
Platelets and the Atherosclerotic Process: An Overview of New Markers of Platelet Activation and Reactivity, and Their Implications in Primary and Secondary Prevention

 ,
,  , ,
, ,  ,
, 
Abstract
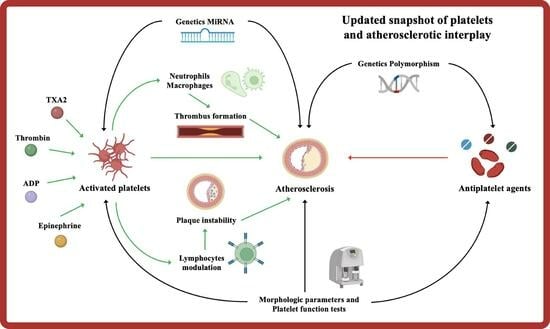
1. Introduction
2. Platelet Adhesion, Activation, and Aggregation
3. Interplay Immune System
4. Morphologic and Structural
5. Function and Reactivity
6. Platelet Reactivity
7. Genetics and miRNAs
8. Future Perspective and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Italiano, J.E.; Shivdasani, R.A. Megakaryocytes and beyond: The Birth of Platelets. J. Thromb. Haemost. 2003, 1, 1174–1182. [Google Scholar] [CrossRef]
- Healy, A.M.; Pickard, M.D.; Pradhan, A.D.; Wang, Y.; Chen, Z.; Croce, K.; Sakuma, M.; Shi, C.; Zago, A.C.; Garasic, J.; et al. Platelet Expression Profiling and Clinical Validation of Myeloid-Related Protein-14 as a Novel Determinant of Cardiovascular Events. Circulation 2006, 113, 2278–2284. [Google Scholar] [CrossRef]
- Hosseini, E.; Mohtashami, M.; Ghasemzadeh, M. Down-Regulation of Platelet Adhesion Receptors Is a Controlling Mechanism of Thrombosis, While Also Affecting Post-Transfusion Efficacy of Stored Platelets. Thromb. J. 2019, 17, 20. [Google Scholar] [CrossRef]
- Hosseini, E.; Nodeh, F.K.; Ghasemzadeh, M. Gamma Irradiation Induces a Pro-Apoptotic State in Longer Stored Platelets, without Progressing to an Overt Apoptosis by Day 7 of Storage. Apoptosis 2023, 28, 1141–1153. [Google Scholar] [CrossRef]
- Baigger, A.; Blasczyk, R.; Figueiredo, C. Towards the Manufacture of Megakaryocytes and Platelets for Clinical Application. Transfus. Med. Hemother. 2017, 44, 165–173. [Google Scholar] [CrossRef]
- Becker, R.C.; Sexton, T.; Smyth, S.S. Translational Implications of Platelets as Vascular First Responders. Circ. Res. 2018, 122, 506–522. [Google Scholar] [CrossRef]
- De Luca, G.; Suryapranata, H.; Stone, G.W.; Antoniucci, D.; Tcheng, J.E.; Neumann, F.-J.; Bonizzoni, E.; Topol, E.J.; Chiariello, M. Relationship between Patient’s Risk Profile and Benefits in Mortality from Adjunctive Abciximab to Mechanical Revascularization for ST-Segment Elevation Myocardial Infarction: A Meta-Regression Analysis of Randomized Trials. J. Am. Coll. Cardiol. 2006, 47, 685–686. [Google Scholar] [CrossRef]
- De Luca, G.; Navarese, E.P.; Cassetti, E.; Verdoia, M.; Suryapranata, H. Meta-Analysis of Randomized Trials of Glycoprotein IIb/IIIa Inhibitors in High-Risk Acute Coronary Syndromes Patients Undergoing Invasive Strategy. Am. J. Cardiol. 2011, 107, 198–203. [Google Scholar] [CrossRef]
- Verdoia, M.; Schaffer, A.; Barbieri, L.; Cassetti, E.; Piccolo, R.; Galasso, G.; Marino, P.; Sinigaglia, F.; De Luca, G. Benefits from New ADP Antagonists as Compared with Clopidogrel in Patients with Stable Angina or Acute Coronary Syndrome Undergoing Invasive Management: A Meta-Analysis of Randomized Trials. J. Cardiovasc. Pharmacol. 2014, 63, 339–350. [Google Scholar] [CrossRef]
- Nieswandt, B.; Pleines, I.; Bender, M. Platelet Adhesion and Activation Mechanisms in Arterial Thrombosis and Ischaemic Stroke. J. Thromb. Haemost. 2011, 9, 92–104. [Google Scholar] [CrossRef]
- Hosseini, E.; Beshkar, P.; Ghasemzadeh, M. Reverse Correlations of Collagen-Dependent Platelet Aggregation and Adhesion with GPVI Shedding during Storage. J. Thromb. Thrombolysis 2018, 46, 534–540. [Google Scholar] [CrossRef] [PubMed]
- Grover, S.P.; Mackman, N. Intrinsic Pathway of Coagulation and Thrombosis. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 331–338. [Google Scholar] [CrossRef]
- Gurbel, P.A.; Kuliopulos, A.; Tantry, U.S. G-Protein-Coupled Receptors Signaling Pathways in New Antiplatelet Drug Development. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 500–512. [Google Scholar] [CrossRef] [PubMed]
- Badimon, L.; Vilahur, G. Thrombosis Formation on Atherosclerotic Lesions and Plaque Rupture. J. Intern. Med. 2014, 276, 618–632. [Google Scholar] [CrossRef] [PubMed]
- Davì, G.; Patrono, C. Platelet Activation and Atherothrombosis. N. Engl. J. Med. 2007, 357, 2482–2494. [Google Scholar] [CrossRef] [PubMed]
- De Luca, G.; Verdoia, M.; Cassetti, E.; Schaffer, A.; Cavallino, C.; Bolzani, V.; Marino, P.; Novara Atherosclerosis Study Group (NAS). High Fibrinogen Level Is an Independent Predictor of Presence and Extent of Coronary Artery Disease among Italian Population. J. Thromb. Thrombolysis 2011, 31, 458–463. [Google Scholar] [CrossRef]
- Verdoia, M.; Schaffer, A.; Barbieri, L.; Aimaretti, G.; Marino, P.; Sinigaglia, F.; Suryapranata, H.; De Luca, G.; Novara Atherosclerosis Study Group (NAS). Impact of Diabetes on Neutrophil-to-Lymphocyte Ratio and Its Relationship to Coronary Artery Disease. Diabetes Metab. 2015, 41, 304–311. [Google Scholar] [CrossRef]
- Henn, V.; Slupsky, J.R.; Gräfe, M.; Anagnostopoulos, I.; Förster, R.; Müller-Berghaus, G.; Kroczek, R.A. CD40 Ligand on Activated Platelets Triggers an Inflammatory Reaction of Endothelial Cells. Nature 1998, 391, 591–594. [Google Scholar] [CrossRef]
- André, P.; Nannizzi-Alaimo, L.; Prasad, S.K.; Phillips, D.R. Platelet-Derived CD40L: The Switch-Hitting Player of Cardiovascular Disease. Circulation 2002, 106, 896–899. [Google Scholar] [CrossRef]
- von Hundelshausen, P.; Weber, C. Platelets as Immune Cells: Bridging Inflammation and Cardiovascular Disease. Circ. Res. 2007, 100, 27–40. [Google Scholar] [CrossRef]
- Semple, J.W.; Italiano, J.E.; Freedman, J. Platelets and the Immune Continuum. Nat. Rev. Immunol. 2011, 11, 264–274. [Google Scholar] [CrossRef]
- Lievens, D.; Zernecke, A.; Seijkens, T.; Soehnlein, O.; Beckers, L.; Munnix, I.C.A.; Wijnands, E.; Goossens, P.; van Kruchten, R.; Thevissen, L.; et al. Platelet CD40L Mediates Thrombotic and Inflammatory Processes in Atherosclerosis. Blood 2010, 116, 4317–4327. [Google Scholar] [CrossRef]
- Shiraki, R.; Inoue, N.; Kawasaki, S.; Takei, A.; Kadotani, M.; Ohnishi, Y.; Ejiri, J.; Kobayashi, S.; Hirata, K.-I.; Kawashima, S.; et al. Expression of Toll-Like Receptors on Human Platelets. Thromb. Res. 2004, 113, 379–385. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.N.; Ramji, D.P. The Role of Transforming Growth Factor-Beta in Atherosclerosis. Cytokine Growth Factor Rev. 2006, 17, 487–499. [Google Scholar] [CrossRef] [PubMed]
- Lutgens, E.; Daemen, M.J. Transforming Growth Factor-Beta: A Local or Systemic Mediator of Plaque Stability? Circ. Res. 2001, 89, 853–855. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Grainger, D.J.; Wakefield, L.; Bethell, H.W.; Farndale, R.W.; Metcalfe, J.C. Release and Activation of Platelet Latent TGF-Beta in Blood Clots during Dissolution with Plasmin. Nat. Med. 1995, 1, 932–937. [Google Scholar] [CrossRef] [PubMed]
- Blair, P.; Flaumenhaft, R. Platelet α-Granules: Basic Biology and Clinical Correlates. Blood Rev. 2009, 23, 177–189. [Google Scholar] [CrossRef]
- Bevilacqua, M.P.; Stengelin, S.; Gimbrone, M.A.; Seed, B. Endothelial Leukocyte Adhesion Molecule 1: An Inducible Receptor for Neutrophils Related to Complement Regulatory Proteins and Lectins. Science 1989, 243, 1160–1165. [Google Scholar] [CrossRef]
