
Chiari Syndrome: Advances in Epidemiology and Pathogenesis: A Systematic Review

 ,
,  , , and
, , and 
Abstract
:1. Introduction
- -
- Arnold Chiari malformation type I (CM-I): this is the most common. There is a descent of one or both cerebellar tonsils 5 mm or more below the foramen magnum. It is usually accompanied by syringomyelia.
- -
- Arnold Chiari malformation type II (CM-II) presents with herniation of the brainstem, and the cerebellum is impotent. It is usually accompanied by spinal dysraphism/myelomeningocele.
- -
- Arnold Chiari malformation type III (CM-III): the entire hindbrain is herniated (with or without brainstem) within a cervical meningoencephalocele.
- -
- Arnold Chiari malformation type IV (CM-IV): severe cerebellar hypoplasia.
2. Materials and Methods
2.1. Eligibility Criteria
2.2. Sources of Information
2.3. Search Strategy
2.4. Data Extraction Process
2.5. Data Collection Process and Data Collected
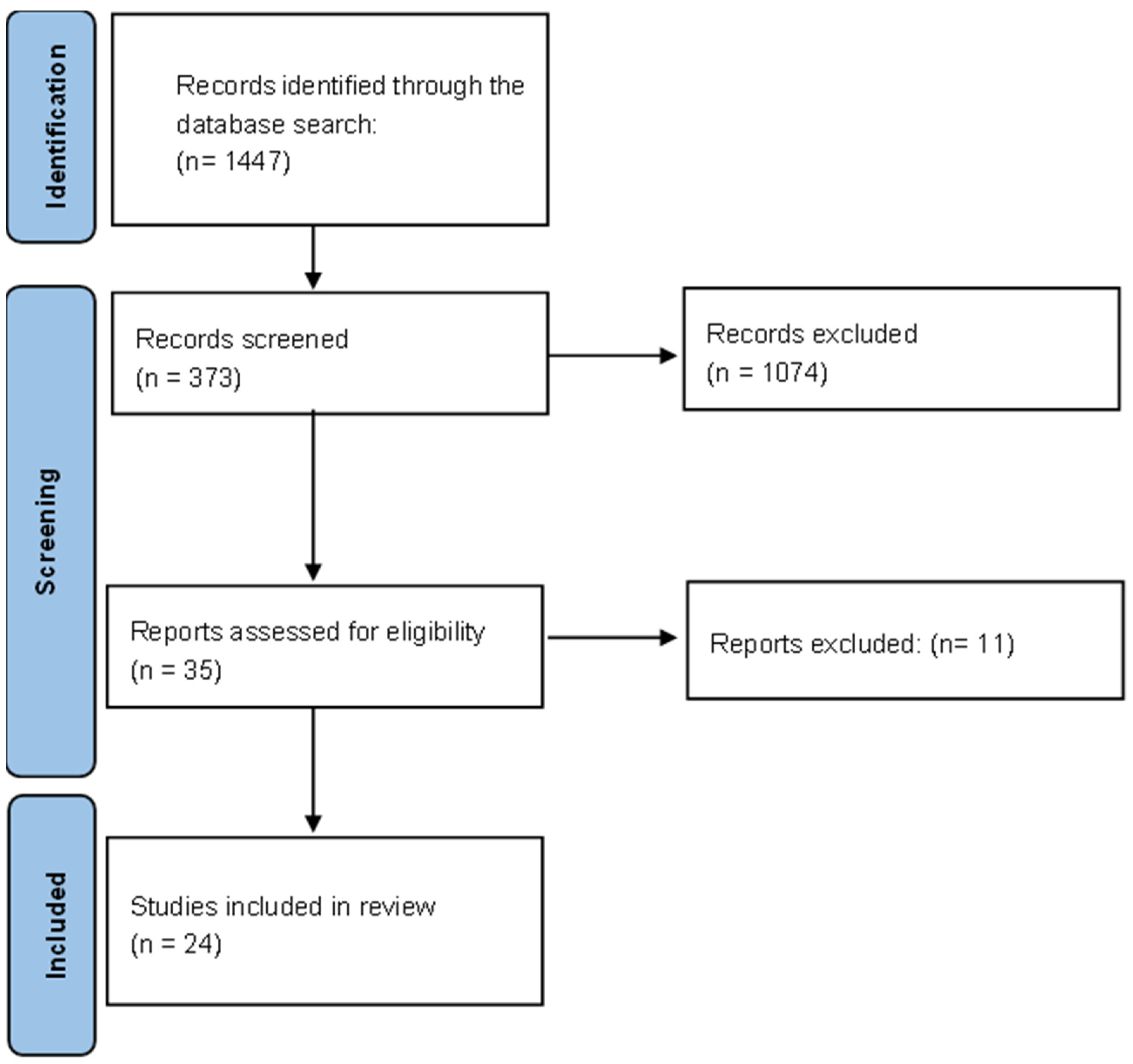
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pearce, J.M.S. Arnold Chiari, or “Cruveilhier Cleland Chiari” Malformation. J. Neurol. Neurosurg. Psychiatry 2000, 68, 13. Available online: https://jnnp.bmj.com/content/68/1/13.short (accessed on 10 July 2023). [CrossRef] [PubMed]
- Hidalgo, J.; Tork, C.; Varacallo, M. Arnold Chiari Malformation. StatPearls. Treasure Island. 2022. Available online: https://www.ncbi.nlm.nih.gov/books/NBK431076/ (accessed on 10 July 2023).
- Fernandes, Y.; Ramina, R.; Campos-Herrera, R.; Borges, G. Evolutionary Hypothesis for Chiari Type I Malformation. Med. Hypotheses 2013, 81, 715–719. Available online: https://www.sciencedirect.com/science/article/abs/pii/S0306987713003599?casa_token=SEhsTobxqdAAAAAA:viA4FKM3iWQa85EYVNYjkx_2H2A0on2ChlRywHS8V0OJ0T6x-Vs7sper52gLJaqwbM6ems54Cw1 (accessed on 10 July 2023). [CrossRef] [PubMed]
- Carrillo-Esper, R.; Vázquez-Elizondo, G.; Gutiérrez-Delgado, L.; Guevara-Arnal, L.; Méndez-Sánchez, N. Malformación de Arnold-Chiari Tipo I, Siringomielia, Siringobulbia y Atrapamiento del Ventrículo IV. Gac. Médica México 2008, 144, 351–354. Available online: https://www.medigraphic.com/cgi-bin/new/resumen.cgi?IDARTICULO=19336 (accessed on 10 July 2023).
- Kular, S.; Cascella, M. Chiari I Malformation. StatPearls. 2022. Available online: https://www.ncbi.nlm.nih.gov/books/NBK554609/ (accessed on 10 July 2023).
- Orphanet. Malformación de Arnold-Chiari Tipo I. Available online: https://www.orpha.net/consor/cgi-bin/Disease_Search.php?lng=ES&data_id=20220&Disease (accessed on 10 July 2023).
- Sahuquillo, J.; Poca, M. Actualizaciones en el tratamiento quirúrgico de la malformación de Chiari tipo I y del complejo Chiari-I/siringomielia. Neurología 1998, 13, 223–245. [Google Scholar]
- De La Peña-León, B.; Dávalos-Alcázar, A.; Valdez-Labastida, R. Intervenciones de la enfermería en un agente de cuidado dependiente con malformación de Arnold-Chiari. Rev. Enfermería Neurológica 2014, 13, 87–94. [Google Scholar] [CrossRef]
