
Temporal Changes in the Skin Microbiome of Epidermolysis Bullosa Patients following the Application of Wound Dressings

 , , and
, , and
Abstract
:1. Introduction
2. Patients and Methods
2.1. Study Design
2.2. Study Population
2.3. Sample Collection
2.4. DNA Extraction and 16S rRNA Gene Amplicon Sequencing
2.5. Data Processing and Analysis
3. Results
3.1. General Description of the Cohort
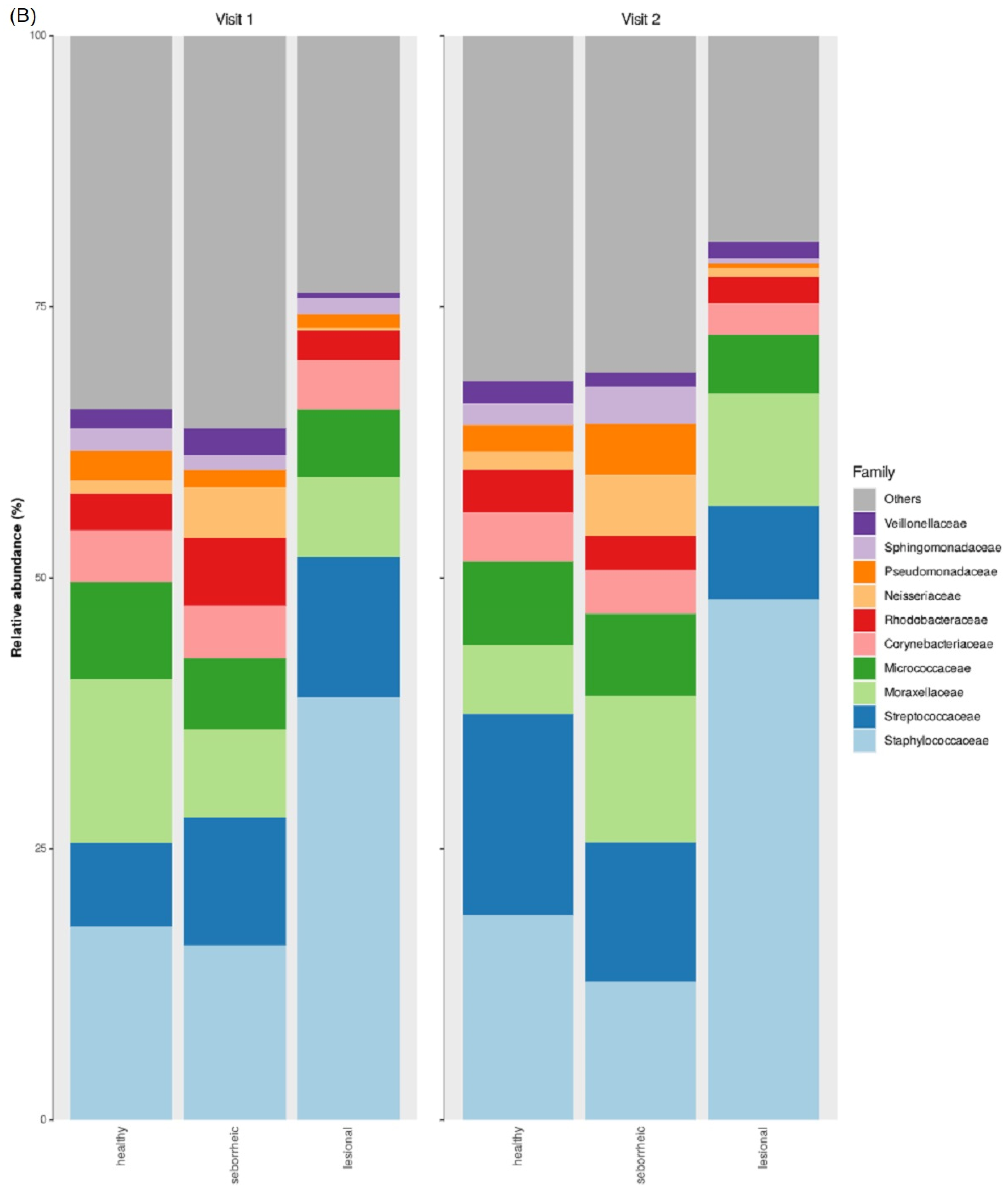
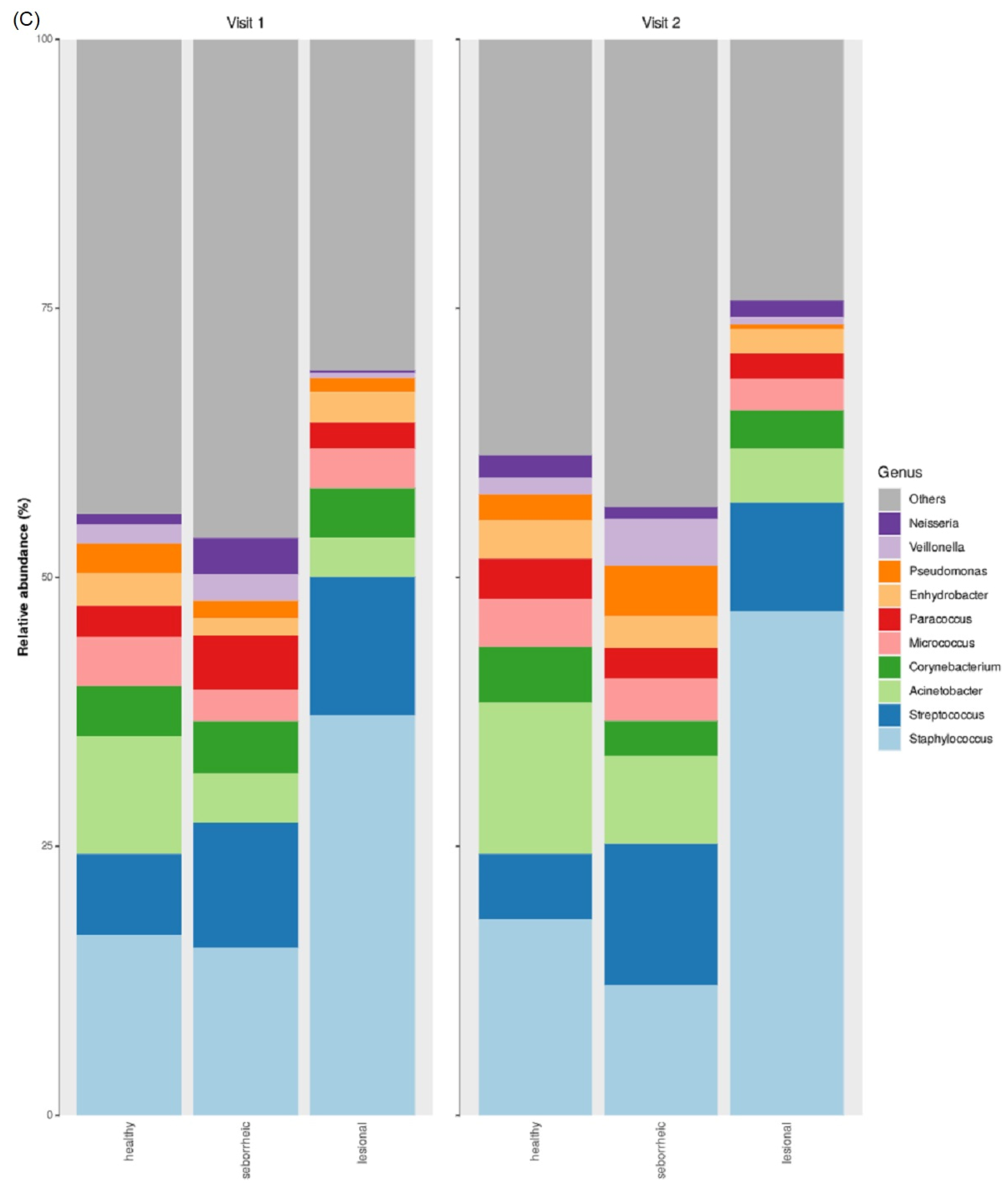
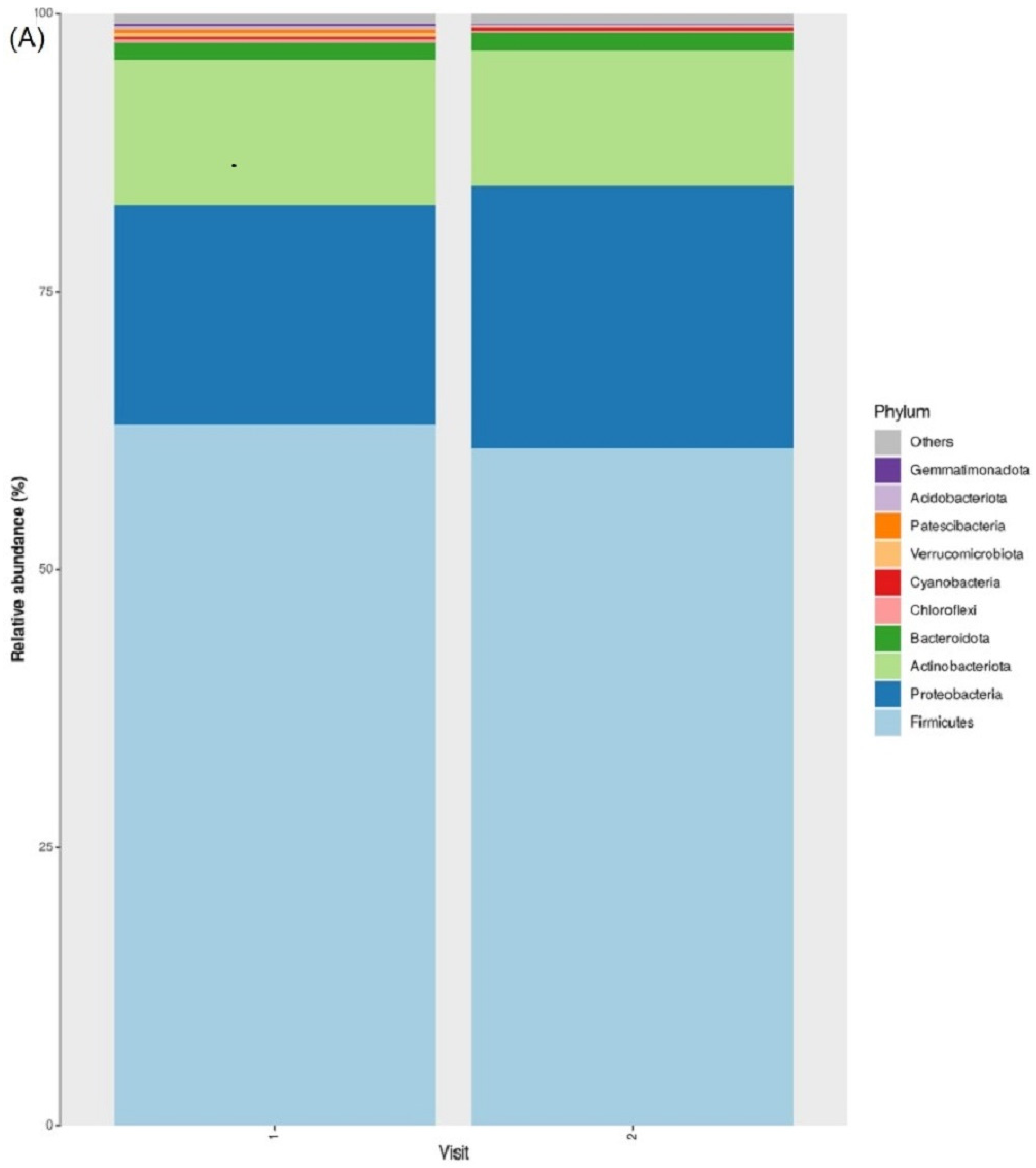
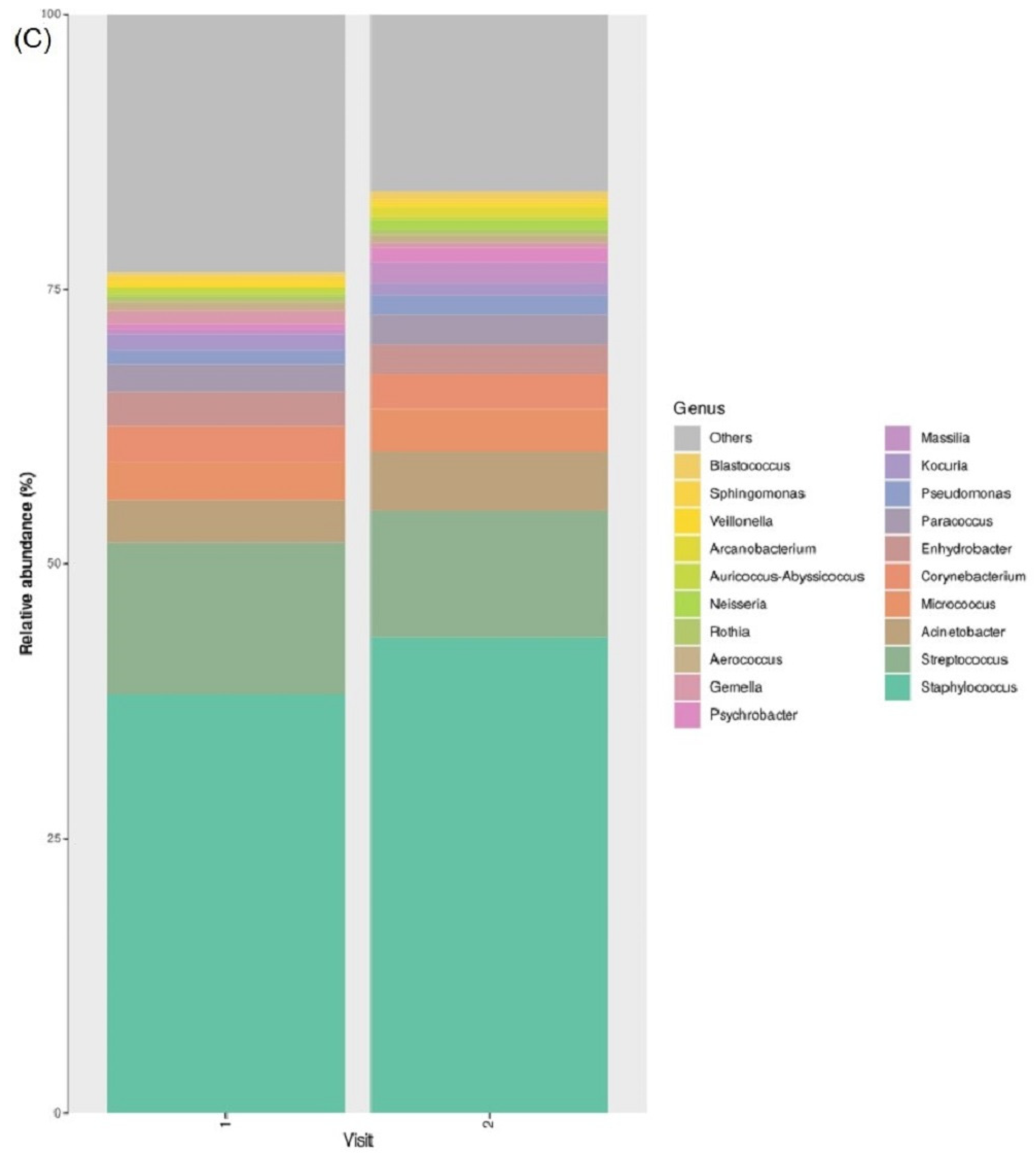
3.2. Taxonomic Abundance
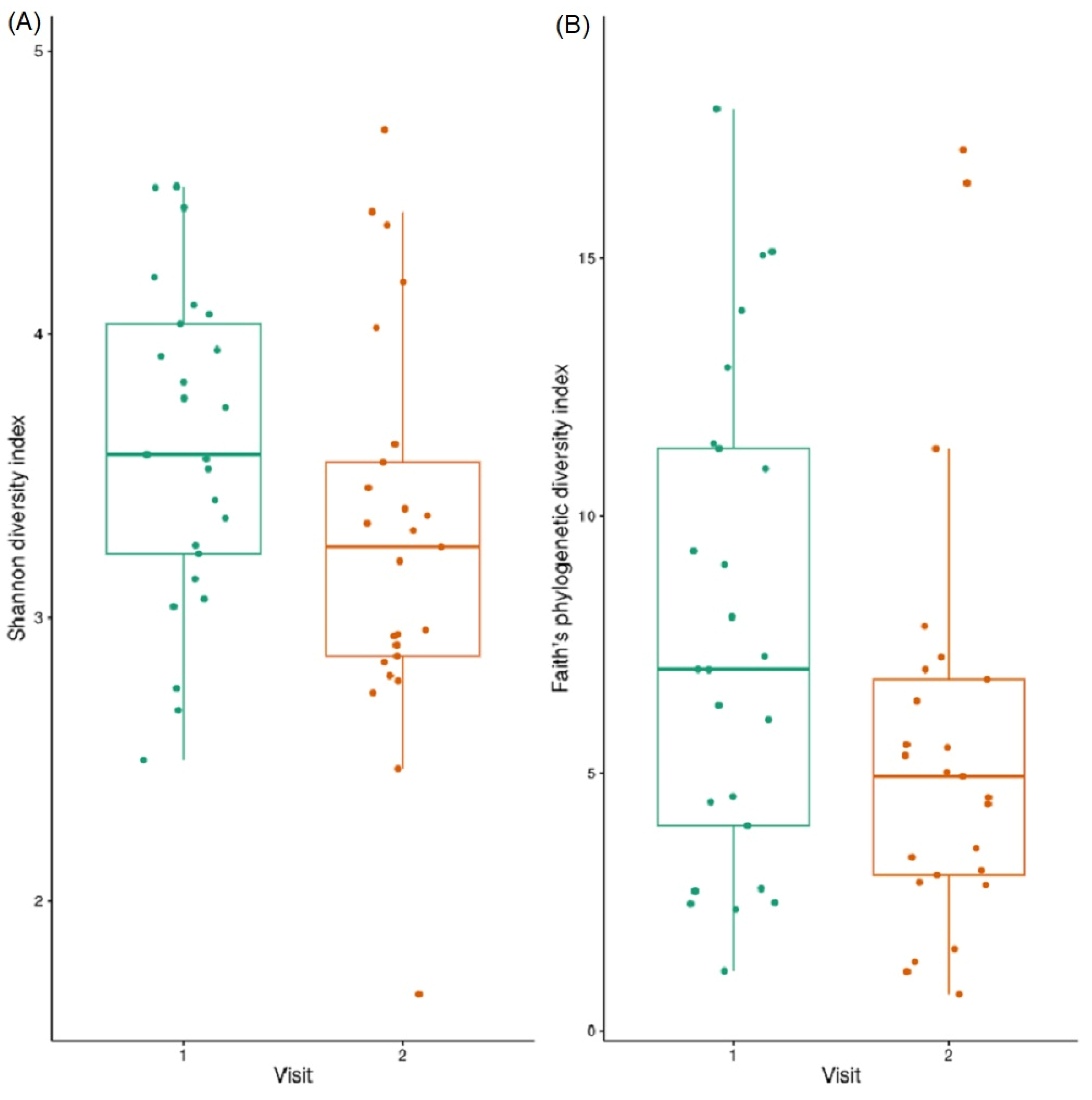
3.3. Alpha Diversity
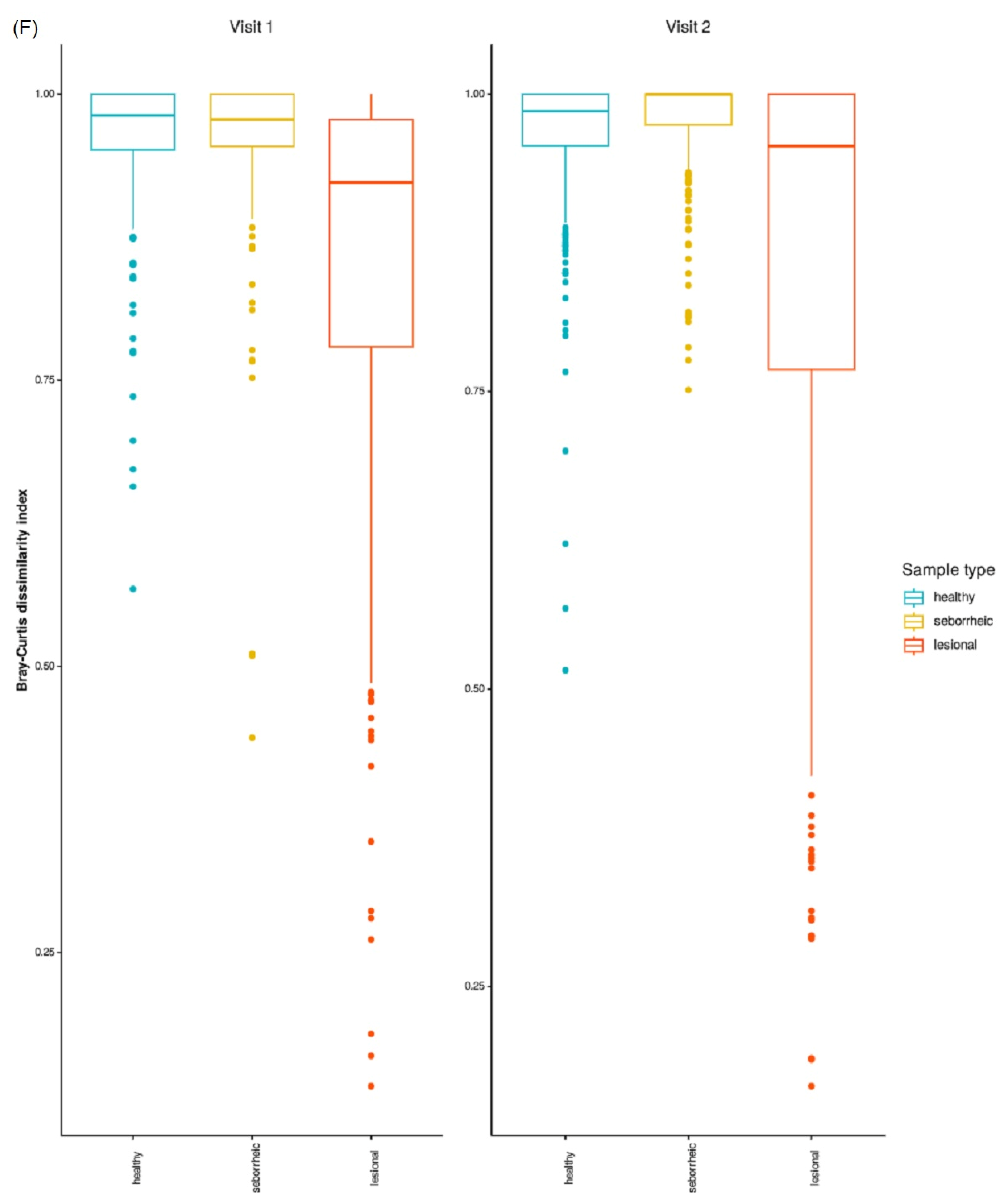
3.4. Beta Diversity
3.5. Differences in Microbiome Taxonomic Composition Per Sample Type
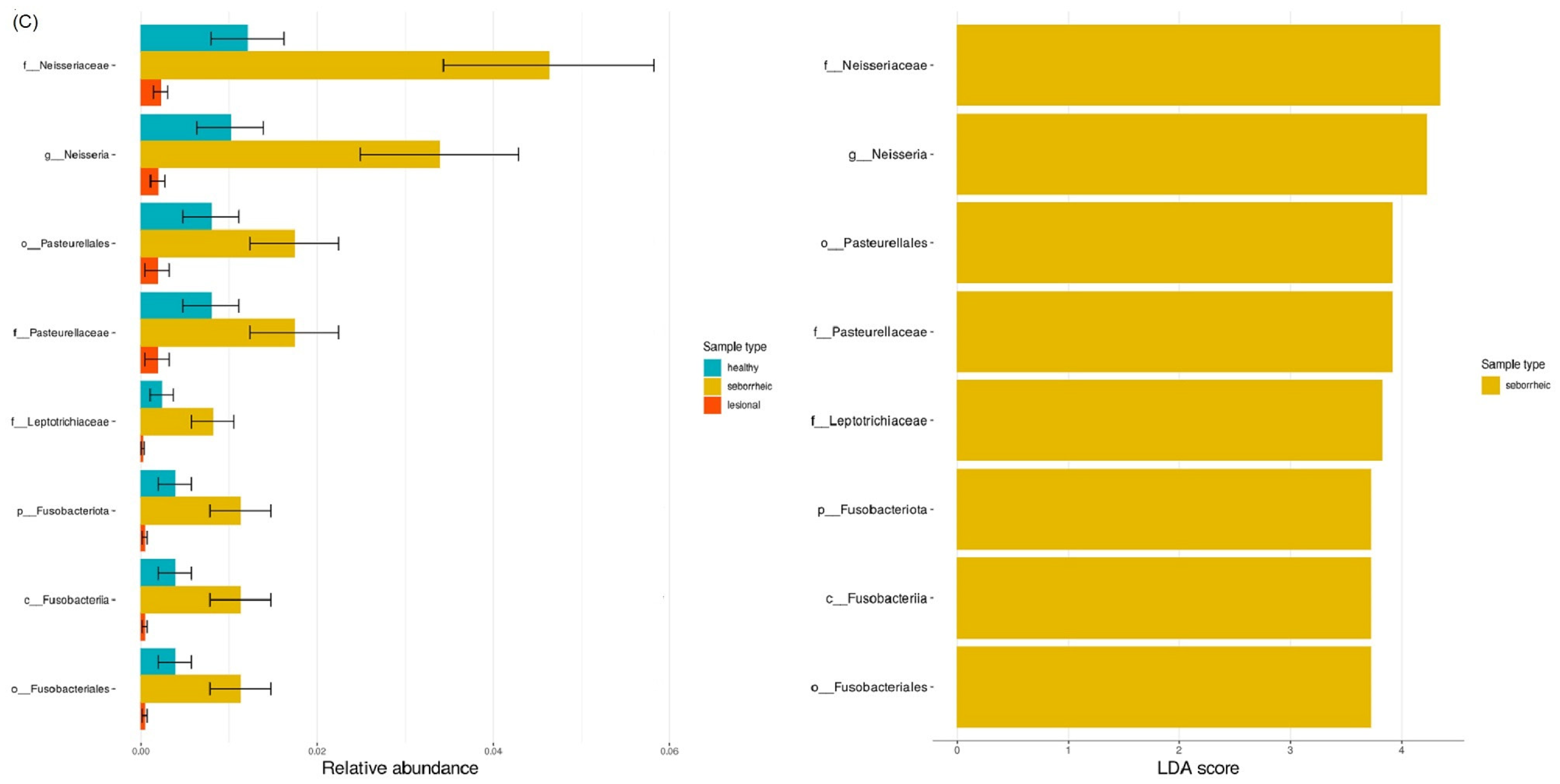
3.6. LefSE Biomarker-Based Differential Abundance between Sample Types
3.7. Taxonomic Abundance
3.8. Alpha Diversity
3.9. Beta Diversity
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pânzaru, M.-C.; Caba, L.; Florea, L.; Braha, E.E.; Gorduza, E.V. Epidermolysis Bullosa—A Different Genetic Approach in Correlation with Genetic Heterogeneity. Diagnostics 2022, 12, 1325. [Google Scholar] [CrossRef]
