
The Somatic Mutational Landscape of Mismatch Repair Deficient Prostate Cancer

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
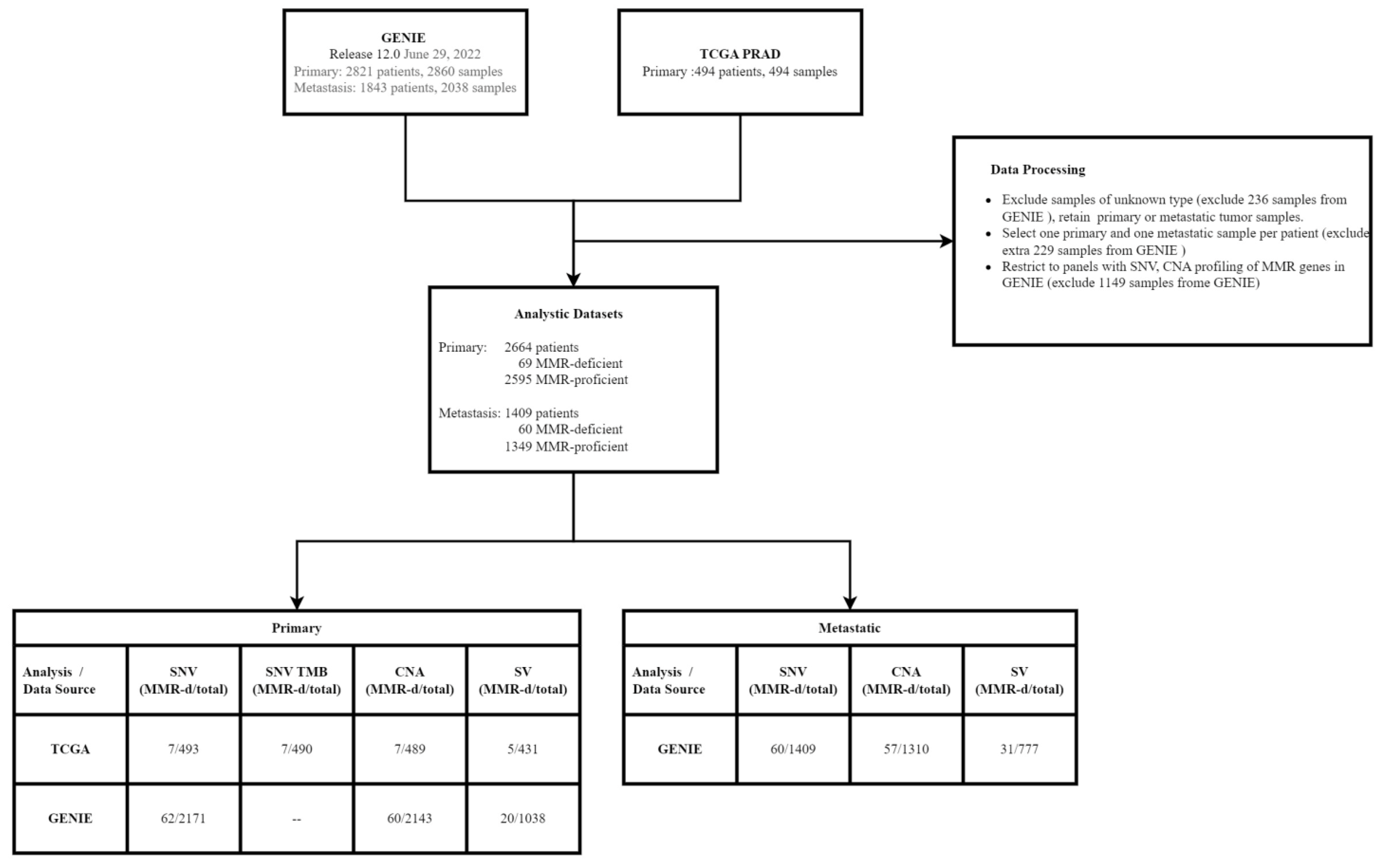
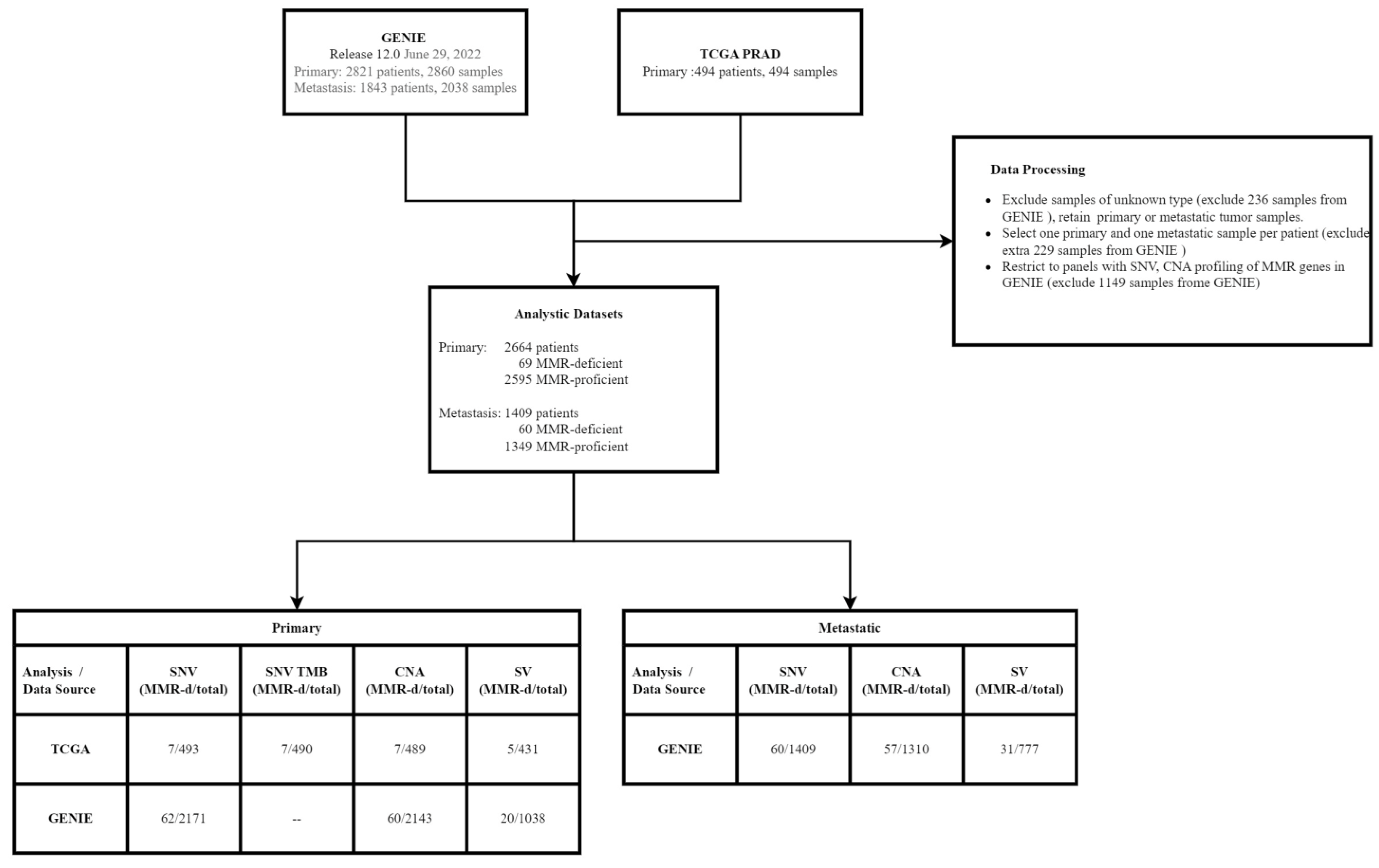
2.1. Samples and Data Processing
2.2. Identification of MMR-Deficient (MMR-d) Tumors
2.3. Genomic Profiling Analyses
3. Statistical Analysis
4. Results
4.1. MMR-d Prostate Tumors
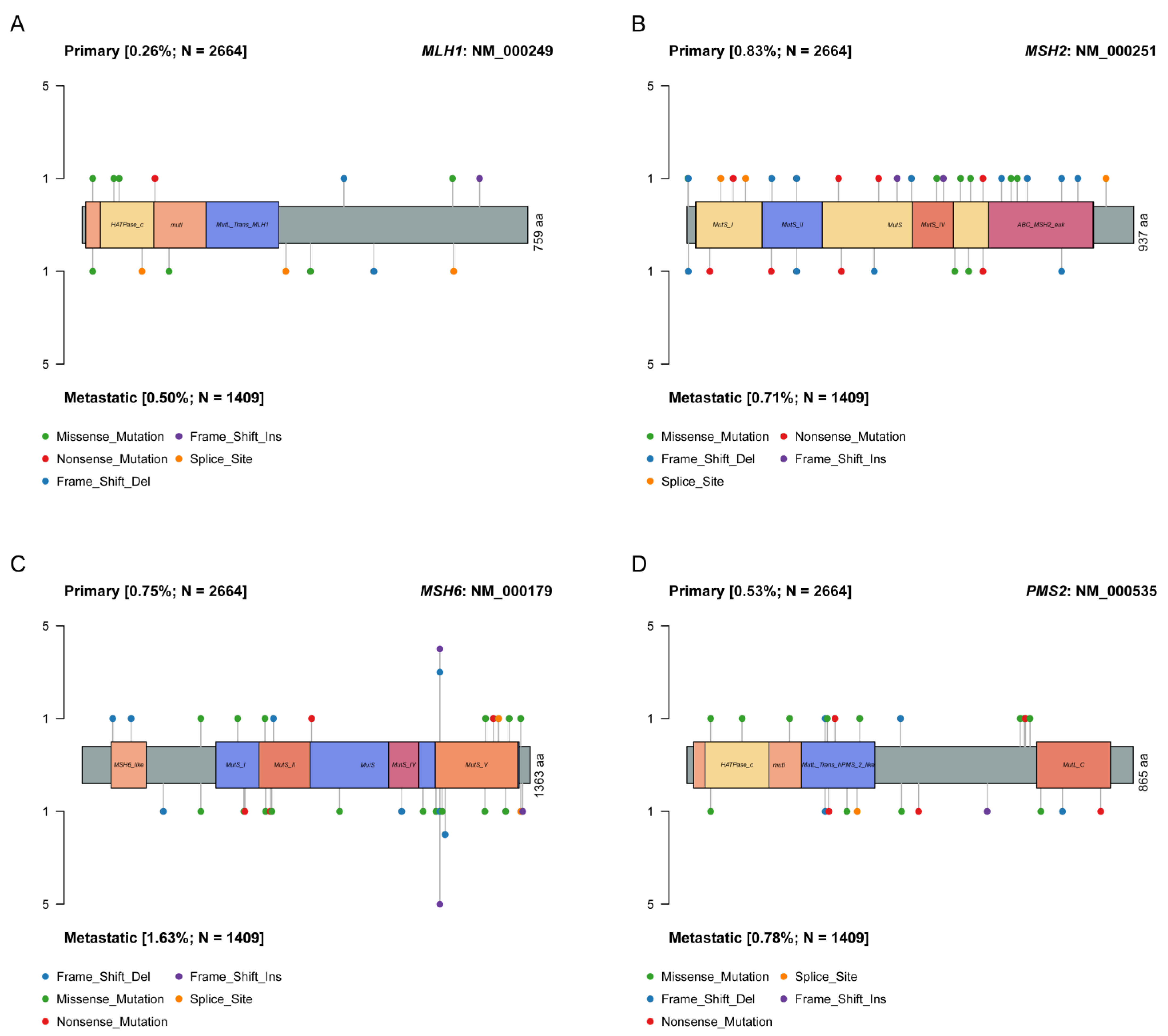
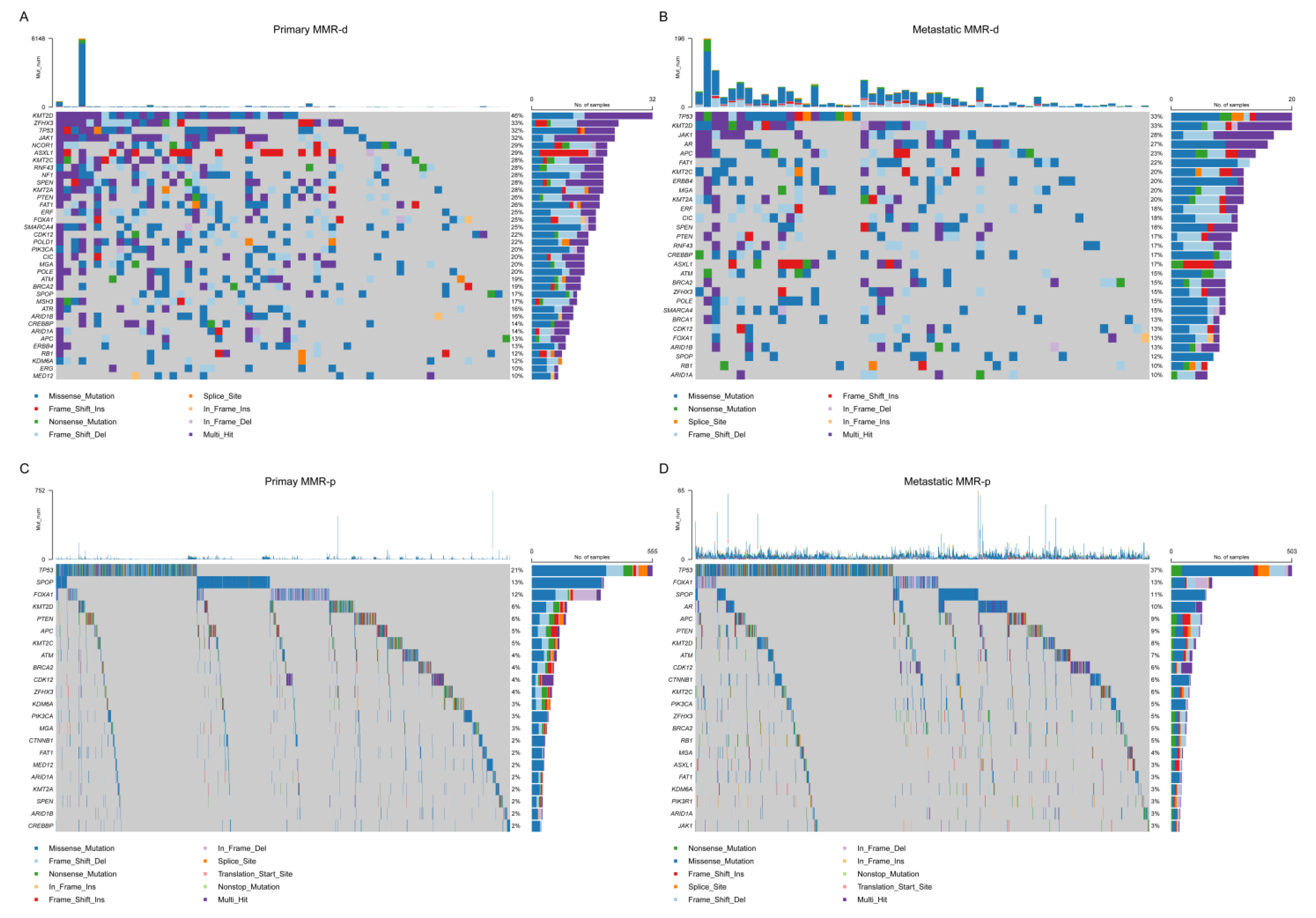
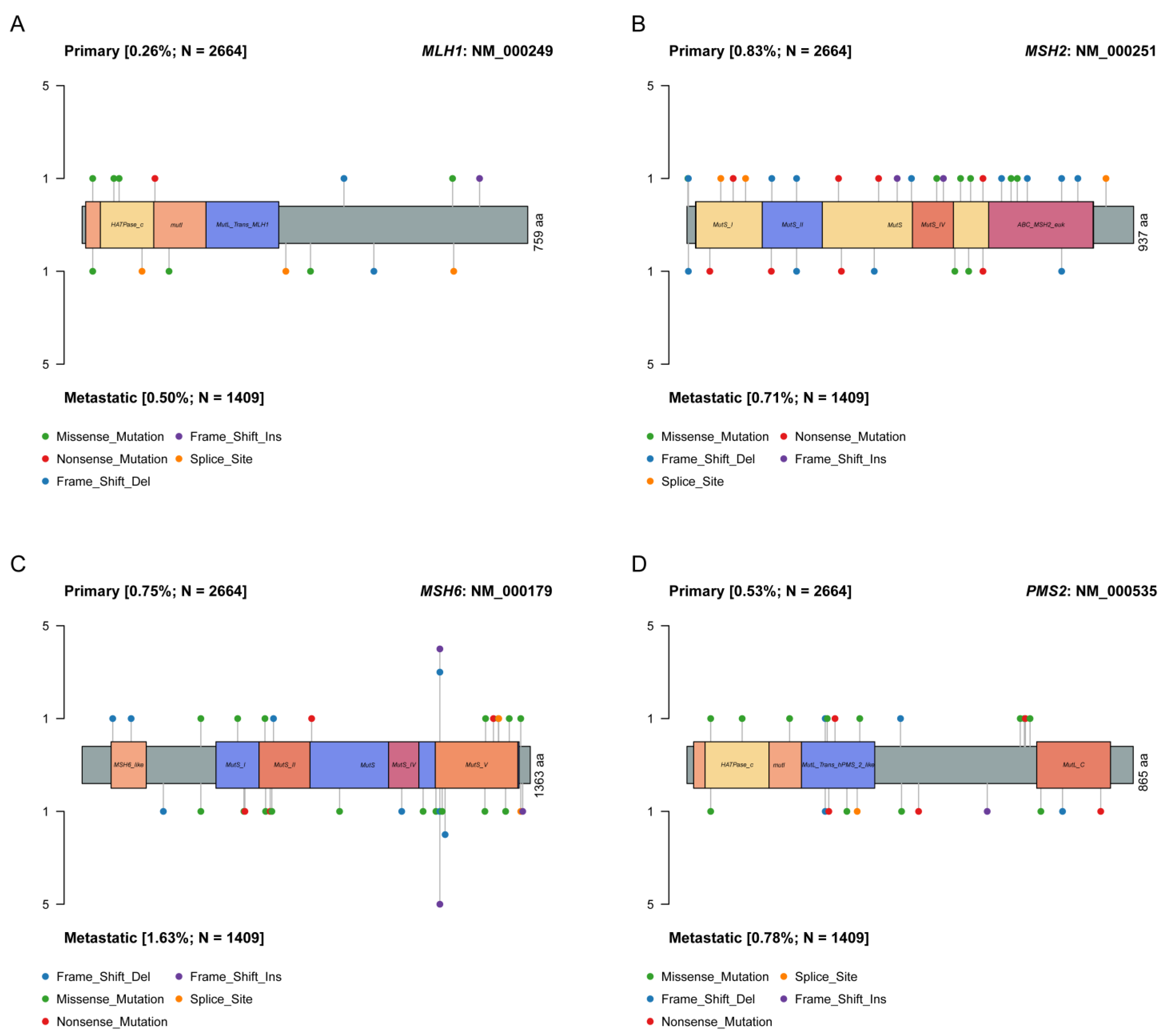
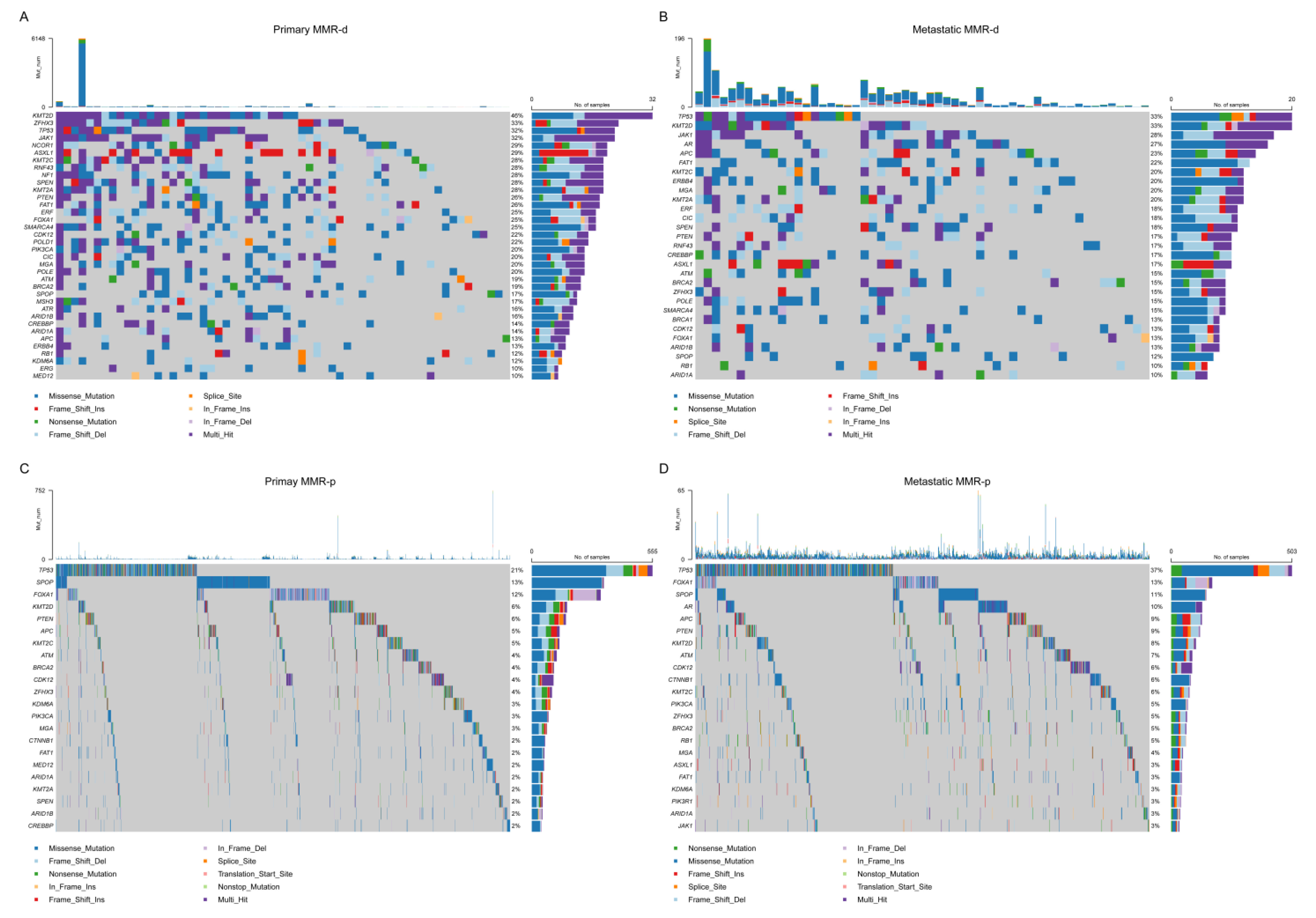
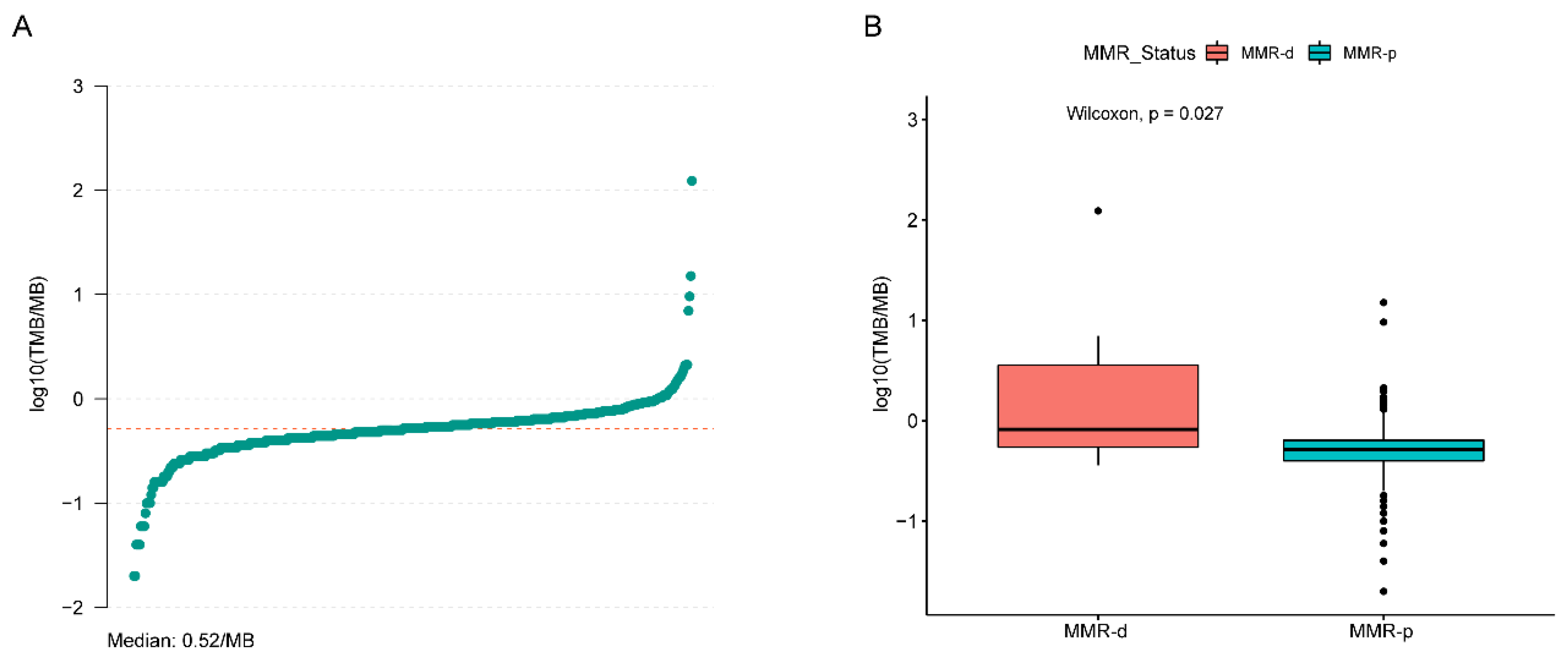
4.2. Single Nucleotide Variant (SNV) Analyses
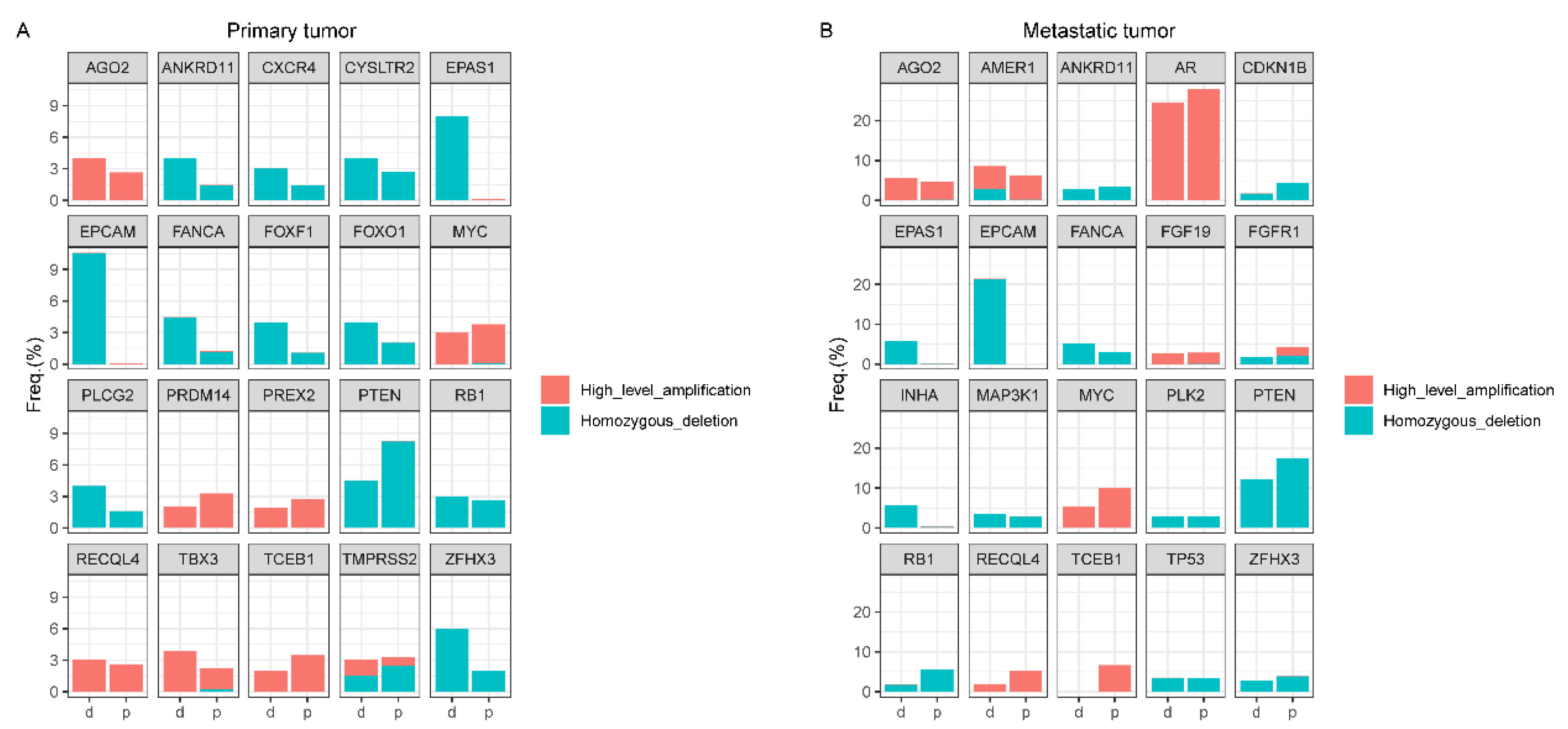
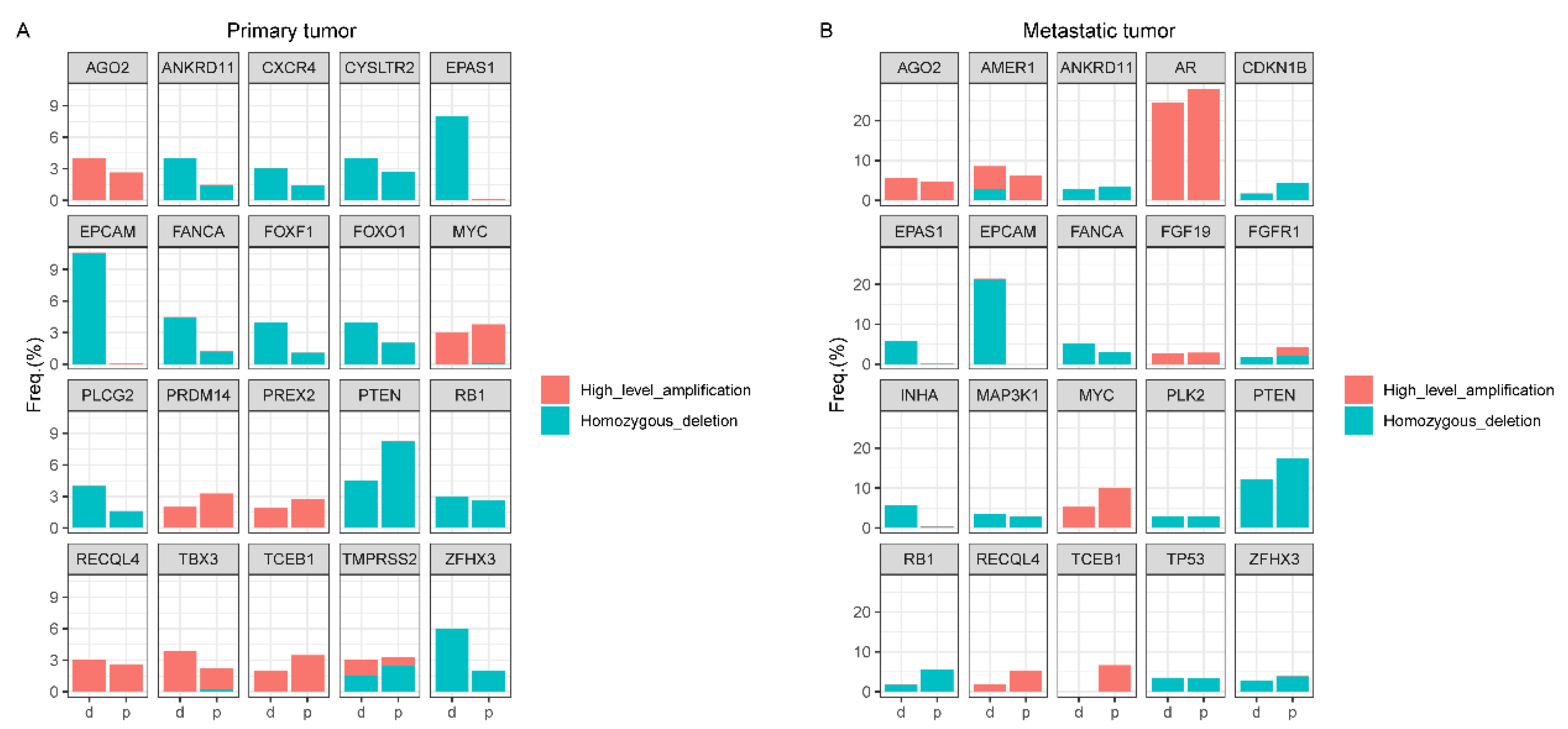
4.3. Copy Number Alteration (CNA) Analyses
4.4. Structural Variant (SV) Analyses
5. Discussion
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Strand, M.; Prolla, T.A.; Liskay, R.M.; Petes, T.D. Destabilization of tracts of simple repetitive DNA in yeast by mutations affecting DNA mismatch repair. Nature 1993, 365, 274–276. [Google Scholar] [CrossRef] [PubMed]
- Shia, J. Evolving approach and clinical significance of detecting DNA mismatch repair deficiency in colorectal carcinoma. Semin. Diagn. Pathol. 2015, 32, 352–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sedhom, R.; Antonarakis, E.S. Clinical implications of mismatch repair deficiency in prostate cancer. Future Oncol. 2019, 15, 2395–2411. [Google Scholar] [CrossRef] [PubMed]