- Ciaramitaro, P.; Garbossa, D.; Peretta, P.; Piatelli, G.; Massimi, L.; Valentini, L.G.; Migliaretti, G.; Baldovino, S.; Roccatello, D.; Kodra, Y.; et al. Syringomyelia and Chiari Syndrome Registry: Advances in epidemiology, clinical phenotypes and natural history based on a North Western Italy cohort. Ann. Ist. Super. Sanita 2020, 56, 48–58. [Google Scholar]
- Provenzano, A.; La Barbera, A.; Scagnet, M.; Pagliazzi, A.; Traficante, G.; Pantaleo, M.; Tiberi, L.; Vergani, D.; Kurtas, N.E.; Guarducci, S.; et al. Chiari 1 malformation and exome sequencing in 51 trios: The emerging role of rare missense variants in chromatin-remodeling genes. Hum. Genet. 2021, 140, 625–647. [Google Scholar] [CrossRef]
- Sadler, B.; Wilborn, J.; Antunes, L.; Kuensting, T.; Hale, A.T.; Gannon, S.R.; McCall, K.; Cruchaga, C.; Harms, M.; Voisin, N.; et al. Rare and de novo coding variants in chromodomain genes in Chiari I malformation. Am. J. Hum. Genet. 2021, 108, 100–114. [Google Scholar] [CrossRef]
- Musolf, A.M.; Ho, W.S.; Long, K.A.; Zhuang, Z.; Argersinger, D.P.; Sun, H.; Moiz, B.A.; Simpson, C.L.; Mendelevich, E.G.; Bogdanov, E.I.; et al. Small posterior fossa in Chiari I malformation affected families is significantly linked to 1q43-44 and 12q23-24.11 using whole exome sequencing. Eur. J. Hum. Genet. 2019, 27, 1599–1610. [Google Scholar] [CrossRef]
- Urbizu, A.; Garrett, M.E.; Soldano, K.; Drechsel, O.; Loth, D.; Marcé-Grau, A.; Mestres i Soler, O.; Poca, M.A.; Ossowski, S.; Macaya, A.; et al. Rare functional genetic variants in COL7A1, COL6A5, COL1A2 and COL5A2 frequently occur in Chiari Malformation Type 1. PLoS ONE 2021, 16, e0251289. [Google Scholar] [CrossRef]
- Martínez-Gil, N.; Mellibovsky, L.; González, D.M.L.; Patiño, J.D.; Cozar, M.; Rabionet, R.; Grinberg, D.; Balcells, S. On the association between Chiari malformation type 1, bone mineral density and bone related genes. Bone Rep. 2022, 15, 16. [Google Scholar] [CrossRef] [PubMed]
- Aydin, S.; Ozoner, B. Comparative volumetric analysis of the brain and cerebrospinal fluid in chiari type I malformation patients: A morphological study. Brain Sci. 2019, 9, 260. [Google Scholar] [CrossRef] [PubMed]
- Heffez, D.S.; Broderick, J.; Connor, M.; Mitchell, M.; Galezowska, J.; Golchini, R.; Ghorai, J. Is there a relationship between the extent of tonsillar ectopia and the severity of the clinical Chiari syndrome? Acta Neurochir. 2020, 162, 1531–1538. [Google Scholar] [CrossRef] [PubMed]
- Lázaro, E.; García, M.; Ibarrola, A.; Amayra, I.; López-Paz, J.F.; Martínez, O.; Pérez, M.; Berrocoso, S.; Al-Rashaida, M.; Rodríguez, A.A.; et al. Chiari type i malformation associated with verbal fluency impairment. J. Speech Lang. Hear. Res. 2018, 61, 2458–2466. [Google Scholar] [CrossRef]