- Alexeev, V.; Huitema, L.; Phillips, T.; Cepeda, R.; Cobos, D.d.L.; Perez, R.I.M.; Salas-Garza, M.; Fajardo-Ramirez, O.R.; Ringpfeil, F.; Uitto, J.; et al. T-cell activation and bacterial infection in skin wounds of recessive dystrophic epidermolysis bullosa patients. Exp. Dermatol. 2022, 31, 1431–1442. [Google Scholar] [CrossRef]
- Frank, D.N.; Wysocki, A.; Specht-Glick, D.D.; Rooney, A.; Feldman, R.A.; Amand, A.L.S.; Pace, N.R.; Trent, J.D. Microbial diversity in chronic open wounds. Wound Repair Regen. 2009, 17, 163–172. [Google Scholar] [CrossRef]
- Grice, E.A.; Segre, J.A. The skin microbiome. Nat. Rev. Microbiol. 2011, 9, 244–253. [Google Scholar] [CrossRef]
- Sandhu, S.S.; Pourang, A.; Sivamani, R.K. A review of next generation sequencing technologies used in the evaluation of the skin microbiome: What a time to be alive. Dermatol. Online J. 2019, 25, 13030/qt3hv5z3q3. [Google Scholar] [CrossRef]
- Naqib, A.; Poggi, S.; Wang, W.; Hyde, M.; Kunstman, K.; Green, S.J. Making and Sequencing Heavily Multiplexed, High-Throughput 16S Ribosomal RNA Gene Amplicon Libraries Using a Flexible, Two-Stage PCR Protocol. Methods Mol. Biol. 2018, 1783, 149–169. [Google Scholar] [CrossRef]
- Walters, W.; Hyde, E.R.; Berg-Lyons, D.; Ackermann, G.; Humphrey, G.; Parada, A.; Gilbert, J.A.; Jansson, J.K.; Caporaso, J.G.; Fuhrman, J.A.; et al. Improved Bacterial 16S rRNA Gene (V4 and V4-5) and Fungal Internal Transcribed Spacer Marker Gene Primers for Microbial Community Surveys. mSystems 2015, 1, e00009–e00015. [Google Scholar] [CrossRef]