- Bancroft, E.K.; Page, E.C.; Brook, M.N.; Thomas, S.; Taylor, N.; Pope, J.; McHugh, J.; Jones, A.B.; Karlsson, Q.; Merson, S.; et al. A prospective prostate cancer screening programme for men with pathogenic variants in mismatch repair genes (IMPACT): Initial results from an international prospective study. Lancet Oncol. 2021, 22, 1618–1631. [Google Scholar] [CrossRef]
- Wyvekens, N.; Tsai, H.K.; Sholl, L.M.; Tucci, J.; Giannico, G.A.; Gordetsky, J.B.; Hirsch, M.S.; Barletta, J.A.; Acosta, A.M. Histopathological and genetic features of mismatch repair-deficient high-grade prostate cancer. Histopathology 2022, 80, 1050–1060. [Google Scholar] [CrossRef]
- Schweizer, M.T.; Cheng, H.H.; Tretiakova, M.S.; Vakar-Lopez, F.; Klemfuss, N.; Konnick, E.Q.; Mostaghel, E.A.; Nelson, P.S.; Yu, E.Y.; Montgomery, B.; et al. Mismatch repair deficiency may be common in ductal adenocarcinoma of the prostate. Oncotarget 2016, 7, 82504–82510. [Google Scholar] [CrossRef] [Green Version]
- Schweizer, M.T.; Antonarakis, E.S.; Bismar, T.A.; Guedes, L.B.; Cheng, H.H.; Tretiakova, M.S.; Vakar-Lopez, F.; Klemfuss, N.; Konnick, E.Q.; Mostaghel, E.A.; et al. Genomic Characterization of Prostatic Ductal Adenocarcinoma Identifies a High Prevalence of DNA Repair Gene Mutations. JCO Precis. Oncol. 2019, 3, 1–9. [Google Scholar] [CrossRef]
- Antonarakis, E.S.; Shaukat, F.; Isaacsson Velho, P.; Kaur, H.; Shenderov, E.; Pardoll, D.M.; Lotan, T.L. Clinical Features and Therapeutic Outcomes in Men with Advanced Prostate Cancer and DNA Mismatch Repair Gene Mutations. Eur. Urol. 2019, 75, 378–382. [Google Scholar] [CrossRef]
- Graham, L.S.; Montgomery, B.; Cheng, H.H.; Yu, E.Y.; Nelson, P.S.; Pritchard, C.; Erickson, S.; Alva, A.; Schweizer, M.T. Mismatch repair deficiency in metastatic prostate cancer: Response to PD-1 blockade and standard therapies. PLoS ONE 2020, 15, e0233260. [Google Scholar] [CrossRef]