- Ciaramitaro, P.; Rota, E.; Ferraris, M.; Stura, I.; Migliaretti, G.; Cocito, D. Migraine in Chiari 1 Malformation: A cross-sectional, single centre study. Acta Neurol Belg. 2022, 22, 947–954. [Google Scholar] [CrossRef]
- Di Rocco, F.; Licci, M.; Garde, A.; Mottolese, C.; Thauvin-Robinet, C.; Chevarin, M.; Faivre, L. Surgical management of Chiari malformation type 1 associated to MCAP syndrome and study of cerebellar and adjacent tissues for PIK3CA mosaicism. Eur. J. Med. Genet. 2023, 66, 104678. [Google Scholar] [CrossRef]
- Eppelheimer, M.; Houston, J.; Bapuraj, J.; Labuda, R.; Loth, D.; Braun, A.M.; Loth, F. A retrospective 2D morphometric analysis of adult female chiari type I patients with commonly reported and related conditions. Front. Neuroanat. 2018, 19, 12. [Google Scholar] [CrossRef]
- Pan, K.; Heiss, J.; Brown, S.; Collins, M.; Boyce, A. Chiari I Malformation and Basilar Invagination in Fibrous Dysplasia: Prevalence, Mechanisms, and Clinical Implications. J. Bone Miner. Res. 2018, 33, 1990–1998. [Google Scholar] [CrossRef]
- Turgut, M.; Turgut, R. Chiari I malformation and craniosynostosis. In The Chiari Malformations; Springer International Publishing: Berlin/Heidelberg, Germany, 2020; pp. 239–259. [Google Scholar]
- García, M.; Amayra, I.; Lázaro, E.; López-Paz, J.; Martínez, O.; Pérez, M.; Al-Rashaida, M. Comparison between decompressed and non-decompressed Chiari Malformation type I patients: A neuropsychological study. Neuropsychologia 2018, 121, 135–143. [Google Scholar] [CrossRef]
- Menezes, A.; Greenlee, J.; Dlouhy, B. Syringobulbia in pediatric patients with Chiari malformation type I. J. Neurosurg. Pediatr. 2018, 22, 52–60. [Google Scholar] [CrossRef]
- Rangari, K.; Das, K.; Singh, S.; Kumar, K.; Bhaisora, K.; Sardhara, J.; Behari, S. Type i chiari malformation without concomitant bony instability: Assessment of different surgical procedures and outcomes in 73 patients. Neurospine 2021, 18, 126–138. [Google Scholar] [CrossRef] [PubMed]
- Spena, G.; Fontanella, M. Management of chiari malformation. In Surgery of the Cranio-Vertebral Junction; Springer International Publishing: Berlin/Heidelberg, Germany, 2019; pp. 483–499. [Google Scholar]
- Goel, A.; Ranjan, S.; Shah, A.; Bhambere, S.; Darji, H. Tethered cord and Chiari formation: Analysis of treatment in a relatively rare clinical situation. J. Craniovertebr. Junction Spine. 2019, 10, 247–249. [Google Scholar] [CrossRef] [PubMed]
- Florou, C.; Andreanos, K.; Georgakoulias, N.; Espinosa, E.; Papakonstantinou, E.; Georgalas, I.; Rotsos, T. Acute visual loss secondary to arnold chiari type i malformation completely resolving after decompressive posterior fossa surgery. Int. Med. Case Rep. J. 2020, 13, 657–661. [Google Scholar] [CrossRef] [PubMed]
- Lorefice, E.; Giovannini, S.; Cervio, A.; Condomí –Alcorta, S.; Mormandi, R. Pseudo Chiari with holocord syringomyelia secondary to cerebrospinal fluid hypotension. Interdiscip. Neurosurg. 2022, 1, 28. [Google Scholar] [CrossRef]