- Hooper, M.J.; Enriquez, G.L.; Veon, F.L.; LeWitt, T.M.; Sweeney, D.; Green, S.J.; Seed, P.C.; Choi, J.; Guitart, J.; Burns, M.B.; et al. Narrowband ultraviolet B response in cutaneous T-cell lymphoma is characterized by increased bacterial diversity and reduced Staphylococcus aureus and Staphylococcus lugdunensis. Front. Immunol. 2022, 13, 1022093. [Google Scholar] [CrossRef]
- Hooper, M.J.; LeWitt, T.M.; Veon, F.L.; Pang, Y.; Chlipala, G.E.; Feferman, L.; Green, S.J.; Sweeney, D.; Bagnowski, K.T.; Burns, M.B.; et al. Nasal Dysbiosis in Cutaneous T-Cell Lymphoma Is Characterized by Shifts in Relative Abundances of Non-Staphylococcus Bacteria. JID Innov. 2022, 2, 100132. [Google Scholar] [CrossRef]
- Kostka, J.E.; Green, S.J.; Rishishwar, L.; Prakash, O.; Katz, L.S.; Mariño-Ramírez, L.; Jordan, I.K.; Munk, C.; Ivanova, N.; Mikhailova, N.; et al. Genome sequences for six rhodanobacter strains, isolated from soils and the terrestrial subsurface, with variable denitrification capabilities. J. Bacteriol. 2012, 194, 4461–4462. [Google Scholar] [CrossRef]