- Nava Rodrigues, D.; Rescigno, P.; Liu, D.; Yuan, W.; Carreira, S.; Lambros, M.B.; Seed, G.; Mateo, J.; Riisnaes, R.; Mullane, S.; et al. Immunogenomic analyses associate immunological alterations with mismatch repair defects in prostate cancer. J. Clin. Investig. 2018, 128, 4441–4453. [Google Scholar] [CrossRef]
- Ritch, E.; Fu, S.Y.F.; Herberts, C.; Wang, G.; Warner, E.W.; Schonlau, E.; Taavitsainen, S.; Murtha, A.J.; Vandekerkhove, G.; Beja, K.; et al. Identification of Hypermutation and Defective Mismatch Repair in ctDNA from Metastatic Prostate Cancer. Clin. Cancer Res. 2020, 26, 1114–1125. [Google Scholar] [CrossRef] [PubMed]
- Abida, W.; Cheng, M.L.; Armenia, J.; Middha, S.; Autio, K.A.; Vargas, H.A.; Rathkopf, D.; Morris, M.J.; Danila, D.C.; Slovin, S.F.; et al. Analysis of the Prevalence of Microsatellite Instability in Prostate Cancer and Response to Immune Checkpoint Blockade. JAMA Oncol. 2019, 5, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Barata, P.; Agarwal, N.; Nussenzveig, R.; Gerendash, B.; Jaeger, E.; Hatton, W.; Ledet, E.; Lewis, B.; Layton, J.; Babiker, H.; et al. Clinical activity of pembrolizumab in metastatic prostate cancer with microsatellite instability high (MSI-H) detected by circulating tumor DNA. J. Immunother. Cancer 2020, 8, e001065. [Google Scholar] [CrossRef]
- Robinson, D.; Van Allen, E.M.; Wu, Y.M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.M.; Montgomery, B.; Taplin, M.E.; Pritchard, C.C.; Attard, G.; et al. Integrative clinical genomics of advanced prostate cancer. Cell 2015, 161, 1215–1228. [Google Scholar] [CrossRef] [Green Version]
