
Vogt-Koyanagi-Harada Disease and COVID


Abstract
:1. Background
2. Epidemiology
3. Pathogenesis
- A direct eye infection caused by a live vaccine may result in intraocular inflammation, especially in immunosuppressed patients. An example of this mechanism is the BCG vaccination against tuberculosis (TB): an in vitro model has shown that BCG can infect the retinal pigment epithelium (RPE) cells. Moreover, the effectiveness of antibiotic therapy in these forms of uveitis supports this hypothesis.
- A molecular mimicry between vaccine particles and ocular structures results in antigen-specific cell and antibody-mediated hypersensitivity reaction.
- A vaccine’s adjuvant or additives may provoke autoimmune uveitis. Adjuvants and additives enhance the host’s innate and adaptive immune response to vaccines. The immune system is directly activated against adjuvants, causing ocular inflammation and a series of clinical manifestations known as ‘Shoenfeld syndrome’, which is characterised by extraocular symptoms, such as myalgia or arthralgia, and has been described after some vaccines such as HPV, measles, mumps and rubella (MMR), influenza, diphtheria-tetanus-pertussis and BCG [27].
4. Clinical Features
- Prodromal stage: This usually lasts for 3–5 days and is characterised by nonspecific symptoms, such as malaise, fever, nausea, headache, meningismus, dizziness and orbital pain, which are sometimes followed by neurological symptoms such as cranial nerve palsies, hemiparesis, transverse myelitis and optic neuritis. During this stage, the patient may also report photophobia and tearing, as well as hair and scalp hypersensitivity [29,30].
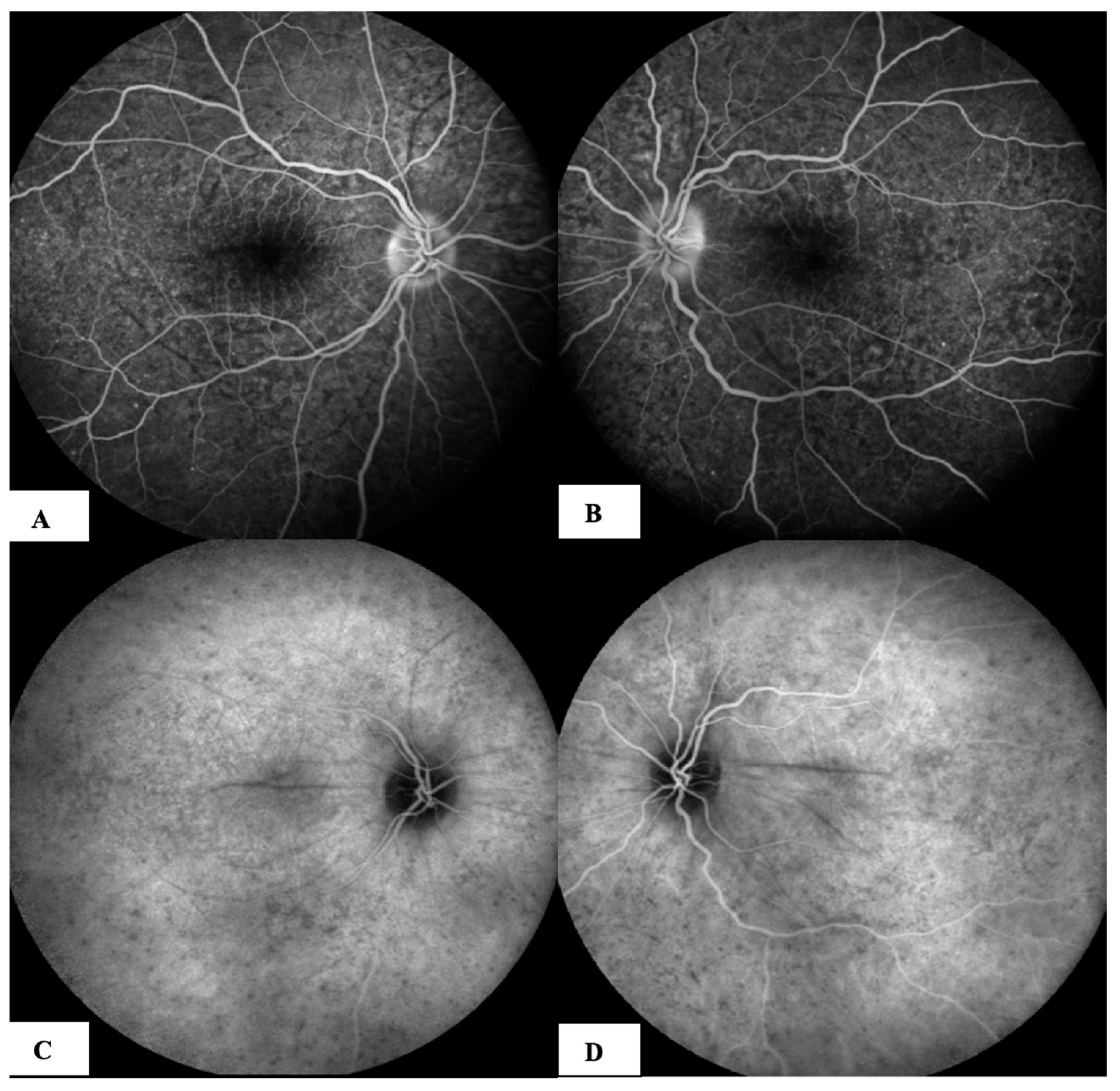
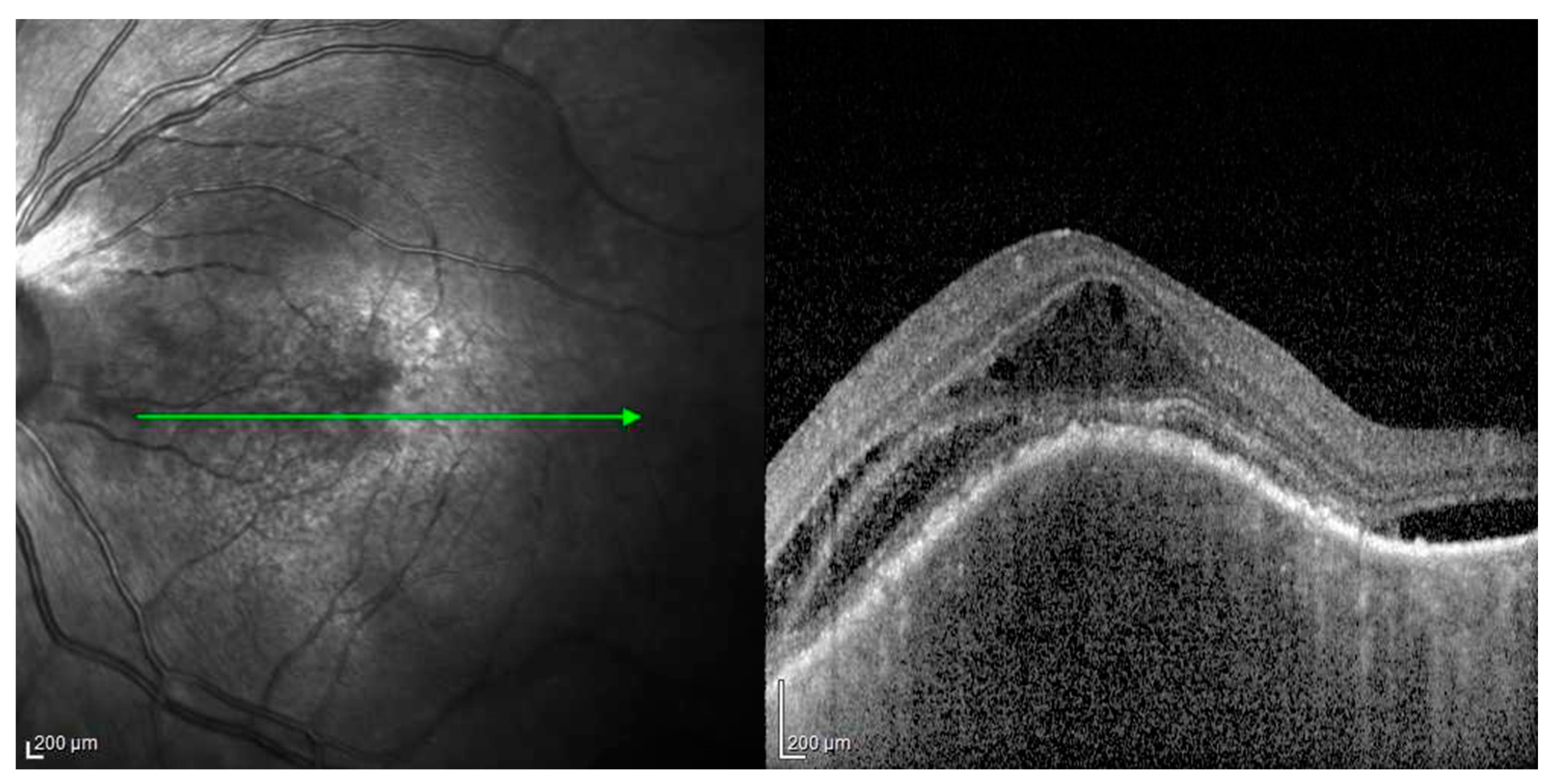
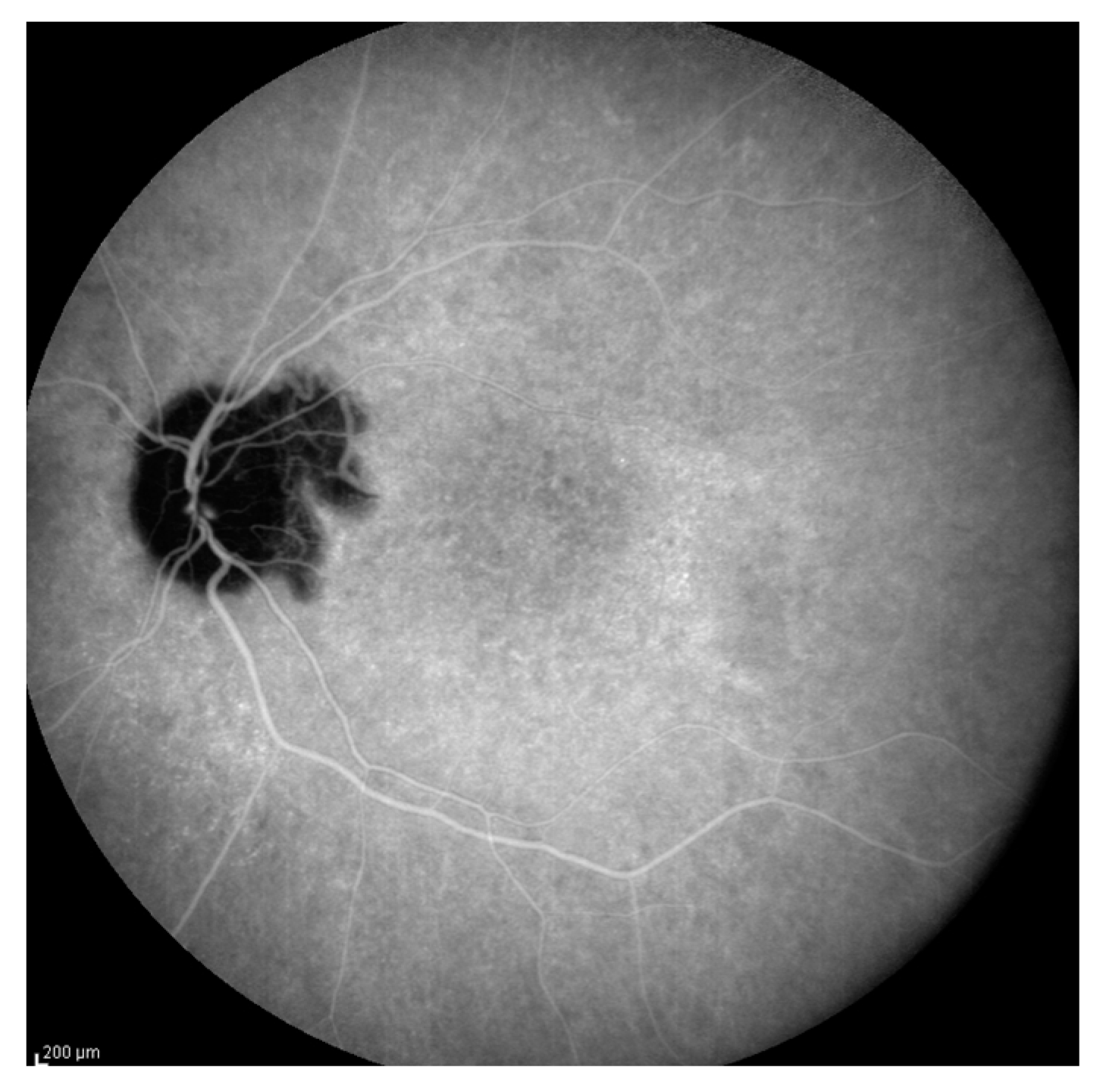
- Acute uveitic stage: This appears a few days after the prodromal phase and lasts for several weeks. During this period, the patient mainly complains of blurred vision, pain and central scotoma, and most patients present with bilateral posterior uveitis. The first sign is the thickening of the posterior choroid manifested as an elevation of the peripapillary retinochoroidal layer, hyperaemia and oedema of the optic disc [31,32] and