- Brandine, G.d.S.; Smith, A.D. Falco: High-speed FastQC emulation for quality control of sequencing data. F1000Research 2019, 8, 1874. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Chakoory, O.; Comtet-Marre, S.; Peyret, P. RiboTaxa: Combined approaches for rRNA genes taxonomic resolution down to the species level from metagenomics data revealing novelties. NAR Genom. Bioinform. 2022, 4, lqac070. [Google Scholar] [CrossRef] [PubMed]
- Robeson, M.S.; O’Rourke, D.R.; Kaehler, B.D.; Ziemski, M.; Dillon, M.R.; Foster, J.T.; Bokulich, N.A. RESCRIPt: Reproducible sequence taxonomy reference database management. PLoS Comput. Biol. 2021, 17, e1009581. [Google Scholar] [CrossRef] [PubMed]
- Bokulich, N.A.; Kaehler, B.D.; Rideout, J.R.; Dillon, M.; Bolyen, E.; Knight, R.; Huttley, G.A.; Caporaso, J.G. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome 2018, 6, 90. [Google Scholar] [CrossRef]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef]
- Liu, C.; Cui, Y.; Li, X.; Yao, M. microeco: An R package for data mining in microbial community ecology. FEMS Microbiol. Ecol. 2021, 97, fiaa255. [Google Scholar] [CrossRef]
- Bar, J.; Sarig, O.; Lotan-Pompan, M.; Dassa, B.; Miodovnik, M.; Weinberger, A.; Sprecher, E.; Segal, E.; Samuelov, L. Evidence for cutaneous dysbiosis in dystrophic epidermolysis bullosa. Clin. Exp. Dermatol. 2021, 46, 1223–1229. [Google Scholar] [CrossRef]