- Geng, J.G.; Bevilacqua, M.P.; Moore, K.L.; McIntyre, T.M.; Prescott, S.M.; Kim, J.M.; Bliss, G.A.; Zimmerman, G.A.; McEver, R.P. Rapid Neutrophil Adhesion to Activated Endothelium Mediated by GMP-140. Nature 1990, 343, 757–760. [Google Scholar] [CrossRef]
- Gross, P.L.; Furie, B.C.; Merrill-Skoloff, G.; Chou, J.; Furie, B. Leukocyte-versus Microparticle-Mediated Tissue Factor Transfer during Arteriolar Thrombus Development. J. Leukoc. Biol. 2005, 78, 1318–1326. [Google Scholar] [CrossRef]
- Futosi, K.; Fodor, S.; Mócsai, A. Neutrophil Cell Surface Receptors and Their Intracellular Signal Transduction Pathways. Int. Immunopharmacol. 2013, 17, 638–650. [Google Scholar] [CrossRef] [PubMed]
- Brandt, E.; Van Damme, J.; Flad, H.D. Neutrophils Can Generate Their Activator Neutrophil-Activating Peptide 2 by Proteolytic Cleavage of Platelet-Derived Connective Tissue-Activating Peptide III. Cytokine 1991, 3, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Bdeir, K.; Gollomp, K.; Stasiak, M.; Mei, J.; Papiewska-Pajak, I.; Zhao, G.; Worthen, G.S.; Cines, D.B.; Poncz, M.; Kowalska, M.A. Platelet-Specific Chemokines Contribute to the Pathogenesis of Acute Lung Injury. Am. J. Respir. Cell Mol. Biol. 2017, 56, 261–270. [Google Scholar] [CrossRef]
- Ghasemzadeh, M.; Ahmadi, J.; Hosseini, E. Platelet-Leukocyte Crosstalk in COVID-19: How Might the Reciprocal Links between Thrombotic Events and Inflammatory State Affect Treatment Strategies and Disease Prognosis? Thromb. Res. 2022, 213, 179–194. [Google Scholar] [CrossRef] [PubMed]
- Stark, K.; Philippi, V.; Stockhausen, S.; Busse, J.; Antonelli, A.; Miller, M.; Schubert, I.; Hoseinpour, P.; Chandraratne, S.; von Brühl, M.-L.; et al. Disulfide HMGB1 Derived from Platelets Coordinates Venous Thrombosis in Mice. Blood 2016, 128, 2435–2449. [Google Scholar] [CrossRef] [PubMed]
- Maugeri, N.; Campana, L.; Gavina, M.; Covino, C.; De Metrio, M.; Panciroli, C.; Maiuri, L.; Maseri, A.; D’Angelo, A.; Bianchi, M.E.; et al. Activated Platelets Present High Mobility Group Box 1 to Neutrophils, Inducing Autophagy and Promoting the Extrusion of Neutrophil Extracellular Traps. J. Thromb. Haemost. 2014, 12, 2074–2088. [Google Scholar] [CrossRef]
- Ghasemzadeh, M.; Hosseini, E. Platelet-Leukocyte Crosstalk: Linking Proinflammatory Responses to Procoagulant State. Thromb. Res. 2013, 131, 191–197. [Google Scholar] [CrossRef]
- Bonow, R.O.; Fonarow, G.C.; O’Gara, P.T.; Yancy, C.W. Association of Coronavirus Disease 2019 (COVID-19) with Myocardial Injury and Mortality. JAMA Cardiol. 2020, 5, 751. [Google Scholar] [CrossRef]
- Middleton, E.A.; He, X.-Y.; Denorme, F.; Campbell, R.A.; Ng, D.; Salvatore, S.P.; Mostyka, M.; Baxter-Stoltzfus, A.; Borczuk, A.C.; Loda, M.; et al. Neutrophil Extracellular Traps Contribute to Immunothrombosis in COVID-19 Acute Respiratory Distress Syndrome. Blood 2020, 136, 1169–1179. [Google Scholar] [CrossRef]
- Brill, A.; Fuchs, T.A.; Savchenko, A.S.; Thomas, G.M.; Martinod, K.; De Meyer, S.F.; Bhandari, A.A.; Wagner, D.D. Neutrophil Extracellular Traps Promote Deep Vein Thrombosis in Mice. J. Thromb. Haemost. 2012, 10, 136–144. [Google Scholar] [CrossRef]
- Massberg, S.; Grahl, L.; von Bruehl, M.-L.; Manukyan, D.; Pfeiler, S.; Goosmann, C.; Brinkmann, V.; Lorenz, M.; Bidzhekov, K.; Khandagale, A.B.; et al. Reciprocal Coupling of Coagulation and Innate Immunity via Neutrophil Serine Proteases. Nat. Med. 2010, 16, 887–896. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, T.A.; Brill, A.; Duerschmied, D.; Schatzberg, D.; Monestier, M.; Myers, D.D.; Wrobleski, S.K.; Wakefield, T.W.; Hartwig, J.H.; Wagner, D.D. Extracellular DNA Traps Promote Thrombosis. Proc. Natl. Acad. Sci. USA 2010, 107, 15880–15885. [Google Scholar] [CrossRef] [PubMed]
- Hally, K.E.; Parker, O.M.; Brunton-O’Sullivan, M.M.; Harding, S.A.; Larsen, P.D. Linking Neutrophil Extracellular Traps and Platelet Activation: A Composite Biomarker Score for Predicting Outcomes after Acute Myocardial Infarction. Thromb. Haemost. 2021, 121, 1637–1649. [Google Scholar] [CrossRef]
- Riegger, J.; Byrne, R.A.; Joner, M.; Chandraratne, S.; Gershlick, A.H.; ten Berg, J.M.; Adriaenssens, T.; Guagliumi, G.; Godschalk, T.C.; Neumann, F.-J.; et al. Histopathological Evaluation of Thrombus in Patients Presenting with Stent Thrombosis. A Multicenter European Study: A Report of the Prevention of Late Stent Thrombosis by an Interdisciplinary Global European Effort Consortium. Eur. Heart J. 2016, 37, 1538–1549. [Google Scholar] [CrossRef] [PubMed]
- Stakos, D.A.; Kambas, K.; Konstantinidis, T.; Mitroulis, I.; Apostolidou, E.; Arelaki, S.; Tsironidou, V.; Giatromanolaki, A.; Skendros, P.; Konstantinides, S.; et al. Expression of Functional Tissue Factor by Neutrophil Extracellular Traps in Culprit Artery of Acute Myocardial Infarction. Eur. Heart J. 2015, 36, 1405–1414. [Google Scholar] [CrossRef]
- Stähli, B.E.; Gebhard, C.; Duchatelle, V.; Cournoyer, D.; Petroni, T.; Tanguay, J.; Robb, S.; Mann, J.; Guertin, M.; Wright, R.S.; et al. Effects of the P-Selectin Antagonist Inclacumab on Myocardial Damage after Percutaneous Coronary Intervention According to Timing of Infusion: Insights from the SELECT-ACS Trial. J. Am. Heart Assoc. 2016, 5, e004255. [Google Scholar] [CrossRef]
- Tardif, J.-C.; Tanguay, J.-F.; Wright, S.R.; Duchatelle, V.; Petroni, T.; Grégoire, J.C.; Ibrahim, R.; Heinonen, T.M.; Robb, S.; Bertrand, O.F.; et al. Effects of the P-Selectin Antagonist Inclacumab on Myocardial Damage after Percutaneous Coronary Intervention for Non–ST-Segment Elevation Myocardial Infarction. J. Am. Coll. Cardiol. 2013, 61, 2048–2055. [Google Scholar] [CrossRef]
- Stähli, B.E.; Tardif, J.-C.; Carrier, M.; Gallo, R.; Emery, R.W.; Robb, S.; Cournoyer, D.; Blondeau, L.; Johnson, D.; Mann, J.; et al. Effects of P-Selectin Antagonist Inclacumab in Patients Undergoing Coronary Artery Bypass Graft Surgery. J. Am. Coll. Cardiol. 2016, 67, 344–346. [Google Scholar] [CrossRef]
- Kaiser, R.; Escaig, R.; Erber, J.; Nicolai, L. Neutrophil-Platelet Interactions as Novel Treatment Targets in Cardiovascular Disease. Front. Cardiovasc. Med. 2022, 8, 824112. [Google Scholar] [CrossRef]
- Chu, S.G.; Becker, R.C.; Berger, P.B.; Bhatt, D.L.; Eikelboom, J.W.; Konkle, B.; Mohler, E.R.; Reilly, M.P.; Berger, J.S. Mean Platelet Volume as a Predictor of Cardiovascular Risk: A Systematic Review and Meta-Analysis. J. Thromb. Haemost. 2010, 8, 148–156. [Google Scholar] [CrossRef]
- Jakubowski, J.A.; Thompson, C.B.; Vaillancourt, R.; Valeri, C.R.; Deykin, D. Arachidonic Acid Metabolism by Platelets of Differing Size. Br. J. Haematol. 1983, 53, 503–511. [Google Scholar] [CrossRef] [PubMed]
- Hosseini, E.; Ghasemzadeh, M.; Atashibarg, M.; Haghshenas, M. ROS Scavenger, N-Acetyl-L-Cysteine and NOX Specific Inhibitor, VAS2870 Reduce Platelets Apoptosis While Enhancing Their Viability during Storage. Transfusion 2019, 59, 1333–1343. [Google Scholar] [CrossRef] [PubMed]
- Kodiatte, T.A.; Manikyam, U.K.; Rao, S.B.; Jagadish, T.M.; Reddy, M.; Lingaiah, H.K.M.; Lakshmaiah, V. Mean Platelet Volume in Type 2 Diabetes Mellitus. J. Lab. Physicians 2012, 4, 5–9. [Google Scholar] [CrossRef]