circumscribed retinal oedema. The choroidal inflammation eventually becomes multifocal with a diffuse breakdown of the RPE causing serous localised elevation of the retina that can rapidly become confluent, leading to a diffuse serous retinal detachment (SRD) [33]. The anterior segment of the eye can be affected immediately after the aforementioned clinical signs in untreated patients. It can be characterised by acute bilateral granulomatous iridocyclitis, mutton-fat keratic precipitates, iris nodules and anterior chamber shallowing due to ciliary body oedema that may lead to acute angle-closure glaucoma.
- Chronic (or convalescent) stage: This lasts for months or even years and results in integumentary and uveal depigmentation. Vitiligo is usually symmetrical, mainly involving the face, eyelids and trunk [1]. A slit-lamp examination may also reveal perilimbal depigmentation, as described by Sugiura, which occurs in the first month after the onset of uveitis and is mainly seen in Japanese subjects (Sugiura’s sign) [34]. During this stage, RPE scars appear in the mid-periphery of the retina. These multiple and well-defined hypopigmented lesions, sometimes surrounded by pigment, express the clinical evolution of Dalen–Fuchs nodules [34]. The natural course of the disease, especially in dark-skinned patients, is characterised by a diffuse pigment loss with a significant colour change in the fundus, which assumes a light orange-reddish appearance called ‘sunset glow fundus.’
- Chronic recurrent stage: This manifests as recurrent, mainly anterior granulomatous uveitis. Nevertheless, a thorough examination of the choroid at this stage with indocyanine green angiography (ICGA) or enhanced-depth imaging (EDI) and optical coherence tomography (OCT) might also find signs of active choroiditis [35]. These episodes of granulomatous uveitis are often resistant to corticosteroid therapy and may be characterised by iris mnodules, focal pigment atrophy of the iris and ocular hypotony. This is the stage where the complications of chronic inflammation, such as glaucoma, cataracts, neovascularisation of the retina and disc, subretinal fibrosis and subretinal neovascularisation, usually develop [36].