- Reimer-Taschenbrecker, A.; Künstner, A.; Hirose, M.; Hübner, S.; Gewert, S.; Ibrahim, S.; Busch, H.; Has, C. Predominance of Staphylococcus Correlates with Wound Burden and Disease Activity in Dystrophic Epidermolysis Bullosa: A Prospective Case-Control Study. J. Investig. Dermatol. 2022, 142, 2117–2127.e8. [Google Scholar] [CrossRef]
- Ha, B.; Kd, M. Common wound colonizers in patients with epidermolysis bullosa. Pediatr. Dermatol. 2010, 27, 25–28. [Google Scholar] [CrossRef]
- van der Kooi-Pol, M.M.; Sadabad, M.S.; Duipmans, J.C.; Sabat, A.J.; Stobernack, T.; Omansen, T.F.; Westerhout-Pluister, G.N.; Jonkman, M.F.; Harmsen, H.J.M.; van Dijl, J.M. Topography of Distinct Staphylococcus aureus Types in Chronic Wounds of Patients with Epidermolysis Bullosa. PLoS ONE 2013, 8, e67272. [Google Scholar] [CrossRef]
- van der Kooi-Pol, M.M.; Duipmans, J.C.; Jonkman, M.F.; van Dijl, J.M. Host–pathogen interactions in epidermolysis bullosa patients colonized with Staphylococcus aureus. Int. J. Med. Microbiol. 2014, 304, 195–203. [Google Scholar] [CrossRef]
- Kooi-Pol, M.M.; Veenstra-Kyuchukova, Y.K.; Duipmans, J.C.; Pluister, G.N.; Schouls, L.M.; Neeling, A.J.; Grundmann, H.; Jonkman, M.F.; Dijl, J.M. High genetic diversity of Staphylococcus aureus strains colonizing patients with epidermolysis bullosa. Exp. Dermatol. 2012, 21, 463–466. [Google Scholar] [CrossRef]