- Chakravarty, D.; Gao, J.; Phillips, S.M.; Kundra, R.; Zhang, H.; Wang, J.; Rudolph, J.E.; Yaeger, R.; Soumerai, T.; Nissan, M.H.; et al. OncoKB: A Precision Oncology Knowledge Base. JCO Precis. Oncol. 2017, 1, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Ng, P.C.; Henikoff, S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003, 31, 3812–3814. [Google Scholar] [CrossRef] [Green Version]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting functional effect of human missense mutations using PolyPhen-2. Curr. Protoc. Hum. Genet. 2013, 76, 7–20. [Google Scholar] [CrossRef] [Green Version]
- Cancer Genome Atlas Research, N. The Molecular Taxonomy of Primary Prostate Cancer. Cell 2015, 163, 1011–1025. [Google Scholar]
- Mao, R.; Krautscheid, P.; Graham, R.P.; Ganguly, A.; Shankar, S.; Ferber, M.; Hegde, M.; Committee, A.L.Q.A. Genetic testing for inherited colorectal cancer and polyposis, 2021 revision: A technical standard of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 2021, 23, 1807–1817. [Google Scholar] [CrossRef]
- Marcus, L.; Lemery, S.J.; Keegan, P.; Pazdur, R. FDA Approval Summary: Pembrolizumab for the Treatment of Microsatellite Instability-High Solid Tumors. Clin. Cancer Res. 2019, 25, 3753–3758. [Google Scholar] [CrossRef] [Green Version]
- Antonarakis, E.S.; Piulats, J.M.; Gross-Goupil, M.; Goh, J.; Ojamaa, K.; Hoimes, C.J.; Vaishampayan, U.; Berger, R.; Sezer, A.; Alanko, T.; et al. Pembrolizumab for Treatment-Refractory Metastatic Castration-Resistant Prostate Cancer: Multicohort, Open-Label Phase II KEYNOTE-199 Study. J. Clin. Oncol. 2020, 38, 395–405. [Google Scholar] [CrossRef] [PubMed]