5. Extra-Ocular Manifestations
6. Diagnosis
7. Therapy
8. Prognosis
9. VKH Disease and COVID Infection
10. VKH Disease and COVID-19 Vaccines
11. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rao, N.A.; Gupta, A.; Dustin, L.; Chee, S.P.; Okada, A.A.; Khairallah, M.; Bodaghi, B.; Lehoang, P.; Accorinti, M.; Mochizuki, M.; et al. Frequency of Distinguishing Clinical Features in Vogt-Koyanagi-Harada Disease. Ophthalmology 2010, 117, 591–599.e1. [Google Scholar] [CrossRef] [PubMed]
- Pivetti-Pezzi, P.; Accorinti, M.; Colabelli-Gisoldi, R.A.; Pirraglia, M.P. Vogt-Koyanagi-Harada Disease and HLA Type in Italian Patients. Am. J. Ophthalmol. 1996, 122, 889–891. [Google Scholar] [CrossRef] [PubMed]
- Martin, T.D.; Rathinam, S.R.; Cunningham, E.T. Prevalence, clinical characteristics, and causes of vision loss in children with Vogt-Koyanagi-Harada disease in South India. Retina 2010, 30, 1113–1121. [Google Scholar] [CrossRef] [PubMed]
- Hamade, I.H.; Shamsi, H.N.A.; Dhibi, H.A.; Chacra, C.B.; El-Asrar, A.M.A.; Tabbara, K.F. Uveitis survey in children. Br. J. Ophthalmol. 2009, 93, 569–572. [Google Scholar] [CrossRef]
- Ikeda, N.; Hayasaka, S.; Hayasaka, Y. Uveitis and pseudouveitis presenting for the first time in Japanese elderly patients. Ophtalmologica 2005, 219, 263–266. [Google Scholar] [CrossRef]
- Kiyomoto, C.; Imaizumi, M.; Kimoto, K.; Abe, H.; Nakano, S.; Nakatsuka, K. Vogt-Koyanagi-Harada disease in elderly Japanese patients. Int. Ophthalmol. 2007, 27, 149–153. [Google Scholar] [CrossRef]
- Pivetti-Pezzi, P.; Accorinti, M.; Pirraglia, M.P.; La Cava, M.; Corradi, R. Vogt-Koyanagi-Harada disease in Italian patients. In Uveitis in the Third Millenium; Doods, E.M., Couto, C.A., Eds.; Elsevier Science Publisher: Amsterdam, The Netherlands, 2000; pp. 163–166. [Google Scholar]
- Lavezzo, M.M.; Sakata, V.M.; Morita, C.; Rodriguez, E.E.C.; Abdallah, S.F.; da Silva, F.T.G.; Hirata, C.E.; Yamamoto, J.H. Vogt-Koyanagi-Harada disease: Review of a rare autoimmune disease targeting antigens of melanocytes. Orphanet J. Rare Dis. 2016, 11, 29. [Google Scholar] [CrossRef]
- Davis, J.L.; Mittal, K.K.; Freidlin, V.; Mellow, S.R.; Optican, D.C.; Palestine, A.G.; Nussenblatt, R.B. HLA associations and ancestry in Vogt-Koyanagi-Harada disease and sympathetic ophthalmia. Ophthalmology. Ophthalmology 1990, 97, 1137–1142. [Google Scholar] [CrossRef]
- Islam, S.M.; Numaga, J.; Fujino, Y.; Hirata, R.; Matsuki, K.; Maeda, H.; Masuda, K. HLA class II genes in Vogt-Koyanagi-Harada disease. Investig. Ophthalmol. Vis. Sci. 1994, 35, 3890–3896. [Google Scholar]
- Weisz, J.M.; Holland, G.N.; Roer, L.N.; Park, M.S.; Yuge, A.J.; Moorthy, R.S.; Forster, D.J.; Rao, N.A.; Terasaki, P.I. Association between Vogt-Koyanagi-Harada syndrome and HLA-DR1 and -DR4 in Hispanic patients living in southern California. Ophthalmology 1995, 102, 1012–1015. [Google Scholar] [CrossRef]
- Hou, S.; Yang, P.; Du, L.; Zhou, H.; Lin, X.; Liu, X.; Kijlstra, A. Small ubiquitin-like modifier 4 (SUMO4) polymorphisms and Vogt-Koyanagi-Harada (VKH) syndrome in the Chinese Han population. Mol. Vis. 2008, 14, 2597–2603. [Google Scholar] [PubMed]
- Tiercy, J.M.; Rathinam, S.R.; Gex-Fabry, M.; Baglivo, E. A shared HLA-DRB1 epitope in the DR beta first domain is associated with Vogt-Koyanagi-Harada syndrome in Indian patients. Mol. Vis. 2010, 16, 353–358. [Google Scholar] [PubMed]
- Shindo, Y.; Inoko, H.; Yamamoto, T.; Ohno, S. HLA-DRB1 typing of Vogt-Koyanagi-Harada’s disease by PCR-RFLP and the strong association with DRB1*0405 and DRB1*0410. Br. J. Ophthalmol. 1994, 78, 223–226. [Google Scholar] [CrossRef] [PubMed]
- Yamaki, K.; Gocho, K.; Hayakawa, K.; Kondo, I.; Sakuragi, S. Tyrosinase Family Proteins Are Antigens Specific to Vogt-Koyanagi-Harada Disease. J. Immunol. 2000, 165, 7323–7329. [Google Scholar] [CrossRef]
- Norose, K.; Yano, A. Melanoma specific Th1 cytotoxic T lymphocyte lines in Vogt-Koyanagi-Harada disease. Br. J. Ophthalmol. 1996, 80, 1002–1008. [Google Scholar] [CrossRef]
- Sugita, S.; Sagawa, K.; Mochizuki, M.; Shichijo, S.; Itoh, K. Melanocyte lysis by cytotoxic T lymphocytes recognizing the MART-1 melanoma antigen in HLA-A2 patients with Vogt–Koyanagi–Harada disease. Int. Immunol. 1996, 8, 799–803. [Google Scholar] [CrossRef]
- Sood, A.B.; O’keefe, G.; Bui, D.; Jain, N. Vogt-Koyanagi-Harada Disease Associated with Hepatitis B Vaccination. Ocul. Immunol. Inflamm. 2019, 27, 524–527. [Google Scholar] [CrossRef]
- Touitou, V.; Bodaghi, B.; Cassoux, N.; Tran, T.H.C.; Rao, N.A.; Cacoub, P.; LeHoang, P. Vogt-Koyanagi-Harada disease in patients with chronic hepatitis C. Am. J. Ophthalmol. 2005, 140, 949–952. [Google Scholar] [CrossRef]
- Sugita, S.; Takase, H.; Kawaguchi, T.; Taguchi, C.; Mochizuki, M. Cross-reaction between tyrosinase peptides and cytomegalovirus antigen by T cells from patients with Vogt-Koyanagi-Harada disease. Int. Ophthalmol. 2007, 27, 87–95. [Google Scholar] [CrossRef]
- Bassili, S.S.; Peyman, G.A.; Gebhardt, B.M.; Daun, M.; Ganiban, G.J.; Rifai, A. Detection of Epstein-Barr virus DNA by polymerase chain reaction in the vitreous from a patient with Vogt-Koyanagi-Harada syndrome. Retina 1996, 16, 160–161. [Google Scholar] [CrossRef]
- Campos, W.R.; Cenachi, S.P.F.; Soares, M.S.; Gonçalves, P.F.; Vasconcelos-Santos, D.V. Vogt-Koyanagi-Harada-like Disease following Yellow Fever Vaccination. Ocul. Immunol. Inflamm. 2021, 29, 124–127. [Google Scholar] [CrossRef] [PubMed]
- Dogan, B.; Erol, M.K.; Cengiz, A. Vogt-Koyanagi-Harada disease following BCG vaccination and tuberculosis. SpringerPlus 2016, 5, 603. [Google Scholar] [CrossRef] [PubMed]
- Murtaza, F.; Pereira, A.; Mandelcorn, M.S.; Kaplan, A.J. Vogt-Koyanagi-Harada disease following influenza vaccination. Am. J. Ophthalmol. Case Rep. 2022, 26, 101516. [Google Scholar] [CrossRef]