- Gabr, M.; Elmataeshy, M.; Abdullah, A. Chiari Type III: Experience of Outcome for 15 Cases. J. Korean Neurosurg. Soc. 2022, 65, 841–845. [Google Scholar] [CrossRef]
- Deleu, T.; Jansen, K.; Van Calenbergh, F. Brain overgrowth associated with megalencephaly-capillary malformation syndrome causing progressive Chiari and syringomyelia. Surg. Neurol. Int. 2022, 13, 211. [Google Scholar] [CrossRef]
- Chu, E.; Trager, R.; Nam, G.; Shum, J. Neck pain and Headache Complicated by Persistent Syringomyelia After Foramen Magnum Decompression for Chiari I Malformation: Improvement with Multimodal Chiropractic Therapies. Am. J. Case Rep. 2022, 23, e937826-1. [Google Scholar] [CrossRef]

{kind=link}
| Database | Search Strategy |
|---|---|
| PubMed | ((“arnold chiari malformation”[MeSH Terms] OR (“arnold chiari”[All Fields] AND “malformation”[All Fields]) OR “arnold chiari malformation”[All Fields] OR (“arnold”[All Fields] AND “chiari”[All Fields] AND “syndrome”[All Fields]) OR “arnold chiari syndrome”[All Fields]) AND (“rare diseases”[MeSH Terms] OR (“rare”[All Fields] AND “diseases”[All Fields]) OR “rare diseases”[All Fields] OR (“rare”[All Fields] AND “disease”[All Fields]) OR “rare disease”[All Fields])) AND (2018:2023[pdat]) NOT (review) |
| Scopus | (Arnold AND Chiari AND syndrome) AND (rare AND disease) AND (LIMIT-TO (PUBYEAR, 2023) OR LIMIT-TO (PUBYEAR, 2022) OR LIMIT-TO (PUBYEAR, 2021) OR LIMIT-TO (PUBYEAR, 2020) OR LIMIT-TO (PUBYEAR, 2019) OR LIMIT-TO (PUBYEAR, 2018)) AND (EXCLUDE (DOCTYPE, “re”)) |
| CINAHL | (Arnold Chiari syndrome) AND (rare disease) |
| Cochrane Library | (Arnold Chiari syndrome) AND (rare disease) |
| SciELO | (Arnold Chiari syndrome [Todos los indices] and rare disease [Todos los indices]) |
| Author | Year | Title | Aims | Results |
|---|---|---|---|---|
| Ciaramitaro et al. [9] | 2020 | “Syringomyelia and Chiari Syndrome Registry: advances in epidemiology, clinical phenotypes and natural history based on a North Western Italy cohort”. | To estimate the incidence and prevalence of CM and syringomyelia in northeastern northeastern Italy. | The prevalence of syringomyelia was estimated at 4.84 CI 95% (4.124–5.527), while that of Chiari was 7.74 CI 95% (6.965–8.596); incidences were 0.82 CI 95%(0.599–1.137) and 3.08 CI 95% (2.605–3.635), respectively. |
| Provenzano et al. [10] | 2021 | “Chiari 1 malformation and exome sequencing in 51 trios: the emerging role of rare missense variants in chromatin-remodeling genes”. | To discover and validate the genes genes associated with CM-I. | It shows that CM-I cases are mainly dependent on variants in chromatin remodeling genes; in most variants in chromatin remodeling genes, in most cases, they are nonsense mutations, and in very rare nonsense mutations, very few of them lead to a truncated protein. |