- Hekimsoy, Z.; Payzin, B.; Ornek, T.; Kandoğan, G. Mean Platelet Volume in Type 2 Diabetic Patients. J. Diabetes Complicat. 2004, 18, 173–176. [Google Scholar] [CrossRef] [PubMed]
- Colwell, J.A.; Nesto, R.W. The Platelet in Diabetes: Focus on Prevention of Ischemic Events. Diabetes Care 2003, 26, 2181–2188. [Google Scholar] [CrossRef] [PubMed]
- Briggs, C. Quality Counts: New Parameters in Blood Cell Counting. Int. J. Lab. Hematol. 2009, 31, 277–297. [Google Scholar] [CrossRef]
- Thompson, C.B.; Eaton, K.A.; Princiotta, S.M.; Rushin, C.A.; Valeri, C.R. Size Dependent Platelet Subpopulations: Relationship of Platelet Volume to Ultrastructure, Enzymatic Activity, and Function. Br. J. Haematol. 1982, 50, 509–519. [Google Scholar] [CrossRef]
- Tsiara, S.; Elisaf, M.; Jagroop, I.A.; Mikhailidis, D.P. Platelets as Predictors of Vascular Risk: Is There a Practical Index of Platelet Activity? Clin. Appl. Thromb. Hemost. 2003, 9, 177–190. [Google Scholar] [CrossRef]
- Jagroop, I.A.; Clatworthy, I.; Lewin, J.; Mikhailidis, D.P. Shape Change in Human Platelets: Measurement with a Channelyzer and Visualisation by Electron Microscopy. Platelets 2000, 11, 28–32. [Google Scholar]
- Park, Y.; Schoene, N.; Harris, W. Mean Platelet Volume as an Indicator of Platelet Activation: Methodological Issues. Platelets 2002, 13, 301–306. [Google Scholar] [CrossRef]
- Schoene, N.W. Design Criteria: Tests Used to Assess Platelet Function. Am. J. Clin. Nutr. 1997, 65, 1665S–1668S. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.F.; Trowbridge, E.A.; Salmon, G.; Plumb, J. The Biological Significance of Platelet Volume: Its Relationship to Bleeding Time, Platelet Thromboxane B2 Production and Megakaryocyte Nuclear DNA Concentration. Thromb. Res. 1983, 32, 443–460. [Google Scholar] [CrossRef]
- Giles, H.; Smith, R.E.; Martin, J.F. Platelet Glycoprotein IIb-IIIa and Size Are Increased in Acute Myocardial Infarction. Eur. J. Clin. Investig. 1994, 24, 69–72. [Google Scholar] [CrossRef] [PubMed]
- Pizzulli, L.; Yang, A.; Martin, J.F.; Lüderitz, B. Changes in Platelet Size and Count in Unstable Angina Compared to Stable Angina or Non-Cardiac Chest Pain. Eur. Heart J. 1998, 19, 80–84. [Google Scholar] [CrossRef] [PubMed]
- Huczek, Z.; Kochman, J.; Filipiak, K.J.; Horszczaruk, G.J.; Grabowski, M.; Piatkowski, R.; Wilczynska, J.; Zielinski, A.; Meier, B.; Opolski, G. Mean Platelet Volume on Admission Predicts Impaired Reperfusion and Long-Term Mortality in Acute Myocardial Infarction Treated with Primary Percutaneous Coronary Intervention. J. Am. Coll. Cardiol. 2005, 46, 284–290. [Google Scholar] [CrossRef]
- Murat, S.N.; Duran, M.; Kalay, N.; Gunebakmaz, O.; Akpek, M.; Doger, C.; Elcik, D.; Ocak, A.; Vatankulu, M.A.; Turfan, M.; et al. Relation between Mean Platelet Volume and Severity of Atherosclerosis in Patients with Acute Coronary Syndromes. Angiology 2013, 64, 131–136. [Google Scholar] [CrossRef] [PubMed]
- De Luca, G.; Verdoia, M.; Cassetti, E.; Schaffer, A.; Di Giovine, G.; Bertoni, A.; Di Vito, C.; Sampietro, S.; Aimaretti, G.; Bellomo, G.; et al. Mean Platelet Volume Is Not Associated with Platelet Reactivity and the Extent of Coronary Artery Disease in Diabetic Patients. Blood Coagul. Fibrinolysis 2013, 24, 619–624. [Google Scholar] [CrossRef] [PubMed]
- Verdoia, M.; Camaro, C.; Barbieri, L.; Schaffer, A.; Marino, P.; Bellomo, G.; Suryapranata, H.; De Luca, G. Mean Platelet Volume and the Risk of Periprocedural Myocardial Infarction in Patients Undergoing Coronary Angioplasty. Atherosclerosis 2013, 228, 136–141. [Google Scholar] [CrossRef]
- Halbmayer, W.M.; Haushofer, A.; Radek, J.; Schön, R.; Deutsch, M.; Fischer, M. Platelet Size, Fibrinogen and Lipoprotein(a) in Coronary Heart Disease. Coron. Artery Dis. 1995, 6, 397–402. [Google Scholar] [CrossRef]
- De Luca, G.; Santagostino, M.; Secco, G.G.; Cassetti, E.; Giuliani, L.; Franchi, E.; Coppo, L.; Iorio, S.; Venegoni, L.; Rondano, E.; et al. Mean Platelet Volume and the Extent of Coronary Artery Disease: Results from a Large Prospective Study. Atherosclerosis 2009, 206, 292–297. [Google Scholar] [CrossRef]
- Verdoia, M.; Pergolini, P.; Rolla, R.; Nardin, M.; Barbieri, L.; Schaffer, A.; Bellomo, G.; Marino, P.; Suryapranata, H.; De Luca, G. Mean Platelet Volume and High-Residual Platelet Reactivity in Patients Receiving Dual Antiplatelet Therapy with Clopidogrel or Ticagrelor. Expert Opin. Pharmacother. 2015, 16, 1739–1747. [Google Scholar] [CrossRef]
- Sansanayudh, N.; Numthavaj, P.; Muntham, D.; Yamwong, S.; McEvoy, M.; Attia, J.; Sritara, P.; Thakkinstian, A. Prognostic Effect of Mean Platelet Volume in Patients with Coronary Artery Disease. A Systematic Review and Meta-Analysis. Thromb. Haemost. 2015, 114, 1299–1309. [Google Scholar] [CrossRef]
- Vagdatli, E.; Gounari, E.; Lazaridou, E.; Katsibourlia, E.; Tsikopoulou, F.; Labrianou, I. Platelet Distribution Width: A Simple, Practical and Specific Marker of Activation of Coagulation. Hippokratia 2010, 14, 28–32. [Google Scholar]
- Vatankulu, M.A.; Sonmez, O.; Ertas, G.; Bacaksiz, A.; Turfan, M.; Erdogan, E.; Tasal, A.; Kul, S.; Uyarel, H.; Goktekin, O. A New Parameter Predicting Chronic Total Occlusion of Coronary Arteries. Angiology 2014, 65, 60–64. [Google Scholar] [CrossRef]
- Ege, M.R.; Guray, U.; Guray, Y.; Acıkgoz, S.; Demirkan, B. Platelet Distribution Width and Saphenous Vein Disease in Patients after CABG. Association with Graft Occlusion. Herz 2013, 38, 197–201. [Google Scholar] [CrossRef] [PubMed]
- Ihara, A.; Kawamoto, T.; Matsumoto, K.; Shouno, S.; Hirahara, C.; Morimoto, T.; Noma, Y. Relationship between Platelet Indexes and Coronary Angiographic Findings in Patients with Ischemic Heart Disease. Pathophysiol. Haemost. Thromb. 2006, 35, 376–379. [Google Scholar] [CrossRef] [PubMed]
- De Luca, G.; Venegoni, L.; Iorio, S.; Secco, G.G.; Cassetti, E.; Verdoia, M.; Schaffer, A.; Coppo, L.; Bellomo, G.; Marino, P.; et al. Platelet Distribution Width and the Extent of Coronary Artery Disease: Results from a Large Prospective Study. Platelets 2010, 21, 508–514. [Google Scholar] [CrossRef] [PubMed]
- Bessman, J.D.; Williams, L.J.; Gilmer, P.R. Mean Platelet Volume. The Inverse Relation of Platelet Size and Count in Normal Subjects, and an Artifact of Other Particles. Am. J. Clin. Pathol. 1981, 76, 289–293. [Google Scholar] [CrossRef] [PubMed]
- Lv, H.-C.; Wu, H.-Y.; Yin, J.-S.; Ge, J.-B. Thrombin Induced Platelet-Fibrin Clot Strength in Relation to Platelet Volume Indices and Inflammatory Markers in Patients with Coronary Artery Disease. Oncotarget 2017, 8, 64217–64223. [Google Scholar] [CrossRef]
- Larsen, S.B.; Grove, E.L.; Hvas, A.-M.; Kristensen, S.D. Platelet Turnover in Stable Coronary Artery Disease—Influence of Thrombopoietin and Low-Grade Inflammation. PLoS ONE 2014, 9, e85566. [Google Scholar] [CrossRef]
- De Luca, G.; Santagostino, M.; Secco, G.G.; Cassetti, E.; Giuliani, L.; Coppo, L.; Schaffer, A.; Fundaliotis, A.; Iorio, S.; Venegoni, L.; et al. Platelet-Large Cell Ratio and the Extent of Coronary Artery Disease: Results from a Large Prospective Study. J. Thromb. Thrombolysis 2010, 30, 426–433. [Google Scholar] [CrossRef] [PubMed]