- Kuniyoshi, K.; Hatsukawa, Y.; Kimura, S.; Fujino, T.; Ohguro, H.; Nakai, R.; Sunami, K.; Mishima, S.-I.; Sato, T.; Kusaka, S.; et al. Acute Bilateral Photoreceptor Degeneration in an Infant After Vaccination Against Measles and Rubella. JAMA Ophthalmol. 2017, 135, 478–482. [Google Scholar] [CrossRef]
- Benage, M.; Fraunfelder, F.W. Vaccine-Associated Uveitis. Mo. Med. 2016, 113, 48–52. [Google Scholar]
- Abdalla Elsayed, M.E.A.; Kozak, I. Pharmacologically induced uveitis. Surv. Ophthalmol. 2021, 66, 781–801. [Google Scholar] [CrossRef] [PubMed]
- Naranjo, C.A.; Busto, U.; Sellers, E.M.; Sandor, P.; Ruiz, I.; Roberts, E.A.; Janecek, E.; Domecq, C.; Greenblatt, D.J. A method for estimating the probability of adverse drug reactions. Clin. Pharmacol. Ther. 1981, 30, 239–245. [Google Scholar] [CrossRef]
- Du, L.; Kijlstra, A.; Yang, P. Vogt-Koyanagi-Harada disease: Novel insights into pathophysiology, diagnosis and treatment. Prog. Retin. Eye Res. 2016, 52, 84–111. [Google Scholar] [CrossRef]
- Ohno, S.; Minakawa, R.; Matsuda, H. Clinical studies of Vogt-Koyanagi-Harada’s disease. Jpn. J. Ophthalmol. 1988, 32, 334–343. [Google Scholar]
- Bordaberry, M.F. Vogt-Koyanagi-Harada disease: Diagnosis and treatments update. Curr. Opin. Ophthalmol. 2010, 21, 430–435. [Google Scholar] [CrossRef]
- Yang, P.; Ren, Y.; Li, B.; Fang, W.; Meng, Q.; Kijlstra, A. Clinical characteristics of Vogt-Koyanagi-Harada syndrome in Chinese patients. Ophthalmology 2007, 114, 606–614. [Google Scholar] [CrossRef] [PubMed]
- Moorthy, R.S.; Inomata, H.; Rao, N.A. Vogt-Koyanagi-Harada syndrome. Surv. Ophthalmol. 1995, 39, 265–292. [Google Scholar] [CrossRef] [PubMed]
- Sugiura, S. Some observations on uveitis in Japan, with special reference to Vogt-Koyanagi-Harada and Behçet diseases. Nippon Ganka Gakkai Zasshi 1976, 80, 1285–1326. [Google Scholar] [PubMed]
- Sachdev, N.; Gupta, V.; Gupta, A.; Singh, R. Posterior segment recurrences in Vogt-Koyanagi-Harada disease. Int. Ophthalmol. 2008, 28, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Urzua, C.A.; Herbort, C.; Valenzuela, R.A.; Abu El-Asrar, A.M.; Arellanes-Garcia, L.; Schlaen, A.; Yamamoto, J.; Pavesio, C. Initial-onset acute and chronic recurrent stages are two distinctive courses of Vogt-Koyanagi-Harada disease. J. Ophthalmic Inflamm. Infect. 2020, 10, 23. [Google Scholar] [CrossRef] [PubMed]
- Dahbour, S.S. MRI documented acute myelitis in a patient with Vogt–Koyanagi–Harada syndrome: First report. Clin. Neurol. Neurosurg. 2009, 111, 200–202. [Google Scholar] [CrossRef] [PubMed]
- Tagawa, Y.; Sugiura, S.; Yakura, H.; Wakisaka, A.; Aizawa, M. Letter: HLA and Vogt-Koyanagh-Harada syndrome. N. Engl. J. Med. 1976, 295, 173. [Google Scholar] [PubMed]
- Snyder, D.A.; Tessler, H.H. Vogt-Koyanagi-Harada syndrome. Am. J. Ophthalmol. 1980, 90, 69–75. [Google Scholar] [CrossRef]
- Read, R.W.; Holland, G.N.; Rao, N.A.; Tabbara, K.F.; Ohno, S.; Arellanes-Garcia, L.; Pivetti-Pezzi, P.; Tessler, H.H.; Usui, M. Revised diagnostic criteria for Vogt-Koyanagi-Harada disease: Report of an in- ternational committee on nomenclature. Am. J. Ophthalmol. 2001, 131, 647–652. [Google Scholar] [CrossRef]
- Yang, P.; Zhong, Y.; Du, L.; Chi, W.; Chen, L.; Zhang, R.; Zhang, M.; Wang, H.; Lu, H.; Yang, L.; et al. Development and Evaluation of Diagnostic Criteria for Vogt-Koyanagi-Harada Disease. JAMA Ophthalmol. 2018, 136, 1025–1031. [Google Scholar] [CrossRef]
- Standardization of Uveitis Nomenclature (SUN) Working Group. Classification Criteria for Vogt-Koyanagi-Harada Disease. Am. J. Ophthalmol. 2021, 228, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Herbort, C.P.; Mantovani, A.; Bouchenaki, N. Indocyanine green angiography in Vogt–Koyanagi–Harada disease: Angiographic signs and utility in patient follow-up. Int. Ophthalmol. 2007, 27, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Fardeau, C.; Tran, T.H.C.; Gharbi, B.; Cassoux, N.; Bodaghi, B.; LeHoang, P. Retinal fluorescein and indocyanine green angiography and optical coherence tomography in successive stages of Vogt-Koyanagi-Harada disease. Int. Ophthalmol. 2007, 27, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Erba, S.; Govetto, A.; Scialdone, A.; Casalino, G. Role of optical coherence tomography angiography in Vogt-Koyanagi-Harada disease. GMS Ophthalmol. Cases 2021, 11, Doc06. [Google Scholar] [PubMed]
- Hirooka, K.; Saito, W.; Namba, K.; Takemoto, Y.; Mizuuchi, K.; Uno, T.; Tagawa, Y.; Hashimoto, Y.; Ishida, S. Relationship between choroidal blood flow velocity and choroidal thickness during systemic corticosteroid therapy for Vogt-Koyanagi-Harada disease. Graefe’s Arch. Clin. Exp. Ophthalmol. 2015, 253, 609–617. [Google Scholar] [CrossRef] [PubMed]
- Abe, S.; Nakamura, T.; Okumura, E.; Oiwake, T.; Okada, A.A.; Hayashi, A. Long-term changes of choroidal blood flow velocity in Vogt-Koyanagi-Harada disease. Graefe’s Arch. Clin. Exp. Ophthalmol. 2022, 260, 1933–1939. [Google Scholar] [CrossRef]
- Liang, A.; Jia, S.; Gao, F.; Han, X.; Pei, M.; Qu, Y.; Xiao, J.; Zhao, C.; Zhang, M. Decrease of choriocapillary vascular density measured by optical coherence tomography angiography in Vogt-Koyanagi-Harada disease. Graefe’s Arch. Clin. Exp. Ophthalmol. 2021, 259, 3395–3404. [Google Scholar] [CrossRef]
- Abu El-Asrar, A.M.; Al Tamimi, M.; Hemachandran, S.; Al-Mezaine, H.S.; Al-Muammar, A.; Kangave, D. Prognostic factors for clinical outcomes in patients with Vogt-Koyanagi-Harada disease treated with high-dose corticosteroids. Acta Ophthalmol. 2013, 91, e486–e493. [Google Scholar] [CrossRef]
- Lai, T.Y.Y.; Chan, R.P.S.; Chan, C.K.M.; Lam, D.S.C. Effects of the duration of initial oral corticosteroid treatment on the recurrence of inflammation in Vogt-Koyanagi-Harada disease. Eye Lond. Engl. 2009, 23, 543–548. [Google Scholar] [CrossRef]
- Rubsamen, P.E.; Gass, J.D. Vogt-Koyanagi-Harada syndrome. Clinical course, therapy, and long-term visual outcome. Arch. Ophthalmol. Chic. Ill 1960 1991, 109, 682–687. [Google Scholar] [CrossRef]