- Sokol, E.S.; Jin, D.X.; Fine, A.; Trabucco, S.E.; Maund, S.; Frampton, G.; Molinero, L.; Antonarakis, E.S. PARP Inhibitor Insensitivity to BRCA1/2 Monoallelic Mutations in Microsatellite Instability-High Cancers. JCO Precis. Oncol. 2022, 6, e2100531. [Google Scholar] [CrossRef] [PubMed]
- Safran, M.; Dalah, I.; Alexander, J.; Rosen, N.; Iny Stein, T.; Shmoish, M.; Nativ, N.; Bahir, I.; Doniger, T.; Krug, H.; et al. GeneCards Version 3: The human gene integrator. Database 2010, 2010, baq020. [Google Scholar] [CrossRef] [Green Version]
- Kantidakis, T.; Saponaro, M.; Mitter, R.; Horswell, S.; Kranz, A.; Boeing, S.; Aygun, O.; Kelly, G.P.; Matthews, N.; Stewart, A.; et al. Mutation of cancer driver MLL2 results in transcription stress and genome instability. Genes Dev. 2016, 30, 408–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.; Chow, R.D.; Zhu, L.; Bai, Z.; Ye, L.; Zhang, F.; Renauer, P.A.; Dong, M.B.; Dai, X.; Zhang, X.; et al. CRISPR-GEMM Pooled Mutagenic Screening Identifies KMT2D as a Major Modulator of Immune Checkpoint Blockade. Cancer Discov. 2020, 10, 1912–1933. [Google Scholar] [CrossRef]
- Lv, S.; Ji, L.; Chen, B.; Liu, S.; Lei, C.; Liu, X.; Qi, X.; Wang, Y.; Lai-Han Leung, E.; Wang, H.; et al. Histone methyltransferase KMT2D sustains prostate carcinogenesis and metastasis via epigenetically activating LIFR and KLF4. Oncogene 2018, 37, 1354–1368. [Google Scholar] [CrossRef] [Green Version]
- Gruber, C.N.; Calis, J.J.A.; Buta, S.; Evrony, G.; Martin, J.C.; Uhl, S.A.; Caron, R.; Jarchin, L.; Dunkin, D.; Phelps, R.; et al. Complex Autoinflammatory Syndrome Unveils Fundamental Principles of JAK1 Kinase Transcriptional and Biochemical Function. Immunity 2020, 53, 672–684.e11. [Google Scholar] [CrossRef]