- Verdoia, M.; Pergolini, P.; Rolla, R.; Nardin, M.; Barbieri, L.; Schaffer, A.; Bellomo, G.; Marino, P.; Suryapranata, H.; De Luca, G.; et al. Platelet Larger Cell Ratio and High-on Treatment Platelet Reactivity during Dual Antiplatelet Therapy. Cardiovasc. Drugs Ther. 2015, 29, 443–450. [Google Scholar] [CrossRef] [PubMed]
- Ault, K.A.; Rinder, H.M.; Mitchell, J.; Carmody, M.B.; Vary, C.P.H.; Hillman, R.S. The Significance of Platelets with Increased RNA Content (Reticulated Platelets) A Measure of the Rate of Thrombopoiesis. Am. J. Clin. Pathol. 1992, 98, 637–646. [Google Scholar] [CrossRef]
- Dusse, L.M.S.; Freitas, L.G. Clinical Applicability of Reticulated Platelets. Clin. Chim. Acta 2015, 439, 143–147. [Google Scholar] [CrossRef]
- McBane, R.D.; Gonzalez, C.; Hodge, D.O.; Wysokinski, W.E. Propensity for Young Reticulated Platelet Recruitment into Arterial Thrombi. J. Thromb. Thrombolysis 2014, 37, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, J.J.M.L. Reticulated Platelets: Analytical Aspects and Clinical Utility. Clin. Chem. Lab. Med. 2014, 52, 1107–1117. [Google Scholar] [CrossRef]
- López-Jiménez, R.A.; Martín-Herrero, F.; González-Porras, J.R.; Sánchez-Barba, M.; Martín-Luengo, C.; Pabón-Osuna, P. Fracción de Plaquetas Inmaduras, Un Nuevo Marcador Pronóstico En El Síndrome Coronario Agudo. Rev. Esp. Cardiol. 2013, 66, 147–148. [Google Scholar] [CrossRef]
- Cesari, F.; Marcucci, R.; Gori, A.; Caporale, R.; Fanelli, A.; Casola, G.; Balzi, D.; Barchielli, A.; Valente, S.; Giglioli, C.; et al. Reticulated Platelets Predict Cardiovascular Death in Acute Coronary Syndrome Patients. Thromb. Haemost. 2013, 109, 846–853. [Google Scholar] [CrossRef]
- Berny-Lang, M.A.; Darling, C.E.; Frelinger, A.L.; Barnard, M.R.; Smith, C.S.; Michelson, A.D. Do Immature Platelet Levels in Chest Pain Patients Presenting to the Emergency Department Aid in the Diagnosis of Acute Coronary Syndrome? Int. J. Lab. Hematol. 2015, 37, 112–119. [Google Scholar] [CrossRef]
- Bernlochner, I.; Goedel, A.; Plischke, C.; Schüpke, S.; Haller, B.; Schulz, C.; Mayer, K.; Morath, T.; Braun, S.; Schunkert, H.; et al. Impact of Immature Platelets on Platelet Response to Ticagrelor and Prasugrel in Patients with Acute Coronary Syndrome. Eur. Heart J. 2015, 36, 3202–3210. [Google Scholar] [CrossRef]
- Perl, L.; Lerman-Shivek, H.; Rechavia, E.; Vaduganathan, M.; Leshem-Lev, D.; Zemer-Wassercug, N.; Dadush, O.; Codner, P.; Bental, T.; Battler, A.; et al. Response to Prasugrel and Levels of Circulating Reticulated Platelets in Patients with ST-Segment Elevation Myocardial Infarction. J. Am. Coll. Cardiol. 2014, 63, 513–517. [Google Scholar] [CrossRef] [PubMed]
- Verdoia, M.; Sartori, C.; Pergolini, P.; Nardin, M.; Rolla, R.; Barbieri, L.; Schaffer, A.; Marino, P.; Bellomo, G.; Suryapranata, H.; et al. Immature Platelet Fraction and High-on Treatment Platelet Reactivity with Ticagrelor in Patients with Acute Coronary Syndromes. J. Thromb. Thrombolysis 2016, 41, 663–670. [Google Scholar] [CrossRef] [PubMed]
- Verdoia, M.; Nardin, M.; Negro, F.; Rolla, R.; Carriero, A.; De Luca, G. Impact of Long-Term Therapy with Acetylsalicylic Acid on Immature Platelet Count. J. Cardiovasc. Med. 2019, 20, 306–312. [Google Scholar] [CrossRef] [PubMed]
- Duke, W.W. The Relation of Blood Platelets to Hemorrhagic Disease. J. Am. Med. Assoc. 1910, 55, 1185. [Google Scholar] [CrossRef]
- Born, G.V. Aggregation of Blood Platelets by Adenosine Diphosphate and Its Reversal. Nature 1962, 194, 927–929. [Google Scholar] [CrossRef]
- O’Brien, J. Platelet Aggregation: Part II Some Results from a New Method of Study. J. Clin. Pathol. 1962, 15, 452–455. [Google Scholar] [CrossRef]
- Tóth, O.; Calatzis, A.; Penz, S.; Losonczy, H.; Siess, W. Multiple Electrode Aggregometry: A New Device to Measure Platelet Aggregation in Whole Blood. Thromb. Haemost. 2006, 96, 781–788. [Google Scholar]
- Smith, J.W.; Steinhubl, S.R.; Lincoff, A.M.; Coleman, J.C.; Lee, T.T.; Hillman, R.S.; Coller, B.S. Rapid Platelet-Function Assay: An Automated and Quantitative Cartridge-Based Method. Circulation 1999, 99, 620–625. [Google Scholar] [CrossRef]
- Paniccia, R.; Antonucci, E.; Gori, A.M.; Marcucci, R.; Giglioli, C.; Antoniucci, D.; Gensini, G.F.; Abbate, R.; Prisco, D. Different Methodologies for Evaluating the Effect of Clopidogrel on Platelet Function in High-Risk Coronary Artery Disease Patients. J. Thromb. Haemost. 2007, 5, 1839–1847. [Google Scholar] [CrossRef]
- Kundu, S.K.; Heilmann, E.J.; Sio, R.; Garcia, C.; Davidson, R.M.; Ostgaard, R.A. Description of an in Vitro Platelet Function Analyzer—PFA-100. Semin. Thromb. Hemost. 1995, 21 (Suppl. S2), 106–112. [Google Scholar] [CrossRef]
- Varon, D.; Dardik, R.; Shenkman, B.; Kotev-Emeth, S.; Farzame, N.; Tamarin, I.; Savion, N. A New Method for Quantitative Analysis of Whole Blood Platelet Interaction with Extracellular Matrix under Flow Conditions. Thromb. Res. 1997, 85, 283–294. [Google Scholar] [CrossRef]
- Luddington, R.J. Thrombelastography/Thromboelastometry. Clin. Lab. Haematol. 2005, 27, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Spurgeon, B.E.J.; Aburima, A.; Oberprieler, N.G.; Taskén, K.; Naseem, K.M. Multiplexed Phosphospecific Flow Cytometry Enables Large-Scale Signaling Profiling and Drug Screening in Blood Platelets. J. Thromb. Haemost. 2014, 12, 1733–1743. [Google Scholar] [CrossRef] [PubMed]
- Siller-Matula, J.M.; Francesconi, M.; Dechant, C.; Jilma, B.; Maurer, G.; Delle-Karth, G.; Gouya, G.; Ruzicka, K.; Podczeck-Schweighofer, A.; Christ, G. Personalized Antiplatelet Treatment after Percutaneous Coronary Intervention: The MADONNA Study. Int. J. Cardiol. 2013, 167, 2018–2023. [Google Scholar] [CrossRef] [PubMed]
- Collet, J.-P.; Thiele, H.; Barbato, E.; Barthélémy, O.; Bauersachs, J.; Bhatt, D.L.; Dendale, P.; Dorobantu, M.; Edvardsen, T.; Folliguet, T.; et al. 2020 ESC Guidelines for the Management of Acute Coronary Syndromes in Patients Presenting without Persistent ST-Segment Elevation. Eur. Heart J. 2021, 42, 1289–1367. [Google Scholar] [CrossRef] [PubMed]
- Cattaneo, M. High On-Treatment Platelet Reactivity—Definition and Measurement. Thromb. Haemost. 2013, 109, 792–798. [Google Scholar] [CrossRef]
- Stone, G.W.; Witzenbichler, B.; Weisz, G.; Rinaldi, M.J.; Neumann, F.-J.; Metzger, D.C.; Henry, T.D.; Cox, D.A.; Duffy, P.L.; Mazzaferri, E.; et al. Platelet Reactivity and Clinical Outcomes after Coronary Artery Implantation of Drug-Eluting Stents (ADAPT-DES): A Prospective Multicentre Registry Study. Lancet 2013, 382, 614–623. [Google Scholar] [CrossRef]
- Montalescot, G.; Rangé, G.; Silvain, J.; Bonnet, J.-L.; Boueri, Z.; Barthélémy, O.; Cayla, G.; Belle, L.; Van Belle, E.; Cuisset, T.; et al. High On-Treatment Platelet Reactivity as a Risk Factor for Secondary Prevention after Coronary Stent Revascularization: A Landmark Analysis of the ARCTIC Study. Circulation 2014, 129, 2136–2143. [Google Scholar] [CrossRef]