- Keino, H.; Goto, H.; Usui, M. Sunset glow fundus in Vogt-Koyanagi-Harada disease with or without chronic ocular inflammation. Graefe’s Arch. Clin. Exp. Ophthalmol. 2002, 240, 878–882. [Google Scholar] [CrossRef]
- Chee, S.-P.; Jap, A.; Bacsal, K. Spectrum of Vogt-Koyanagi-Harada disease in Singapore. Int. Ophthalmol. 2007, 27, 137–142.e1. [Google Scholar] [CrossRef]
- Sakata, V.M.; da Silva, F.T.; Hirata, C.E.; Marin, M.L.C.; Rodrigues, H.; Kalil, J.; Costa, R.A.; Yamamoto, J.H. High rate of clinical recurrence in patients with Vogt-Koyanagi-Harada disease treated with early high-dose corticosteroids. Graefe’s Arch. Clin. Exp. Ophthalmol. 2015, 253, 785–790. [Google Scholar] [CrossRef]
- Paredes, I.; Ahmed, M.; Foster, C.S. Immunomodulatory therapy for Vogt-Koyanagi-Harada patients as first-line therapy. Ocul. Immunol. Inflamm. 2006, 14, 87–90. [Google Scholar] [CrossRef] [PubMed]
- Abu El-Asrar, A.M.; Hemachandran, S.; Al-Mezaine, H.S.; Kangave, D.; Al-Muammar, A.M. The outcomes of mycophenolate mofetil therapy combined with systemic corticosteroids in acute uveitis associated with Vogt-Koyanagi-Harada disease. Acta Ophthalmol. 2012, 90, e603–e608. [Google Scholar] [CrossRef] [PubMed]
- Abu El-Asrar, A.M.; Dosari, M.; Hemachandran, S.; Gikandi, P.W.; Al-Muammar, A. Mycophenolate mofetil combined with systemic corticosteroids prevents progression to chronic recurrent inflammation and development of “sunset glow fundus” in initial-onset acute uveitis associated with Vogt-Koyanagi-Harada disease. Acta Ophthalmol. 2017, 95, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Papasavvas, I.; Tugal-Tutkun, I.; Herbort, C.P. Vogt-Koyanagi-Harada is a Curable Autoimmune Disease: Early Diagnosis and Immediate Dual Steroidal and Non-Steroidal Immunosuppression are Crucial Prerequisites. J. Curr. Ophthalmol. 2020, 32, 310–314. [Google Scholar] [PubMed]
- Nakayama, M.; Keino, H.; Watanabe, T.; Okada, A.A. Clinical features and visual outcomes of 111 patients with new-onset acute Vogt-Koyanagi-Harada disease treated with pulse intravenous corticosteroids. Br. J. Ophthalmol. 2019, 103, 274–278. [Google Scholar] [CrossRef]
- Miyanaga, M.; Kawaguchi, T.; Shimizu, K.; Miyata, K.; Mochizuki, M. Influence of early cerebrospinal fluid-guided diagnosis and early high-dose corticosteroid therapy on ocular outcomes of Vogt-Koyanagi-Harada disease. Int. Ophthalmol. 2007, 27, 183–188. [Google Scholar] [CrossRef]
- O’Keefe, G.A.D.; Rao, N.A. Vogt-Koyanagi-Harada disease. Surv. Ophthalmol. 2017, 62, 1–25. [Google Scholar] [CrossRef]
- Accorinti, M.; Saturno, M.C.; Iannetti, L.; Manni, P.; Mastromarino, D.; Pirraglia, M.P. Treatment and Prognosis of Vogt–Koyanagi–Harada Disease: Real-Life Experience in Long-Term Follow-Up. J. Clin. Med. 2022, 11, 3632. [Google Scholar] [CrossRef] [PubMed]
- Al-Kharashi, A.S.; Aldibhi, H.; Al-Fraykh, H.; Kangave, D.; Abu El-Asrar, A.M. Prognostic factors in Vogt-Koyanagi-Harada disease. Int. Ophthalmol. 2007, 27, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, T.; Horie, S.; Bouchenaki, N.; Ohno-Matsui, K.; Mochizuki, M.; Herbort, C.P. Suboptimal therapy controls clinically apparent disease but not subclinical progression of Vogt-Koyanagi-Harada disease. Int. Ophthalmol. 2010, 30, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Jap, A.; Luu, C.D.; Yeo, I.; Chee, S.-P. Correlation between peripapillary atrophy and corticosteroid therapy in patients with Vogt–Koyanagi–Harada disease. Eye 2008, 22, 240–245. [Google Scholar] [CrossRef]
- Chee, S.-P.; Jap, A.; Bacsal, K. Prognostic Factors of Vogt-Koyanagi-Harada Disease in Singapore. Am. J. Ophthalmol. 2009, 147, 154–161.e1. [Google Scholar] [CrossRef]
- Read, R.W.; Rechodouni, A.; Butani, N.; Johnston, R.; LaBree, L.D.; Smith, R.E.; Rao, N.A. Complications and prognostic factors in Vogt-Koyanagi-Harada disease. Am. J. Ophthalmol. 2001, 131, 599–606. [Google Scholar] [CrossRef]
- Tabbara, K.F.; Chavis, P.S.; Freeman, W.R. Vogt-Koyanagi-Harada syndrome in children compared to adults: Vogt-Koyanagi-Harada syndrome in children compared to adults. Acta Ophthalmol. Scand. 1998, 76, 723–726. [Google Scholar] [CrossRef]
- Islam, S.M.; Numaga, J.; Matsuki, K.; Fujino, Y.; Maeda, H.; Masuda, K. Influence of HLA-DRB1 gene variation on the clinical course of Vogt-Koyanagi-Harada disease. Investig. Ophthalmol. Vis. Sci. 1994, 35, 752–756. [Google Scholar]
- Seyed Hosseini, E.; Riahi Kashani, N.; Nikzad, H.; Azadbakht, J.; Hassani Bafrani, H.; Haddad Kashani, H. The novel coronavirus Disease-2019 (COVID-19): Mechanism of action, detection and recent therapeutic strategies. Virology 2020, 551, 1–9. [Google Scholar] [CrossRef]
- Anthony, E.; Rajamani, A.; Baskaran, P.; Rajendran, A. Vogt Koyanagi Harada disease following a recent COVID-19 infection. Indian J. Ophthalmol. 2022, 70, 670–672. [Google Scholar]
- Santamaria, A.; Chang, J.; Savarain, C. SARS-CoV-2 among the Potential Viral Triggers for Vogt-Konayagi-Harada Disease: First Case Report and Literature Review. Ocul. Immunol. Inflamm. 2021, 26, 1869–1875. [Google Scholar] [CrossRef]
- Saraceno, J.J.F.; Souza, G.M.; Dos Santos Finamor, L.P.; Nascimento, H.M.; Belfort, R. Vogt-Koyanagi-Harada Syndrome following COVID-19 and ChAdOx1 nCoV-19 (AZD1222) vaccine. Int. J. Retin. Vitr. 2021, 7, 49. [Google Scholar] [CrossRef] [PubMed]
- Yepez, J.B.; Murati, F.A.; Petitto, M.; De Yepez, J.; Galue, J.M.; Revilla, J.; Petitto, A.; Vinardell, S.; Arevalo, J.F. Vogt-Koyanagi-Harada Disease Following COVID-19 Infection. Case Rep. Ophthalmol. 2021, 12, 804–808. [Google Scholar] [CrossRef] [PubMed]
- Fischer, J.; Jung, N.; Robinson, N.; Lehmann, C. Sex differences in immune responses to infectious diseases. Infection 2015, 43, 399–403. [Google Scholar] [CrossRef] [PubMed]
- Klein, S.L.; Flanagan, K.L. Sex differences in immune responses. Nat. Rev. Immunol. 2016, 16, 626–638. [Google Scholar] [CrossRef] [PubMed]