- Fuentes, I.; Yubero, M.J.; Morandé, P.; Varela, C.; Oróstica, K.; Acevedo, F.; Rebolledo-Jaramillo, B.; Arancibia, E.; Porte, L.; Palisson, F. Longitudinal study of wound healing status and bacterial colonisation of Staphylococcus aureus and Corynebacterium diphtheriae in epidermolysis bullosa patients. Int. Wound J. 2023, 20, 774–783. [Google Scholar] [CrossRef]
- Kalan, L.R.; Brennan, M.B. The role of the microbiome in nonhealing diabetic wounds. Ann. N. Y. Acad. Sci. 2019, 1435, 79–92. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient No. | Age (Years) | Sex | Ethnicity | EB Type | Diagnosis | Genetic Mutation |
|---|---|---|---|---|---|---|
| 1 | 16 | Female | Jewish | Simplex localized | Clinical | N/A |
| 2 | 5 | Female | Bedouin | Simplex localized | Clinical | N/A |
| 3 | 7 | Male | Bedouin | Simplex localized | Clinical | N/A |
| 4 | 11 | Female | Bedouin | Simplex localized | Clinical | N/A |
| 5 | 9 | Male | Bedouin | Simplex localized | Clinical | N/A |
| 6 | 7 | Male | Bedouin | Simplex localized | Clinical | N/A |
| 7 | 9 | Female | Bedouin | Simplex localized | Clinical | N/A |
| 8 | 6 | Male | Bedouin | Simplex localized | Clinical | N/A |
| 9 | 3 | Male | Bedouin | Simplex localized | Clinical | N/A |
| 10 | 0.1 | Male | Bedouin | Simplex localized | Clinical | N/A |