- Zaretsky, J.M.; Garcia-Diaz, A.; Shin, D.S.; Escuin-Ordinas, H.; Hugo, W.; Hu-Lieskovan, S.; Torrejon, D.Y.; Abril-Rodriguez, G.; Sandoval, S.; Barthly, L.; et al. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N. Engl. J. Med. 2016, 375, 819–829. [Google Scholar] [CrossRef]
- Sena, L.A.; Fountain, J.; Isaacsson Velho, P.; Lim, S.J.; Wang, H.; Nizialek, E.; Rathi, N.; Nussenzveig, R.; Maughan, B.L.; Velez, M.G.; et al. Tumor Frameshift Mutation Proportion Predicts Response to Immunotherapy in Mismatch Repair-Deficient Prostate Cancer. Oncologist 2021, 26, e270–e278. [Google Scholar] [CrossRef]
- Chaudry, M.A.; Sales, K.; Ruf, P.; Lindhofer, H.; Winslet, M.C. EpCAM an immunotherapeutic target for gastrointestinal malignancy: Current experience and future challenges. Br. J. Cancer 2007, 96, 1013–1019. [Google Scholar] [CrossRef]
- Ligtenberg, M.J.; Kuiper, R.P.; Chan, T.L.; Goossens, M.; Hebeda, K.M.; Voorendt, M.; Lee, T.Y.; Bodmer, D.; Hoenselaar, E.; Hendriks-Cornelissen, S.J.; et al. Heritable somatic methylation and inactivation of MSH2 in families with Lynch syndrome due to deletion of the 3’ exons of TACSTD1. Nat. Genet. 2009, 41, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Kloor, M.; Voigt, A.Y.; Schackert, H.K.; Schirmacher, P.; von Knebel Doeberitz, M.; Blaker, H. Analysis of EPCAM protein expression in diagnostics of Lynch syndrome. J. Clin. Oncol. 2011, 29, 223–227. [Google Scholar] [CrossRef]