- Brandão-De-Resende, C.; Cunha, L.H.M.; Oliveira, S.L.; Pereira, L.S.; Oliveira, J.G.F.; Santos, T.A.; Vasconcelos-Santos, D.V. Characterization of Retinopathy Among Patients With Yellow Fever During 2 Outbreaks in Southeastern Brazil. JAMA Ophthalmol. 2019, 137, 996–1002. [Google Scholar] [CrossRef]
- Khoshnood, S.; Arshadi, M.; Akrami, S.; Koupaei, M.; Ghahramanpour, H.; Shariati, A.; Sadeghifard, N.; Heidary, M. An overview on inactivated and live-attenuated SARS-CoV-2 vaccines. J. Clin. Lab. Anal. 2022, 36, e24418. [Google Scholar] [CrossRef]
- Baird, F.J.; Lopata, A.L. The dichotomy of pathogens and allergens in vaccination approaches. Front. Microbiol. 2014, 5, 365. [Google Scholar] [CrossRef]
- Papasavvas, I.; Herbort, C.P. Reactivation of Vogt-Koyanagi-Harada disease under control for more than 6 years, following anti-SARS-CoV-2 vaccination. J. Ophthalmic Inflamm. Infect. 2021, 11, 21. [Google Scholar] [CrossRef]
- Bolletta, E.; Iannetta, D.; Mastrofilippo, V.; De Simone, L.; Gozzi, F.; Croci, S.; Bonacini, M.; Belloni, L.; Zerbini, A.; Adani, C.; et al. Uveitis and Other Ocular Complications Following COVID-19 Vaccination. J. Clin. Med. 2021, 10, 5960. [Google Scholar] [CrossRef] [PubMed]
- Accorinti, M.; Saturno, M.C.; Manni, P. Vogt-Koyanagi-Harada Relapse after COVID-19 Vaccination. Ocul. Immunol. Inflamm. 2022, 30, 1228–1233. [Google Scholar] [CrossRef]
- De Domingo, B.; López, M.; Lopez-Valladares, M.; Ortegon-Aguilar, E.; Sopeña-Perez-Argüelles, B.; Gonzalez, F. Vogt-Koyanagi-Harada Disease Exacerbation Associated with COVID-19 Vaccine. Cells 2022, 11, 1012. [Google Scholar] [CrossRef]
- Rujkorakarn, P.; Patamatamkul, S. Vogt-Koyanagi-Harada disease following ChAdOx1 nCoV-19 and mRNA-1273 vaccination. J. Français Ophtalmol. 2023, 46, 207–210. [Google Scholar] [CrossRef]
- Koong, L.R.; Chee, W.K.; Toh, Z.H.; Le Ng, X.; Agrawal, R.; Ho, S.L. Vogt-Koyanagi-Harada Disease Associated with COVID-19 mRNA Vaccine. Ocul. Immunol. Inflamm. 2021, 29, 1212–1215. [Google Scholar] [CrossRef]
- Chen, X.; Wang, B.; Li, X. Acute-onset Vogt-Koyanagi-Harada like uveitis following COVID-19 inactivated virus vaccination. Am. J. Ophthalmol. Case Rep. 2022, 26, 101404. [Google Scholar] [CrossRef]
- Yamaguchi, C.; Kunikata, H.; Hashimoto, K.; Yoshida, M.; Ninomiya, T.; Hariya, T.; Abe, T.; Nakazawa, T. De novo Vogt-Koyanagi-Harada disease after vaccination for COVID-19, successfully treated with systemic steroid therapy and monitored with laser speckle flowgraphy. Am. J. Ophthalmol. Case Rep. 2022, 27, 101616. [Google Scholar] [CrossRef]
- Chen, X.; Li, X.; Li, H.; Li, M.; Gong, S. Ocular Adverse Events after Inactivated COVID-19 Vaccination in Xiamen. Vaccines 2022, 10, 482. [Google Scholar] [CrossRef]
- de Courssou, J.-B.B.; Tisseyre, M.; Hadjadj, J.; Chouchana, L.; Broca, F.; Terrier, B.; Duraffour, P.; Henriquez, S. De Novo Vogt-Koyanagi-Harada Disease following COVID-19 Vaccine: A Case Report and Literature Overview. Ocul. Immunol. Inflamm. 2022, 30, 1292–1295. [Google Scholar] [CrossRef]
- Joo, C.W.; Kim, Y.-K.; Park, S.P. Vogt-Koyanagi-Harada Disease following mRNA-1273 (Moderna) COVID-19 Vaccination. Ocul. Immunol. Inflamm. 2022, 30, 1250–1254. [Google Scholar] [CrossRef]
- de Queiroz Tavares Ferreira, F.; Araújo, D.C.; de Albuquerque, L.M.; Bianchini, P.M.; Holanda, E.C.; Pugliesi, A. Possible Association between Vogt-Koyanagi-Harada Disease and Coronavirus Disease Vaccine: A Report of Four Cases. Ocul. Immunol. Inflamm. 2022, 31, 1134–1140. [Google Scholar] [CrossRef]
- Ding, X.; Chang, Q. Probable Vogt–Koyanagi–Harada Disease after COVID-19 Vaccination: Case Report and Literature Review. Vaccines 2022, 10, 783. [Google Scholar] [CrossRef]
- Kim, S.Y.; Kang, M.S.; Kwon, H.J. Bilateral Panuveitis Mimicking Vogt-Koyanagi-Harada Disease following the First Dose of ChAdOx1 nCoV-19 Vaccine. Ocul. Immunol. Inflamm. 2022, 30, 1218–1221. [Google Scholar] [CrossRef]
- Reddy, Y.; Pandey, A.; Ojha, A.; Ramchandani, S. Harada-like syndrome post-Covishield vaccination: A rare adverse effect. Indian J. Ophthalmol. 2022, 70, 321–323. [Google Scholar] [CrossRef]
- Sato, T.; Nihei, R.; Sora, D.; Nishio, Y.; Takeuchi, M. Case report: Bilateral panuveitis resembling Vogt-Koyanagi-Harada disease after second dose of BNT162b2 mRNA COVID-19 vaccine. Front. Immunol. 2022, 13, 967972. [Google Scholar] [CrossRef]
- Shariati, M.M.; Abrishami, M.; Jahani, S.; Bolouki, A.; Ansari-Astaneh, M.-R.; Hosseini, S.M. Uveitis including Vogt-Koyanagi-Harada syndrome following inactive COVID-19 vaccination: A case series. J. Ophthalmic Inflamm. Infect. 2023, 13, 26. [Google Scholar] [CrossRef]
- Ren, J.; Zhang, T.; Li, X.; Liu, G. Ocular Inflammatory Reactions following an Inactivated SARS-CoV-2 Vaccine: A Four Case Series. Ocul. Immunol. Inflamm. 2023, 31, 1128–1133. [Google Scholar] [CrossRef]
- Han, R.; Xu, G.; Ding, X. COVID-19 Vaccine-Related Vogt-Koyanagi-Harada Disease Complicated by Central Serous Chorioretinopathy during Treatment Course: Case Report and Literature Review. Vaccines 2022, 10, 1792. [Google Scholar] [CrossRef]
- Nakayama, M.; Okada, A.A.; Hayashi, I.; Keino, H. COVID-19 vaccination-related intraocular inflammation in Japanese patients. Graefe’s Arch. Clin. Exp. Ophthalmol. 2023, 261, 897–899. [Google Scholar] [CrossRef]
- Pillar, S.; Weinberg, T.; Amer, R. Posterior ocular manifestations following BNT162b2 mRNA COVID-19 vaccine: A case series. Int. Ophthalmol. 2023, 43, 1677–1686. [Google Scholar] [CrossRef]
- Wang, L.-U.; Chen, F.-T.; Wang, J.-K.; Huang, T.-L.; Chang, P.-Y.; Chen, Y.-J. Ocular inflammatory manifestations following COVID-19 vaccinations in Taiwan: A case series. Taiwan J. Ophthalmol. 2022, 12, 465–471. [Google Scholar]