| 11 | 2 | Male | Bedouin | Simplex localized | Clinical | N/A |
| 12 | 7 | Male | Bedouin | Simplex localized | Clinical | N/A |
| 13 | 14 | Male | Bedouin | Simplex localized | Clinical | N/A |
| 14 | 6 | Male | Bedouin | Simplex localized | Clinical | N/A |
| 15 | 5 | Male | Bedouin | Simplex localized | Clinical | N/A |
| 16 | 2 | Female | Bedouin | Simplex localized | Clinical | N/A |
| 17 | 2 | Female | Bedouin | Simplex localized | Clinical | N/A |
| 18 | 3 | Male | Bedouin | Simplex localized | Clinical | N/A |
| 19 | 5 | Female | Bedouin | Simplex localized | Clinical | N/A |
| 20 | 4 | Male | Bedouin | Simplex localized | Genetic | KRT14 |
| 21 | 1 | Female | Bedouin | Simplex generalized | Genetic | KRT14 |
| 22 | 13 | Male | Bedouin | Simplex generalized | Genetic | KRT14 |
| 23 | 0.9 | Male | Jewish | Simplex generalized | Genetic | KRT14 |
| 24 | 1 | Female | Bedouin | Simplex generalized | Genetic | KRT14 |
| 25 | 0.1 | Male | Bedouin | Simplex generalized | Genetic | KRT14 |
| 26 | 1 | Male | Bedouin | Simplex generalized | Genetic | KRT14 |
| 27 | 4 | Male | Bedouin | Simplex generalized | Genetic | PLEC |
| 28 | 1 | Female | Jewish | Simplex generalized | Genetic | PLEC |
| 29 | 0.5 | Male | Bedouin | Dystrophic recessive | Genetic | COL7A |
| 30 | 2 | Male | Bedouin | Dystrophic recessive | Genetic | COL7A |