- Verdoia, M.; Pergolini, P.; Nardin, M.; Rolla, R.; Suryapranata, H.; Kedhi, E.; De Luca, G.; Novara Atherosclerosis Study Group (NAS). Ticagrelor and Prasugrel in Acute Coronary Syndrome: A Single-Arm Crossover Platelet Reactivity Study. J. Cardiovasc. Med. 2021, 22, 686–692. [Google Scholar] [CrossRef]
- Price, M.J.; Berger, P.B.; Angiolillo, D.J.; Teirstein, P.S.; Tanguay, J.-F.; Kandzari, D.E.; Cannon, C.P.; Topol, E.J. Evaluation of Individualized Clopidogrel Therapy after Drug-Eluting Stent Implantation in Patients with High Residual Platelet Reactivity: Design and Rationale of the GRAVITAS Trial. Am. Heart J. 2009, 157, 818–824.e1. [Google Scholar] [CrossRef]
- Aleil, B.; Jacquemin, L.; De Poli, F.; Zaehringer, M.; Collet, J.-P.; Montalescot, G.; Cazenave, J.-P.; Dickele, M.-C.; Monassier, J.-P.; Gachet, C. Clopidogrel 150 Mg/Day to Overcome Low Responsiveness in Patients Undergoing Elective Percutaneous Coronary Intervention: Results from the VASP-02 (Vasodilator-Stimulated Phosphoprotein-02) Randomized Study. JACC Cardiovasc. Interv. 2008, 1, 631–638. [Google Scholar] [CrossRef] [PubMed]
- Bonello, L.; Camoin-Jau, L.; Arques, S.; Boyer, C.; Panagides, D.; Wittenberg, O.; Simeoni, M.-C.; Barragan, P.; Dignat-George, F.; Paganelli, F. Adjusted Clopidogrel Loading Doses According to Vasodilator-Stimulated Phosphoprotein Phosphorylation Index Decrease Rate of Major Adverse Cardiovascular Events in Patients with Clopidogrel Resistance: A Multicenter Randomized Prospective Study. J. Am. Coll. Cardiol. 2008, 51, 1404–1411. [Google Scholar] [CrossRef] [PubMed]
- Cuisset, T.; Frere, C.; Quilici, J.; Morange, P.-E.; Mouret, J.-P.; Bali, L.; Moro, P.-J.; Lambert, M.; Alessi, M.-C.; Bonnet, J.L. Glycoprotein IIb/IIIa Inhibitors Improve Outcome after Coronary Stenting in Clopidogrel Nonresponders: A Prospective, Randomized Study. JACC Cardiovasc. Interv. 2008, 1, 649–653. [Google Scholar] [CrossRef] [PubMed]
- Valgimigli, M.; Campo, G.; de Cesare, N.; Meliga, E.; Vranckx, P.; Furgieri, A.; Angiolillo, D.J.; Sabatè, M.; Hamon, M.; Repetto, A.; et al. Intensifying Platelet Inhibition with Tirofiban in Poor Responders to Aspirin, Clopidogrel, or Both Agents Undergoing Elective Coronary Intervention: Results from the Double-Blind, Prospective, Randomized Tailoring Treatment with Tirofiban in Patients Sho. Circulation 2009, 119, 3215–3222. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, D.; Zhuang, S.; Lai, Y. Modifying Clopidogrel Maintenance Doses According to Vasodilator-Stimulated Phosphoprotein Phosphorylation Index Improves Clinical Outcome in Patients with Clopidogrel Resistance. Clin. Cardiol. 2011, 34, 332–338. [Google Scholar] [CrossRef]
- Price, M.J.; Berger, P.B.; Teirstein, P.S.; Tanguay, J.-F.; Angiolillo, D.J.; Spriggs, D.; Puri, S.; Robbins, M.; Garratt, K.N.; Bertrand, O.F.; et al. Standard- vs. High-Dose Clopidogrel Based on Platelet Function Testing after Percutaneous Coronary Intervention: The GRAVITAS Randomized Trial. JAMA 2011, 305, 1097–1105. [Google Scholar] [CrossRef]
- Aradi, D.; Rideg, O.; Vorobcsuk, A.; Magyarlaki, T.; Magyari, B.; Kónyi, A.; Pintér, T.; Horváth, I.G.; Komócsi, A. Justification of 150 Mg Clopidogrel in Patients with High On-Clopidogrel Platelet Reactivity. Eur. J. Clin. Investig. 2012, 42, 384–392. [Google Scholar] [CrossRef]
- Hazarbasanov, D.; Velchev, V.; Finkov, B.; Postadjian, A.; Kostov, E.; Rifai, N.; Aradi, D. Tailoring Clopidogrel Dose According to Multiple Electrode Aggregometry Decreases the Rate of Ischemic Complications after Percutaneous Coronary Intervention. J. Thromb. Thrombolysis 2012, 34, 85–90. [Google Scholar] [CrossRef]
- Trenk, D.; Stone, G.W.; Gawaz, M.; Kastrati, A.; Angiolillo, D.J.; Müller, U.; Richardt, G.; Jakubowski, J.A.; Neumann, F.-J. A Randomized Trial of Prasugrel versus Clopidogrel in Patients with High Platelet Reactivity on Clopidogrel after Elective Percutaneous Coronary Intervention with Implantation of Drug-Eluting Stents: Results of the TRIGGER-PCI (Testing Platelet Reactivity in Patients Undergoing Elective Stent Placement on Clopidogrel to Guide Alternative Therapy with Prasugrel) Study. J. Am. Coll. Cardiol. 2012, 59, 2159–2164. [Google Scholar] [CrossRef]
- Collet, J.-P.; Cuisset, T.; Rangé, G.; Cayla, G.; Elhadad, S.; Pouillot, C.; Henry, P.; Motreff, P.; Carrié, D.; Boueri, Z.; et al. Bedside Monitoring to Adjust Antiplatelet Therapy for Coronary Stenting. N. Engl. J. Med. 2012, 367, 2100–2109. [Google Scholar] [CrossRef]
- Cayla, G.; Cuisset, T.; Silvain, J.; Leclercq, F.; Manzo-Silberman, S.; Saint-Etienne, C.; Delarche, N.; Bellemain-Appaix, A.; Range, G.; El Mahmoud, R.; et al. Platelet Function Monitoring to Adjust Antiplatelet Therapy in Elderly Patients Stented for an Acute Coronary Syndrome (ANTARCTIC): An Open-Label, Blinded-Endpoint, Randomised Controlled Superiority Trial. Lancet 2016, 388, 2015–2022. [Google Scholar] [CrossRef] [PubMed]
- Sibbing, D.; Aradi, D.; Jacobshagen, C.; Gross, L.; Trenk, D.; Geisler, T.; Orban, M.; Hadamitzky, M.; Merkely, B.; Kiss, R.G.; et al. Guided De-Escalation of Antiplatelet Treatment in Patients with Acute Coronary Syndrome Undergoing Percutaneous Coronary Intervention (TROPICAL-ACS): A Randomised, Open-Label, Multicentre Trial. Lancet 2017, 390, 1747–1757. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.-D.; Wang, W.; Yang, M.; Zhang, K.; Chen, J.; Qiao, S.; Yan, H.; Wu, Y.; Huang, X.; Xu, B.; et al. Randomized Comparisons of Double-Dose Clopidogrel or Adjunctive Cilostazol versus Standard Dual Antiplatelet in Patients with High Posttreatment Platelet Reactivity: Results of the CREATIVE Trial. Circulation 2018, 137, 2231–2245. [Google Scholar] [CrossRef] [PubMed]
- Podrez, E.A.; Byzova, T.V.; Febbraio, M.; Salomon, R.G.; Ma, Y.; Valiyaveettil, M.; Poliakov, E.; Sun, M.; Finton, P.J.; Curtis, B.R.; et al. Platelet CD36 Links Hyperlipidemia, Oxidant Stress and a Prothrombotic Phenotype. Nat. Med. 2007, 13, 1086–1095. [Google Scholar] [CrossRef]
- Togna, A.R.; Latina, V.; Orlando, R.; Togna, G.I. Cigarette Smoke Inhibits Adenine Nucleotide Hydrolysis by Human Platelets. Platelets 2008, 19, 537–542. [Google Scholar] [CrossRef]
- Hioki, H.; Aoki, N.; Kawano, K.; Homori, M.; Hasumura, Y.; Yasumura, T.; Maki, A.; Yoshino, H.; Yanagisawa, A.; Ishikawa, K. Acute Effects of Cigarette Smoking on Platelet-Dependent Thrombin Generation. Eur. Heart J. 2001, 22, 56–61. [Google Scholar] [CrossRef]
- Renaud, S.; Blache, D.; Dumont, E.; Thevenon, C.; Wissendanger, T. Platelet Function after Cigarette Smoking in Relation to Nicotine and Carbon Monoxide. Clin. Pharmacol. Ther. 1984, 36, 389–395. [Google Scholar] [CrossRef]
- Gremmel, T.; Steiner, S.; Seidinger, D.; Koppensteiner, R.; Panzer, S.; Kopp, C.W. Smoking Promotes Clopidogrel-Mediated Platelet Inhibition in Patients Receiving Dual Antiplatelet Therapy. Thromb. Res. 2009, 124, 588–591. [Google Scholar] [CrossRef]
- Crimi, G.; Somaschini, A.; Cattaneo, M.; Angiolillo, D.J.; Piscione, F.; Palmerini, T.; De Servi, S. Cigarette Smoking Reduces Platelet Reactivity Independently of Clopidogrel Treatment in Patients with Non-ST Elevation Acute Coronary Syndromes. Platelets 2018, 29, 309–311. [Google Scholar] [CrossRef]