- Li, Z.; Hu, F.; Li, Q.; Wang, S.; Chen, C.; Zhang, Y.; Mao, Y.; Shi, X.; Zhou, H.; Cao, X.; et al. Ocular Adverse Events after Inactivated COVID-19 Vaccination. Vaccines 2022, 10, 918. [Google Scholar] [CrossRef] [PubMed]
- Yasaka, Y.; Hasegawa, E.; Keino, H.; Usui, Y.; Maruyama, K.; Yamamoto, Y.; Kaburaki, T.; Iwata, D.; Takeuchi, M.; Kusuhara, S.; et al. A multicenter study of ocular inflammation after COVID-19 vaccination. Jpn. J. Ophthalmol. 2023, 67, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Deng, T.; Zhu, L.; Zhong, J. Association of human leukocyte antigen (HLA)-DQ and HLA-DQA1/DQB1 alleles with Vogt–Koyanagi–Harada disease: A systematic review and meta-analysis. Medicine 2018, 97, e9914. [Google Scholar] [CrossRef] [PubMed]
- Bonacini, M.; Cimino, L.; De Simone, L.; Bolletta, E.; Gozzi, F.; Soriano, A.; Muratore, F.; Zerbini, A.; Fontana, L.; Salvarani, C.; et al. Vogt–Koyanagi–Harada patients show higher frequencies of circulating NKG2Dpos NK and NK T cells. Clin. Exp. Immunol. 2021, 204, 41–48. [Google Scholar] [CrossRef]
- Talotta, R. Molecular Mimicry and HLA Polymorphisms May Drive Autoimmunity in Recipients of the BNT-162b2 mRNA Vaccine: A Computational Analysis. Microorganisms 2023, 11, 1686. [Google Scholar] [CrossRef]
- Akinosoglou, K.; Tzivaki, I.; Marangos, M. COVID-19 vaccine and autoimmunity: Awakening the sleeping dragon. Clin. Immunol. 2021, 226, 108721. [Google Scholar] [CrossRef]
- Apaydin, H.; Erden, A.; Güven, S.C.; Armağan, B.; Konak, H.E.; Polat, B.; Afşin, Y.; Kaygisiz, M.; Omma, A.; Kucuksahin, O. Effects of anti-SARS-CoV-2 vaccination on safety and disease exacerbation in patients with Behçet syndrome in a monocentric cohort. Int. J. Rheum. Dis. 2022, 25, 1068–1077. [Google Scholar] [CrossRef]
- Yıldırım, R.; Üsküdar Cansu, D.; Dinler, M.; Korkmaz, C. Reactivation in major organ involvement following SARS-CoV-2 mRNA vaccination in Behçet’s syndrome patient receiving immunosuppressive therapy. Rheumatology 2022, 61, SI197–SI199. [Google Scholar] [CrossRef]
- Tagini, F.; Carrel, L.; Fallet, B.; Gachoud, D.; Ribi, C.; Monti, M. Behçet’s-like adverse event or inaugural Behçet’s disease after SARS-CoV-2 mRNA-1273 vaccination? Rheumatology 2022, 61, e112–e113. [Google Scholar] [CrossRef]
- Bauckneht, M.; Aloè, T.; Tagliabue, E.; Cittadini, G.; Guadagno, A.; Morbelli, S.; Barisione, E. Beyond COVID-19 vaccination-associated pitfalls on [18F]Fluorodeoxyglucose (FDG) PET: A case of a concomitant sarcoidosis. Eur. J. Nucl. Med. Mol. Imaging 2021, 48, 2661–2662. [Google Scholar] [CrossRef]
- Rademacher, J.G.; Tampe, B.; Korsten, P. First Report of Two Cases of Löfgren’s Syndrome after SARS-CoV-2 Vaccination-Coincidence or Causality? Vaccines 2021, 9, 1313. [Google Scholar] [CrossRef]
- Numakura, T.; Murakami, K.; Tamada, T.; Yamaguchi, C.; Inoue, C.; Ohkouchi, S.; Tode, N.; Sano, H.; Aizawa, H.; Sato, K.; et al. A Novel Development of Sarcoidosis Following COVID-19 Vaccination and a Literature Review. Intern. Med. 2022, 61, 3101–3106. [Google Scholar] [CrossRef] [PubMed]
- Accorinti, M.; Manni, P.; Sampalmieri, L.; Saturno, M.C. Ocular Behçet disease and COVID-19 [published online ahead of print, 2022 Jan 21]. Eur. J. Ophthalmol. 2022, 32, NP148–NP149. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, E.T.; Moorthy, R.S. Vaccine-Associated Posterior Uveitis. Retin. Cases Brief Rep. 2022, 16, 595–598. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.B.; Parmar, U.P.S.; Kahale, F.; Agarwal, A.; Tsui, E. Vaccine-Associated Uveitis after COVID-19 Vaccination. Ophthalmology 2022, 130, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Herzum, A.; Micalizzi, C.; Molle, M.F.; Parodi, A. New-onset vitiligo following COVID-19 disease. Ski. Health Dis. 2022, 2, e86. [Google Scholar] [CrossRef]
- Uğurer, E.; Sivaz, O.; Kıvanç Altunay, İ. Newly developed vitiligo following COVID-19 mRNA vaccine. J. Cosmet. Dermatol. 2022, 21, 1350. [Google Scholar] [CrossRef]
- Militello, M.; Ambur, A.B.; Steffes, W. Vitiligo Possibly Triggered by COVID-19 Vaccination. Cureus 2022, 14, e20902. [Google Scholar] [CrossRef]
- Singh, R.; Cohen, J.L.; Astudillo, M.; Harris, J.E.; Freeman, E.E. Vitiligo of the arm after COVID-19 vaccination. JAAD Case Rep. 2022, 28, 142–144. [Google Scholar] [CrossRef]
- Lai, X.; Wichers, H.J.; Soler-Lopez, M.; Dijkstra, B.W. Structure and Function of Human Tyrosinase and Tyrosinase-Related Proteins. Chemistry 2018, 24, 47–55. [Google Scholar] [CrossRef]
- Zhang, J.; Xiao, T.; Cai, Y.; Chen, B. Structure of SARS-CoV-2 spike protein. Curr. Opin. Virol. 2021, 50, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Tenforde, M.W.; Self, W.H.; Adams, K.; Gaglani, M.; Ginde, A.A.; McNeal, T.; Ghamande, S.; Douin, D.J.; Talbot, H.K.; Casey, J.D.; et al. Influenza and Other Viruses in the Acutely Ill (IVY) Network. Association Between mRNA Vaccination and COVID-19 Hospitalization and Disease Severity. JAMA 2021, 326, 2043–2054. [Google Scholar] [CrossRef] [PubMed]
- Chmielewska, B.; Barratt, I.; Townsend, R.; Kalafat, E.; van der Meulen, J.; Gurol-Urganci, I.; O’Brien, P.; Morris, E.; Draycott, T.; Thangaratinam, S.; et al. Effects of the COVID-19 pandemic on maternal and perinatal outcomes: A systematic review and meta-analysis. Lancet Glob. Health 2021, 9, e759–e772. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Question | Yes | No | Do Not Know | Score |
|---|---|---|---|---|
| 1. Are there previous conclusive reports on this reaction? | ||||
| 2. Did the adverse event appear after the suspected drug was administered? | ||||
| 3. Did the adverse reaction improve when the drug was discontinued, or a specific antagonist was administered? | ||||
| 4. Did the adverse event reappear when the drug was re-administered? | ||||
| 5. Are there alternative causes (other than the drug) that could on their own have caused the reaction? | ||||
| 6. Did the reaction reappear when a placebo was given? | ||||
| 7. Was the drug detected in blood (or other fluids) in concentrations known to be toxic? | ||||