| 31 | 2 | Male | Bedouin | Dystrophic recessive | Genetic | COL7A |
| 32 | 0.6 | Male | Bedouin | Dystrophic recessive | Genetic | COL7A |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Horev, A.; Brandwein, M.; Vaknin, A.; Motro, Y.; Moran-Gilad, J. Temporal Changes in the Skin Microbiome of Epidermolysis Bullosa Patients following the Application of Wound Dressings. J. Clin. Med. 2023, 12, 6435. https://doi.org/10.3390/jcm12206435
Horev A, Brandwein M, Vaknin A, Motro Y, Moran-Gilad J. Temporal Changes in the Skin Microbiome of Epidermolysis Bullosa Patients following the Application of Wound Dressings. Journal of Clinical Medicine. 2023; 12(20):6435. https://doi.org/10.3390/jcm12206435
Chicago/Turabian StyleHorev, Amir, Michael Brandwein, Avraham Vaknin, Yair Motro, and Jacob Moran-Gilad. 2023. "Temporal Changes in the Skin Microbiome of Epidermolysis Bullosa Patients following the Application of Wound Dressings" Journal of Clinical Medicine 12, no. 20: 6435. https://doi.org/10.3390/jcm12206435
APA StyleHorev, A., Brandwein, M., Vaknin, A., Motro, Y., & Moran-Gilad, J. (2023). Temporal Changes in the Skin Microbiome of Epidermolysis Bullosa Patients following the Application of Wound Dressings. Journal of Clinical Medicine, 12(20), 6435. https://doi.org/10.3390/jcm12206435