- Ryan, S.; Jenkins, M.A.; Win, A.K. Risk of prostate cancer in Lynch syndrome: A systematic review and meta-analysis. Cancer Epidemiol. Biomark. Prev. 2014, 23, 437–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomlins, S.A.; Rhodes, D.R.; Perner, S.; Dhanasekaran, S.M.; Mehra, R.; Sun, X.W.; Varambally, S.; Cao, X.; Tchinda, J.; Kuefer, R.; et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science 2005, 310, 644–648. [Google Scholar] [CrossRef] [PubMed]
- King, J.C.; Xu, J.; Wongvipat, J.; Hieronymus, H.; Carver, B.S.; Leung, D.H.; Taylor, B.S.; Sander, C.; Cardiff, R.D.; Couto, S.S.; et al. Cooperativity of TMPRSS2-ERG with PI3-kinase pathway activation in prostate oncogenesis. Nat. Genet. 2009, 41, 524–526. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Yuan, Q.; Di, W.; Xia, X.; Liu, Z.; Mao, N.; Li, L.; Li, C.; He, J.; Li, Y.; et al. ERG orchestrates chromatin interactions to drive prostate cell fate reprogramming. J. Clin. Investig. 2020, 130, 5924–5941. [Google Scholar] [CrossRef] [PubMed]
- Deshpande, M.; Romanski, P.A.; Rosenwaks, Z.; Gerhardt, J. Gynecological Cancers Caused by Deficient Mismatch Repair and Microsatellite Instability. Cancers 2020, 12, 3319. [Google Scholar] [CrossRef]
- Mayakonda, A.; Lin, D.C.; Assenov, Y.; Plass, C.; Koeffler, H.P. Maftools: Efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 2018, 28, 1747–1756. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SV Type | Primary MMR-d | Primary MMR-p | p-Value | Metastatic MMR-d | Metastatic MMR-p | p-Value |
|---|---|---|---|---|---|---|
| TMPRSS2-ETS | ||||||
| Present | 8 (32.0%) | 783 (54.2%) | 0.041 | 11 (35.5%) | 360 (48.3%) | 0.200 |