- Nardin, M.; Verdoia, M.; Negro, F.; Rolla, R.; Tonon, F.; De Luca, G. Impact of Active Smoking on the Immature Platelet Fraction and Its Relationship with the Extent of Coronary Artery Disease. Eur. J. Clin. Investig. 2020, 50, e13181. [Google Scholar] [CrossRef]
- De Luca, G.; Dirksen, M.T.; Spaulding, C.; Kelbæk, H.; Schalij, M.; Thuesen, L.; van der Hoeven, B.; Vink, M.A.; Kaiser, C.; Musto, C.; et al. Impact of Diabetes on Long-Term Outcome after Primary Angioplasty. Diabetes Care 2013, 36, 1020–1025. [Google Scholar] [CrossRef] [PubMed]
- Wisman, P.P.; Roest, M.; Asselbergs, F.W.; de Groot, P.G.; Moll, F.L.; van der Graaf, Y.; de Borst, G.J. Platelet-Reactivity Tests Identify Patients at Risk of Secondary Cardiovascular Events: A Systematic Review and Meta-Analysis. J. Thromb. Haemost. 2014, 12, 736–747. [Google Scholar] [CrossRef] [PubMed]
- Nardin, M.; Verdoia, M.; Sartori, C.; Pergolini, P.; Rolla, R.; Barbieri, L.; Schaffer, A.; Bellomo, G.; Suryapranata, H.; De Luca, G. Diabetes Mellitus, Glucose Control Parameters and Platelet Reactivity in Ticagrelor Treated Patients. Thromb. Res. 2016, 143, 45–49. [Google Scholar] [CrossRef] [PubMed]
- Vivas, D.; Garcia-Rubira, J.C.; Bernardo, E.; Angiolillo, D.J.; Martin, P.; Calle-Pascual, A.; Nunez-Gil, I.; Macaya, C.; Fernandez-Ortiz, A. Effects of Intensive Glucose Control on Platelet Reactivity in Patients with Acute Coronary Syndromes. Results of the CHIPS Study (“Control de Hiperglucemia y Actividad Plaquetaria en Pacientes con Sindrome Coronario Agudo”). Heart 2011, 97, 803–809. [Google Scholar] [CrossRef][Green Version]
- Filiopoulos, V.; Hadjiyannakos, D.; Vlassopoulos, D. New Insights into Uric Acid Effects on the Progression and Prognosis of Chronic Kidney Disease. Ren. Fail. 2012, 34, 510–520. [Google Scholar] [CrossRef]
- Elbasan, Z.; Şahin, D.Y.; Gür, M.; Kuloğlu, O.; Kıvrak, A.; İçen, Y.K.; Türkoğlu, C.; Yıldırım, A.; Özdoğru, İ.; Çaylı, M. Contrast-Induced Nephropathy in Patients with ST Elevation Myocardial Infarction Treated with Primary Percutaneous Coronary Intervention. Angiology 2014, 65, 37–42. [Google Scholar] [CrossRef]
- Barbieri, L.; Verdoia, M.; Pergolini, P.; Nardin, M.; Rolla, R.; Marino, P.; Bellomo, G.; Suryapranata, H.; De Luca, G. Uric Acid and High-Residual Platelet Reactivity in Patients Treated with Clopidogrel or Ticagrelor. Nutr. Metab. Cardiovasc. Dis. 2016, 26, 352–358. [Google Scholar] [CrossRef]
- Giovannucci, E.; Liu, Y.; Hollis, B.W.; Rimm, E.B. 25-Hydroxyvitamin D and Risk of Myocardial Infarction in Men: A Prospective Study. Arch. Intern. Med. 2008, 168, 1174–1180. [Google Scholar] [CrossRef]
- Tarcin, O.; Yavuz, D.G.; Ozben, B.; Telli, A.; Ogunc, A.V.; Yuksel, M.; Toprak, A.; Yazici, D.; Sancak, S.; Deyneli, O.; et al. Effect of Vitamin D Deficiency and Replacement on Endothelial Function in Asymptomatic Subjects. J. Clin. Endocrinol. Metab. 2009, 94, 4023–4030. [Google Scholar] [CrossRef]
- Silvagno, F.; De Vivo, E.; Attanasio, A.; Gallo, V.; Mazzucco, G.; Pescarmona, G. Mitochondrial Localization of Vitamin D Receptor in Human Platelets and Differentiated Megakaryocytes. PLoS ONE 2010, 5, e8670. [Google Scholar] [CrossRef]
- Aihara, K.; Azuma, H.; Akaike, M.; Ikeda, Y.; Yamashita, M.; Sudo, T.; Hayashi, H.; Yamada, Y.; Endoh, F.; Fujimura, M.; et al. Disruption of Nuclear Vitamin D Receptor Gene Causes Enhanced Thrombogenicity in Mice. J. Biol. Chem. 2004, 279, 35798–35802. [Google Scholar] [CrossRef] [PubMed]
- López-Farré, A.J.; Mateos-Cáceres, P.J.; Sacristán, D.; Azcona, L.; Bernardo, E.; de Prada, T.P.; Alonso-Orgaz, S.; Fernández-Arquero, M.; Fernández-Ortiz, A.; Macaya, C. Relationship between Vitamin D Binding Protein and Aspirin Resistance in Coronary Ischemic Patients: A Proteomic Study. J. Proteome Res. 2007, 6, 2481–2487. [Google Scholar] [CrossRef] [PubMed]
- Verdoia, M.; Pergolini, P.; Rolla, R.; Sartori, C.; Nardin, M.; Schaffer, A.; Barbieri, L.; Daffara, V.; Marino, P.; Bellomo, G.; et al. Vitamin D Levels and High-Residual Platelet Reactivity in Patients Receiving Dual Antiplatelet Therapy with Clopidogrel or Ticagrelor. Platelets 2016, 27, 576–582. [Google Scholar] [CrossRef] [PubMed]
- Verdoia, M.; Daffara, V.; Pergolini, P.; Rolla, R.; Marino, P.; Bellomo, G.; Carriero, A.; De Luca, G.; Novara Atherosclerosis Study Group (NAS). Vitamin D Binding Protein Rs7041 Polymorphism and High-Residual Platelet Reactivity in Patients Receiving Dual Antiplatelet Therapy with Clopidogrel or Ticagrelor. Vascul. Pharmacol. 2017, 93–95, 42–47. [Google Scholar] [CrossRef]
- Michos, E.D.; Misialek, J.R.; Selvin, E.; Folsom, A.R.; Pankow, J.S.; Post, W.S.; Lutsey, P.L. 25-Hydroxyvitamin D Levels, Vitamin D Binding Protein Gene Polymorphisms and Incident Coronary Heart Disease among Whites and Blacks: The ARIC Study. Atherosclerosis 2015, 241, 12–17. [Google Scholar] [CrossRef]
- Sibbing, D.; Koch, W.; Gebhard, D.; Schuster, T.; Braun, S.; Stegherr, J.; Morath, T.; Schömig, A.; von Beckerath, N.; Kastrati, A. Cytochrome 2C19*17 Allelic Variant, Platelet Aggregation, Bleeding Events, and Stent Thrombosis in Clopidogrel-Treated Patients with Coronary Stent Placement. Circulation 2010, 121, 512–518. [Google Scholar] [CrossRef]
- Mega, J.L.; Simon, T.; Collet, J.-P.; Anderson, J.L.; Antman, E.M.; Bliden, K.; Cannon, C.P.; Danchin, N.; Giusti, B.; Gurbel, P.; et al. Reduced-Function CYP2C19 Genotype and Risk of Adverse Clinical Outcomes among Patients Treated with Clopidogrel Predominantly for PCI: A Meta-Analysis. JAMA 2010, 304, 1821–1830. [Google Scholar] [CrossRef]
- Simon, T.; Verstuyft, C.; Mary-Krause, M.; Quteineh, L.; Drouet, E.; Méneveau, N.; Steg, P.G.; Ferrières, J.; Danchin, N.; Becquemont, L.; et al. Genetic Determinants of Response to Clopidogrel and Cardiovascular Events. N. Engl. J. Med. 2009, 360, 363–375. [Google Scholar] [CrossRef]
- Mega, J.L.; Hochholzer, W.; Frelinger, A.L.; Kluk, M.J.; Angiolillo, D.J.; Kereiakes, D.J.; Isserman, S.; Rogers, W.J.; Ruff, C.T.; Contant, C.; et al. Dosing Clopidogrel Based on CYP2C19 Genotype and the Effect on Platelet Reactivity in Patients with Stable Cardiovascular Disease. JAMA 2011, 306, 2221–2228. [Google Scholar] [CrossRef]
- Carreras, E.T.; Hochholzer, W.; Frelinger, A.L.; Nordio, F.; O’Donoghue, M.L.; Wiviott, S.D.; Angiolillo, D.J.; Michelson, A.D.; Sabatine, M.S.; Mega, J.L. Diabetes Mellitus, CYP2C19 Genotype, and Response to Escalating Doses of Clopidogrel. Insights from the ELEVATE-TIMI 56 Trial. Thromb. Haemost. 2016, 116, 69–77. [Google Scholar] [CrossRef]
- Scott, S.A.; Sangkuhl, K.; Stein, C.M.; Hulot, J.-S.; Mega, J.L.; Roden, D.M.; Klein, T.E.; Sabatine, M.S.; Johnson, J.A.; Shuldiner, A.R.; et al. Clinical Pharmacogenetics Implementation Consortium Guidelines for CYP2C19 Genotype and Clopidogrel Therapy: 2013 Update. Clin. Pharmacol. Ther. 2013, 94, 317–323. [Google Scholar] [CrossRef] [PubMed]
- Royal Dutch Pharmacists Association (KNMP); Dutch Pharmacogenetics Working Group (DPWG). Pharmacogenetic Guidelines. Netherlands. Clopidogrel—CYP2C19. Available online: http://kennisbank.knmp.nl (accessed on 1 May 2023).