| 8. Was the reaction more severe when the dose was increased or less severe when the dose was decreased? | ||||
| 9. Did the patient have a similar reaction to the same or similar drugs in any previous exposure? | ||||
| 10. Was the adverse event confirmed by any objective evidence? |
| American Uveitis Society Criteria for Diagnosis of VKH Disease |
|---|
No history of trauma or surgery and one finding from at least three of the following four groups:
|
| Complete Vogt-Koyanagi-Harada Disease (Criteria 1 to 5 Must Be Present) |
|---|
| 1. No history of penetrating ocular trauma or surgery preceding the initial onset of uveitis |
| 2. No clinical or laboratory evidence suggestive of other ocular disease entities |
3. Bilateral ocular involvement (A or B must be met, depending on the stage of disease when the patient is examined)
|
4. Neurological/auditory findings (may resolve by time of evaluation)
|
5. Integumentary finding (not preceding onset of central nervous system or ocular disease)
|
Incomplete Vogt-Koyanagi-Harada disease (criteria 1 to 3 and either 4 or 5 must be present)
|
| Anthon E. et al. [71]. | Santamaria A. et al. [72]. | Saraceno JJF. et al. [73]. | Yepez J.B. et al. [74]. | |
|---|---|---|---|---|
| Age | 22 | 32 | 37 | 29 |
| Gender | F | F | F | F |
| Time interval between infection and symptoms of VKH | 21 days | 14 days | 14 days | 30 days |
| Treatment | Topical steroids Oral steroids | Topical steroids Oral steroids Immunosuppressive drugs | Oral steroids | Intravenous Steroids Oral Steroids |
| Live Attenuated Vaccine |
|---|
Vaccine with an inactivated virus:
|
Vaccine based on a viral vector:
|
Protein vaccines:
|
Messenger RNA (mRNA) vaccines:
|
| Papasavvas I [80]. | Bolletta E [81]. | Accorinti M [82]. | De Domingo B [83]. | Rujkorakarn P [84]. | |
|---|---|---|---|---|---|
| Age | 43 |
| 55 | 46 | 31 |
| Gender | F |
| F | F | F |
| Interval between vaccination and VKH symptoms onset | 6 weeks | unknown | 11 days | 2 days | 1 week |
| Type of vaccine | Tozinarem |
| Tozinarem | Tozinarem | Spikevax |
| Treatment at time of vaccination | Infliximab 5 mg/kg/10 weeks |
| Azathioprine 1.5 mg/kg/day | none | azathioprine 2 mg/kg/day, cyclosporine 4 mg/kg/day, mycophenolate mofetil 500 mg and prednisolone 2.5 mg. |
| Treatments for VKH relapses | Oral steroids + Immunosuppressive drugs | unknown | Intravenous, oral and local steroids + Immunosuppressive drugs | Topical steroids after the first vaccination Intravenous and oral steroids after the relapse occurred following the second vaccination | Topical corticosteroids |
| Koong L.R [85]. | Chen X [86]. | Yamaguchi C [87]. | Chen X [88]. | Brunet de Courssou J.B [89]. | Joo C.W [90]. | de Queiroz Tavares Ferreira F [91]. | Ding X [92]. | Kim S.Y [93]. | Ferreira Saraceno JJ et al. [73]. | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Age | 54 | 19 | 30 | 57 21 28 | 57 | 50 | 27 39 38 32 | 33 | 72 | 62 | |
| Gender | M | F | F | F M F | F | F | M M F F | M | F | F | |
| Time interval between vaccination and VKH symptoms onset | 1 day | 12 h | 2 weeks | 10 days 1 day 3 days | 3 weeks | 35 days | 2 weeks 1 week 5 days some weeks | 1 day | 3 days | 2 days | |
| Type of vaccine | Tozinameran | CoronaVac | Tozinameran | Sinopharm CoronaVac Sinopharm | Tozinameran | Spikevax | Vaxzevria Vaxzevria Tozinameran CoronaVac | Sinopharm | Vaxzevria | Vaxzevria | |
| Treatment after the disease onset | Systemic steroids (intravenous, then oral) | One periocular injection of triamcino-lone acetonide 40 mg in each eye | Systemic steroids (intravenous, then oral) | Oral steroids. Periocular steroids | Systemic steroids (intravenous, then oral) Local steroids | Oral steroids | Systemic steroids (intravenous, then oral) with immunosuppressive therapy (3 azathioprine, 1 methotrexate) | Oral steroids | Systemic steroids (intravenous and then oral) | Oral steroids | |
| Reddy [94] | Rujkorakarn P [84]. | Sato T [95]. | Shariati MM [96] | Ren J. [97] | Han R [98] | Nakayama M [99] | Pillar S [100] | Wang LU [101] | Li Z [102] | Yasaka Y * [103] | |
| Age | 30 | 59 | 45 | 23 | 46 | 62 | 53 45 29 52 | 24 | 52 | 48 41 | 78 71 |
| Gender | F | M | M | F | F | M | M F M F | M | F | F M | F M |
| Time interval between vaccination and VKH symptoms onset | 7 days | 2 weeks | 1 day | 2 weeks | 4 h | 6 days | 10 days 12 days 2 weeks 6 days | 3 weeks | 3 days | 14 days 5 days | 9 days 13 days |
| Type of vaccine | Vaxzevria | Vaxzevria | Tozinameran | Sinopharm | CoronaVac | Sinopharm | Tozinameran Tozinameran Spikevax Tozinameran | Tozinameran | Vaxzevria | CoronaVac CoronaVac | Tozinameran Tozinameran |
| Treatment after the disease onset | Oral steroids | Systemic steroids (intravenous, then oral) with immunosuppressive therapy (methotrexate) | Systemic steroids (intravenous, then oral) | Systemic steroids (intravenous) Local steroids | Systemic steroids (intravenous, then oral) | Oral steroids | Systemic steroids (intravenous) and local | Oral steroids | Systemic steroids and cyclosporine A. | Unspecified | Systemic steroids (intravenous, then oral) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Manni, P.; Saturno, M.C.; Accorinti, M. Vogt-Koyanagi-Harada Disease and COVID. J. Clin. Med. 2023, 12, 6242. https://doi.org/10.3390/jcm12196242
Manni P, Saturno MC, Accorinti M. Vogt-Koyanagi-Harada Disease and COVID. Journal of Clinical Medicine. 2023; 12(19):6242. https://doi.org/10.3390/jcm12196242
Chicago/Turabian StyleManni, Priscilla, Maria Carmela Saturno, and Massimo Accorinti. 2023. "Vogt-Koyanagi-Harada Disease and COVID" Journal of Clinical Medicine 12, no. 19: 6242. https://doi.org/10.3390/jcm12196242
APA StyleManni, P., Saturno, M. C., & Accorinti, M. (2023). Vogt-Koyanagi-Harada Disease and COVID. Journal of Clinical Medicine, 12(19), 6242. https://doi.org/10.3390/jcm12196242