BiTE) | Relapsed/Refractory Acute Myeloid Leukemia; Myelodysplastic Syndrome | Phase I | N/A |
| MCLA-117 | NCT03038230 | CD3 × CLEC12A bispecific antibody (BiTE) | Relapsed/Refractory Acute Myeloid Leukemia; newly diagnosed elderly Acute Myeloid Leukemia | Phase I | N/A |
| SAR443579 | NCT05086315 | Trifunctional Natural Killer Cell Engager (NKCE) targeting the CD123 tumor antigen on cancer cells and co-engaging NKp46 and CD16a on NK cells | Relapsed/Refractory Acute Myeloid Leukemia; high risk Myelodysplastic Syndrome; B-cell Acute Lymphoblastic Leukemia | Phase I/II | N/A |
| MP0533 | NCT05673057 | Multispecific DARPin CD3 Engager Targeting CD33, CD123 and CD70 | Relapsed/Refractory Acute Myeloid Leukemia; Myelodysplastic Syndrome | Phase I/II | N/A |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Riva, C.; Vernarecci, C.; Minetto, P.; Goda, R.; Greppi, M.; Pesce, S.; Chies, M.; Zecchetti, G.; Ferro, B.; Maio, E.; et al. Harnessing Immune Response in Acute Myeloid Leukemia. J. Clin. Med. 2023, 12, 5824. https://doi.org/10.3390/jcm12185824
Riva C, Vernarecci C, Minetto P, Goda R, Greppi M, Pesce S, Chies M, Zecchetti G, Ferro B, Maio E, et al. Harnessing Immune Response in Acute Myeloid Leukemia. Journal of Clinical Medicine. 2023; 12(18):5824. https://doi.org/10.3390/jcm12185824
Chicago/Turabian StyleRiva, Carola, Chiara Vernarecci, Paola Minetto, Rayan Goda, Marco Greppi, Silvia Pesce, Maria Chies, Giada Zecchetti, Beatrice Ferro, Elena Maio, and et al. 2023. "Harnessing Immune Response in Acute Myeloid Leukemia" Journal of Clinical Medicine 12, no. 18: 5824. https://doi.org/10.3390/jcm12185824
APA StyleRiva, C., Vernarecci, C., Minetto, P., Goda, R., Greppi, M., Pesce, S., Chies, M., Zecchetti, G., Ferro, B., Maio, E., Cea, M., Lemoli, R. M., Marcenaro, E., & Guolo, F. (2023). Harnessing Immune Response in Acute Myeloid Leukemia. Journal of Clinical Medicine, 12(18), 5824. https://doi.org/10.3390/jcm12185824