- Pereira, N.L.; Farkouh, M.E.; So, D.; Lennon, R.; Geller, N.; Mathew, V.; Bell, M.; Bae, J.-H.; Jeong, M.H.; Chavez, I.; et al. Effect of Genotype-Guided Oral P2Y12 Inhibitor Selection vs. Conventional Clopidogrel Therapy on Ischemic Outcomes after Percutaneous Coronary Intervention. JAMA 2020, 324, 761. [Google Scholar] [CrossRef] [PubMed]
- Tavenier, A.H.; Mehran, R.; Chiarito, M.; Cao, D.; Pivato, C.A.; Nicolas, J.; Beerkens, F.; Nardin, M.; Sartori, S.; Baber, U.; et al. Guided and Unguided De-Escalation from Potent P2Y12 Inhibitors among Patients with Acute Coronary Syndrome: A Meta-Analysis. Eur. Heart J. Cardiovasc. Pharmacother. 2022, 8, 492–502. [Google Scholar] [CrossRef] [PubMed]
- Mega, J.L.; Close, S.L.; Wiviott, S.D.; Shen, L.; Hockett, R.D.; Brandt, J.T.; Walker, J.R.; Antman, E.M.; Macias, W.L.; Braunwald, E.; et al. Cytochrome P450 Genetic Polymorphisms and the Response to Prasugrel: Relationship to Pharmacokinetic, Pharmacodynamic, and Clinical Outcomes. Circulation 2009, 119, 2553–2560. [Google Scholar] [CrossRef]
- Varenhorst, C.; Eriksson, N.; Johansson, Å.; Barratt, B.J.; Hagström, E.; Åkerblom, A.; Syvänen, A.-C.; Becker, R.C.; James, S.K.; Katus, H.A.; et al. Effect of Genetic Variations on Ticagrelor Plasma Levels and Clinical Outcomes. Eur. Heart J. 2015, 36, 1901–1912. [Google Scholar] [CrossRef]
- Nardin, M.; Verdoia, M.; Pergolini, P.; Rolla, R.; Barbieri, L.; Marino, P.; Bellomo, G.; Kedhi, E.; Suryapranata, H.; Carriero, A.; et al. Impact of Adenosine A2a Receptor Polymorphism Rs5751876 on Platelet Reactivity in Ticagrelor Treated Patients. Pharmacol. Res. 2018, 129, 27–33. [Google Scholar] [CrossRef]
- Laffont, B.; Corduan, A.; Plé, H.; Duchez, A.-C.; Cloutier, N.; Boilard, E.; Provost, P. Activated Platelets Can Deliver MRNA Regulatory Ago2•microRNA Complexes to Endothelial Cells via Microparticles. Blood 2013, 122, 253–261. [Google Scholar] [CrossRef]
- Kaudewitz, D.; Skroblin, P.; Bender, L.H.; Barwari, T.; Willeit, P.; Pechlaner, R.; Sunderland, N.P.; Willeit, K.; Morton, A.C.; Armstrong, P.C.; et al. Association of MicroRNAs and YRNAs with Platelet Function. Circ. Res. 2016, 118, 420–432. [Google Scholar] [CrossRef]
- Willeit, P.; Zampetaki, A.; Dudek, K.; Kaudewitz, D.; King, A.; Kirkby, N.S.; Crosby-Nwaobi, R.; Prokopi, M.; Drozdov, I.; Langley, S.R.; et al. Circulating MicroRNAs as Novel Biomarkers for Platelet Activation. Circ. Res. 2013, 112, 595–600. [Google Scholar] [CrossRef]
- Nagalla, S.; Shaw, C.; Kong, X.; Kondkar, A.A.; Edelstein, L.C.; Ma, L.; Chen, J.; McKnight, G.S.; López, J.A.; Yang, L.; et al. Platelet MicroRNA-MRNA Coexpression Profiles Correlate with Platelet Reactivity. Blood 2011, 117, 5189–5197. [Google Scholar] [CrossRef]
- Landry, P.; Plante, I.; Ouellet, D.L.; Perron, M.P.; Rousseau, G.; Provost, P. Existence of a MicroRNA Pathway in Anucleate Platelets. Nat. Struct. Mol. Biol. 2009, 16, 961–966. [Google Scholar] [CrossRef] [PubMed]
- Elgheznawy, A.; Shi, L.; Hu, J.; Wittig, I.; Laban, H.; Pircher, J.; Mann, A.; Provost, P.; Randriamboavonjy, V.; Fleming, I. Dicer Cleavage by Calpain Determines Platelet MicroRNA Levels and Function in Diabetes. Circ. Res. 2015, 117, 157–165. [Google Scholar] [CrossRef]
- Fejes, Z.; Póliska, S.; Czimmerer, Z.; Káplár, M.; Penyige, A.; Gál Szabó, G.; Debreceni, I.B.; Kunapuli, S.P.; Kappelmayer, J.; Nagy, B. Hyperglycaemia Suppresses MicroRNA Expression in Platelets to Increase P2RY12 and SELP Levels in Type 2 Diabetes Mellitus. Thromb. Haemost. 2017, 117, 529–542. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Liu, J.; Qin, L.; Liu, J.; Xi, S.; Lu, C.; Yin, T. Interaction between Platelet-Derived MicroRNAs and CYP2C19*2 Genotype on Clopidogrel Antiplatelet Responsiveness in Patients with ACS. Thromb. Res. 2017, 157, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Barwari, T.; Eminaga, S.; Mayr, U.; Lu, R.; Armstrong, P.C.; Chan, M.V.; Sahraei, M.; Fernández-Fuertes, M.; Moreau, T.; Barallobre-Barreiro, J.; et al. Inhibition of Profibrotic MicroRNA-21 Affects Platelets and Their Releasate. JCI Insight 2018, 3, e123335. [Google Scholar] [CrossRef]
- Wendel Garcia, P.D.; Fumeaux, T.; Guerci, P.; Heuberger, D.M.; Montomoli, J.; Roche-Campo, F.; Schuepbach, R.A.; Hilty, M.P.; Alfaro Farias, M.; Margarit, A.; et al. Prognostic Factors Associated with Mortality Risk and Disease Progression in 639 Critically Ill Patients with COVID-19 in Europe: Initial Report of the International RISC-19-ICU Prospective Observational Cohort. eClinicalMedicine 2020, 25, 100449. [Google Scholar] [CrossRef]
- Zapilko, V.; Fish, R.J.; Garcia, A.; Reny, J.-L.; Dunoyer-Geindre, S.; Lecompte, T.; Neerman-Arbez, M.; Fontana, P. MicroRNA-126 Is a Regulator of Platelet-Supported Thrombin Generation. Platelets 2020, 31, 746–755. [Google Scholar] [CrossRef]
- Becker, K.C.; Kwee, L.C.; Neely, M.L.; Grass, E.; Jakubowski, J.A.; Fox, K.A.A.; White, H.D.; Gregory, S.G.; Gurbel, P.A.; Carvalho, L.d.P.; et al. Circulating MicroRNA Profiling in Non-ST Elevated Coronary Artery Syndrome Highlights Genomic Associations with Serial Platelet Reactivity Measurements. Sci. Rep. 2020, 10, 6169. [Google Scholar] [CrossRef]
- Braza-Boïls, A.; Barwari, T.; Gutmann, C.; Thomas, M.R.; Judge, H.M.; Joshi, A.; Pechlaner, R.; Shankar-Hari, M.; Ajjan, R.A.; Sabroe, I.; et al. Circulating MicroRNA Levels Indicate Platelet and Leukocyte Activation in Endotoxemia Despite Platelet P2Y12 Inhibition. Int. J. Mol. Sci. 2020, 21, 2897. [Google Scholar] [CrossRef]
- Tsui, N.B.Y.; Ng, E.K.O.; Lo, Y.M.D. Stability of Endogenous and Added RNA in Blood Specimens, Serum, and Plasma. Clin. Chem. 2002, 48, 1647–1653. [Google Scholar] [CrossRef]
- Gutmann, C.; Mayr, M. Circulating MicroRNAs as Biomarkers and Mediators of Platelet Activation. Platelets 2022, 33, 512–519. [Google Scholar] [CrossRef]
- Jansen, F.; Yang, X.; Proebsting, S.; Hoelscher, M.; Przybilla, D.; Baumann, K.; Schmitz, T.; Dolf, A.; Endl, E.; Franklin, B.S.; et al. MicroRNA Expression in Circulating Microvesicles Predicts Cardiovascular Events in Patients with Coronary Artery Disease. J. Am. Heart Assoc. 2014, 3, e001249. [Google Scholar] [CrossRef]
- Zampetaki, A.; Willeit, P.; Tilling, L.; Drozdov, I.; Prokopi, M.; Renard, J.-M.; Mayr, A.; Weger, S.; Schett, G.; Shah, A.; et al. Prospective Study on Circulating MicroRNAs and Risk of Myocardial Infarction. J. Am. Coll. Cardiol. 2012, 60, 290–299. [Google Scholar] [CrossRef]
- Widera, C.; Gupta, S.K.; Lorenzen, J.M.; Bang, C.; Bauersachs, J.; Bethmann, K.; Kempf, T.; Wollert, K.C.; Thum, T. Diagnostic and Prognostic Impact of Six Circulating MicroRNAs in Acute Coronary Syndrome. J. Mol. Cell. Cardiol. 2011, 51, 872–875. [Google Scholar] [CrossRef]
- Capodanno, D.; Mehran, R.; Krucoff, M.W.; Baber, U.; Bhatt, D.L.; Capranzano, P.; Collet, J.-P.; Cuisset, T.; De Luca, G.; De Luca, L.; et al. Defining Strategies of Modulation of Antiplatelet Therapy in Patients with Coronary Artery Disease: A Consensus Document from the Academic Research Consortium. Circulation 2023, 147, 1933–1944. [Google Scholar] [CrossRef] [PubMed]
- De Servi, S.; Landi, A.; Savonitto, S.; De Luca, L.; De Luca, G.; Morici, N.; Montalto, C.; Crimi, G.; Cattaneo, M. Tailoring Oral Antiplatelet Therapy in Acute Coronary Syndromes: From Guidelines to Clinical Practice. J. Cardiovasc. Med. 2023, 24, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Costa, F.; Montalto, C.; Branca, M.; Hong, S.-J.; Watanabe, H.; Franzone, A.; Vranckx, P.; Hahn, J.-Y.; Gwon, H.-C.; Feres, F.; et al. Dual Antiplatelet Therapy Duration after Percutaneous Coronary Intervention in High Bleeding Risk: A Meta-Analysis of Randomized Trials. Eur. Heart J. 2023, 44, 954–968. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Methods | Sample | Application | Principle | Advantages | Disadvantages |
|---|---|---|---|---|---|
| Tests based on platelet aggregation | |||||
| Light transmission platelet aggregation (LTA); also named optical platelet aggregation | Citrated PRP | Screening test for bleeding propensity Diagnostic for platelet defects, both congenital and acquired Monitoring antiplatelet treatment effect | Photo-optical measurement of light transmission increase in relation to specific agonist-induced platelet aggregation | Historical gold standard Diagnostic method Different platelet pathways investigated Sensitive to different antiplatelet drugs therapy | Manual and long sample processing Pre-analytic and analytic variables High sample volume Time consuming |
| Impedance platelet aggregation | Citrated WB | Screening test for bleeding propensity Diagnostic for platelet defects, both congenital and acquired Monitoring antiplatelet treatment effect | Measurement of electrical impedance between two electrodes after induction of platelet aggregation through a specific agonist | No sample processing Diagnostic method Flexible Different platelet pathways investigated Sensitive to anti-platelet therapy Close to LTA | Sample preparation Time consuming |