| Absent | 17 (68.0%) | 661 (45.8%) | 20 (64.5%) | 386 (51.7%) | ||
| TMPRSS2-ERG | ||||||
| Present | 8 (32.0%) | 755 (52.3%) | 0.067 | 10 (32.3%) | 353 (47.3%) | 0.141 |
| Absent | 17 (68.0%) | 689 (47.7%) | 21 (67.7%) | 393 (52.7%) | ||
| ETS-others | ||||||
| Present | 0 (0%) | 62 (4.3%) | 0.622 | 1 (3.2%) | 22 (2.9%) | 0.613 |
| Absent | 25 (100%) | 1382 (95.7%) | 30 (96.8%) | 724 (97.1%) | ||
| ERG-others | ||||||
| Present | 0 (0%) | 32 (2.2%) | 1.000 | 1 (3.2%) | 6 (0.8%) | 0.249 |
| Absent | 25 (100%) | 1412 (97.8%) | 30 (96.8%) | 740 (99.2%) | ||
| TMPRSS2-intragenic | ||||||
| Present | 1 (5.0%) | 75 (7.4%) | 1.000 | 1 (3.2%) | 54 (7.2%) | 0.718 |
| Absent | 19 (95.0%) | 943 (92.6%) | 30 (96.8%) | 692 (92.8%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fang, B.; Wei, Y.; Pan, J.; Zhang, T.; Ye, D.; Zhu, Y. The Somatic Mutational Landscape of Mismatch Repair Deficient Prostate Cancer. J. Clin. Med. 2023, 12, 623. https://doi.org/10.3390/jcm12020623
Fang B, Wei Y, Pan J, Zhang T, Ye D, Zhu Y. The Somatic Mutational Landscape of Mismatch Repair Deficient Prostate Cancer. Journal of Clinical Medicine. 2023; 12(2):623. https://doi.org/10.3390/jcm12020623
Chicago/Turabian StyleFang, Bangwei, Yu Wei, Jian Pan, Tingwei Zhang, Dingwei Ye, and Yao Zhu. 2023. "The Somatic Mutational Landscape of Mismatch Repair Deficient Prostate Cancer" Journal of Clinical Medicine 12, no. 2: 623. https://doi.org/10.3390/jcm12020623
APA StyleFang, B., Wei, Y., Pan, J., Zhang, T., Ye, D., & Zhu, Y. (2023). The Somatic Mutational Landscape of Mismatch Repair Deficient Prostate Cancer. Journal of Clinical Medicine, 12(2), 623. https://doi.org/10.3390/jcm12020623