| Lumiaggregometry (i.e., VerifyNow) | Citrated WB | Modified aggregometry for detection of storage/release disorders | LTA or WB aggregometry combined with luminescence | No WB processing Quick and easy to do methods Monitoring antiplatelet therapy | Nonflexible Very expensive Limited hematocrit and platelet count |
| Plateletworks | Citrated WB | Monitoring of the platelet response to antiplatelet agents | Platelets’ counting and aggregates pre- and post-activation (use of ADP or arachidonic acid) in whole blood based in GP IIb/IIIa-dependent aggregation | Minimal sample preparation Easy, rapid screening test | Indirect assay Performance within few minutes after sample collection Required adjunctive platelet count Scarce clinical data |
| Platelet function methods combined with viscoelastic test | |||||
| TEG/r-TEG platelet mapping system | Citrated WB | Assessment of global hemostasis plus monitoring antiplatelet treatments effect | Assessment of the rate and the strength of clot formation based on low shear-induced and agonist addition | Point of care for viscoelastic test Global hemostasis test Measure clot properties Reduces blood transfusions | More studies are needed |
| ROTEM platelet | Citrated WB | Assessment of global hemostasis plus diagnostic of platelet defects plus monitoring antiplatelet treatments effect | Measurement of electrical impedance increase in relation to agonist-induced platelet aggregation | Adaptation from TEG: results are identical Predicts bleeding Reduces blood transfusions Improves clinical outcome Global hemostasis test WB platelet aggregometry | Limited hematocrit and platelet count range (for platelet system) Lack of clinical studies |
| Tests based on platelet adhesion under shear stress | |||||
| PFA-100; Innovance PFA-200 | Citrated WB | Assessment of bleeding risk and drug effects Searching severe platelet dysfunctions, revealing of vWF disease | Time evaluation of high shear WB flow blocked by platelet plug into a hole in activated surface. Use of combination of collagen/epinephrine or collagen/ADP | In vitro standardized bleeding test Quick and easy to do Sensitive to severe platelet defects | Rigid closed system Dependent on hematocrit, platelet count and vWF Not sensitive to platelet granules defects Contrasting evidence in thienopyridines treatment, especially for PFA-100 |
| IMPACT—Cone and Plate(let) Analyzer (CPA) | Citrated WB | Screening of primary hemostasis and platelet defects | Shear-induced platelet adhesion–aggregation upon specific surface covered by polystyrene. Ongoing studies on addition ADP and arachidonic acid for antiplatelet therapy monitoring | Global platelet method Small sample volume | Expensive Experienced staff Lack of clinical studies Not widely available |
| Platelet analysis based on flow cytometry | |||||
| Flow cytometry | Citrated WB, PRP, washed platelets | Cell counting, detection platelet activation by extent of expression of surface and/or cytoplasmic biomarkers | Engineering laser-based detection of suspending fluorescent label platelets in a flowing solution | Useful into diagnose inherited platelet disorders | Expensive Experienced staff Not widely available |
| Vasodilator Stimulated Phosphoprotein (VASP) | Citrated WB | Intracellular platelet pathway | Immunofluorescence on assay with a specific monoclonal antibody | Useful into monitoring antiplatelet drug | Expensive |
| Evaluation of Thromboxane metabolites | |||||
| Radio- or enzyme-linked immune assays | Serum, urine, citrated plasma | Measurement of TXA2 metabolites (and β-TG, PF4, soluble P-selectin) | Ligand-binding assays | Directly related to COX-1, the aspirin’s target | Indirect measure No platelet specific |
| Study | Year | PFT | Intervention | Subjects | Study design | Findings |
| VASP-02 [111] | 2008 | VASP | VASP-guided switch after 2 weeks in low responders | 150 patients undergoing elective PCI | Randomized to 150 mg vs. 75 mg clopidogrel | Greater platelet inhibition with 150 mg clopidogrel in poor responders |
| Bonello et al. [112] | 2008 | VASP | VASP-based adjustment of clopidogrel loading dose | 162 patients undergoing PCI with basal VASP > 50% | Randomized to VASP-guided vs. standard of care | Lower rate of 1-month MACE in VASP-guided group |
| Cuisset et al. [113] | 2008 | LTA | PFT-based identification of poor responder before randomization | 149 clopidogrel non-responders undergoing elective PCI | Randomized to administration of additional GP IIb/IIIa antagonist vs. standard of care | Lower rate of 1-month CV event in intervention group |
| 3T/2R [114] | 2009 | VerifyNow | PFT-based identification of poor responder before randomization | 263 ASA and/or clopidogrel non-responders undergoing PCI | Randomized to tirofiban administration vs. standard of care | Lower MI and 1-months MACE in intervention group |
| Wang et al. [115] | 2011 | VASP | PFT-based drug adjustment in tailored strategy | 306 patients undergoing PCI with basal VASP > 50% | Randomized to VASP-guided vs. standard of care | Lower rate of 1-year MACE in VASP-guided group |
| GRAVITAS [116] | 2011 | VerifyNow | PFT-based identification of poor responder before randomization | 2796 clopidogrel non-responder patients undergoing PCI | Randomized to higher dose of clopidogrel vs. standard of care | No differences in 6-month MACE rate |
| Aradi et al. [117] | 2012 | LTA | PFT-based identification of poor responder before randomization | 200 clopidogrel non-responders undergoing elective PCI | Randomized to higher dose of clopidogrel vs. standard of care | Lower MI and 1-months MACE in intervention group |
| Hazarbasanov et al. [118] | 2012 | Impedance aggregometry | PFT-based drug adjustment in tailored strategy | 192 patients undergoing PCI | Randomization to tailored strategy vs. standard of care | Lower rate of 6-months MACCE in intervention group |
| TRIGGER-PCI [119] | 2012 | VerifyNow | PFT-based identification of poor responder before randomization | 423 clopidogrel non-responders undergoing PCI | Randomized to switch to prasugrel vs. clopidogrel maintenance | Higher platelet inhibition in prasugrel group. |
| ARTIC [120] | 2012 | VerifyNow | PFT-based administration of additional bolus of clopidogrel, prasugrel or ASA along with GP IIb/IIIa antagonist | 2440 patients undergoing elective PCI | Randomization to tailored strategy vs. standard of care | No differences in 1-year MACE |
| MADONNA study [104] | 2013 | Impedance aggregometry | PFT-based additional antiplatelet drug loading dose | 798 patients undergoing PCI | Randomization to tailored strategy vs. standard of care | Lower stent thrombosis and ACS rate in tailored strategy |
| ANTARTIC [121] | 2016 | VerifyNow | PFT-based dose or drug adjustment in tailored strategy | 877 patients undergoing PCI for an ACS | Randomization to tailored strategy vs. standard of care | No differences in 1-year MACE |
| TROPICAL-ACS [122] | 2017 | Impedance aggregometry | PFT-based de-escalation strategy after 14 days | 2610 patients undergoing PCI for an ACS | Randomization to tailored strategy vs. standard of care | Tailored de-escalation strategy non-inferior to standard of care |
| CREATIVE [123] | 2018 | TEG | PFT-based identification of poor responder before randomization | 1087 clopidogrel poor responders undergoing PCI | Randomized to higher dose of clopidogrel vs. standard dose of clopidogrel plus cilostazol vs. standard of care | Lower rate of 18-months MACCE with the adjunct of cilostazol |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nardin, M.; Verdoia, M.; Cao, D.; Nardin, S.; Kedhi, E.; Galasso, G.; van ‘t Hof, A.W.J.; Condorelli, G.; De Luca, G. Platelets and the Atherosclerotic Process: An Overview of New Markers of Platelet Activation and Reactivity, and Their Implications in Primary and Secondary Prevention. J. Clin. Med. 2023, 12, 6074. https://doi.org/10.3390/jcm12186074
Nardin M, Verdoia M, Cao D, Nardin S, Kedhi E, Galasso G, van ‘t Hof AWJ, Condorelli G, De Luca G. Platelets and the Atherosclerotic Process: An Overview of New Markers of Platelet Activation and Reactivity, and Their Implications in Primary and Secondary Prevention. Journal of Clinical Medicine. 2023; 12(18):6074. https://doi.org/10.3390/jcm12186074
Chicago/Turabian StyleNardin, Matteo, Monica Verdoia, Davide Cao, Simone Nardin, Elvin Kedhi, Gennaro Galasso, Arnoud W. J. van ‘t Hof, Gianluigi Condorelli, and Giuseppe De Luca. 2023. "Platelets and the Atherosclerotic Process: An Overview of New Markers of Platelet Activation and Reactivity, and Their Implications in Primary and Secondary Prevention" Journal of Clinical Medicine 12, no. 18: 6074. https://doi.org/10.3390/jcm12186074
APA StyleNardin, M., Verdoia, M., Cao, D., Nardin, S., Kedhi, E., Galasso, G., van ‘t Hof, A. W. J., Condorelli, G., & De Luca, G. (2023). Platelets and the Atherosclerotic Process: An Overview of New Markers of Platelet Activation and Reactivity, and Their Implications in Primary and Secondary Prevention. Journal of Clinical Medicine, 12(18), 6074. https://doi.org/10.3390/jcm12186074