| Sadler et al. [11] | 2021 | “Rare and the novo coding variants in chromodomain genes in Chiari I malformation”. | Identify genes and genetic variants genetic variants associated with CM-I risk. | Significant enrichment of rare non-synonymous variants and de novo variants in chromodomain genes was observed in patients with CM-I, including three de novo loss-of-function variants in CHD8 and a notable burden of rare, transmitted variants in CHD3. Overall, individuals with CM-I exhibited larger head circumference, with many carrying rare CHD variants associated with macrocephaly. Haploinsufficiency for CHD8 resulted in macrocephaly and posterior brain displacement reminiscent of CM-I; this highlights the involvement of chromodomain genes and excessive brain growth in the pathogenesis of CM-I. |
| Musolf et al. [12] | 2019 | “Small posterior fossa in Chiari I malformation affected families is significantly linked to 1q43-44 and 12q23-24.11 using whole exome sequencing”. | Demonstrate the relationship between the measurement of the posterior fossa and sequencing of the exome sequencing in patients with CM-I. | Two novel linked haplotypes are identified at 1q43-44 and 12q23 for small posterior fossa, one of the underlying causes of CM-I. Both haplotypes encompassed large genomic regions, thereby incorporating a substantial number of candidate genes. |
| Urbizu et al. [13] | 2021 | “Rare functional genetic variants in COL7A1, COL6A5, COL1A2 and COL5A2 frequently occur in Chiari Malformation Type 1”. | Identify genetic risk variants of CM-I. | Rare variants in collagen genes were observed to be common in CM-I. Some of these genes have previously been associated with musculoskeletal phenotypes but never with this pathology until now. These findings underscore the contribution of rare genetic variants in these genes to CM-I and the association between the presence of connective tissue-related symptoms in CM-I patients and the occurrence of such variants. |
| Martínez-Gil et al. [14] | 2022 | “On the association between Chiari malformation type 1, bone mineral density and bone related genes”. | Identifying rare variants in genes associated with bone mineral density (BMD) and their association with CM-I. | Genetic variants have been discovered in genes related to bone mineral density (BMD) in certain patients with CM-I. It has been observed that these variants contribute to craniofacial development in various ways or have been previously associated with the condition. Further investigation is warranted to better understand the relationship between bone-related genes and CM-I and to gather more evidence of their contribution to the etiology. A direct correlation between bone mineral density and CM-I was not observed. |
| Aydin et al. [15] | 2019 | “Comparative Volumetric Analysis of the Brain and Cerebrospinal Fluid in Chiari Type I Malformation Patients: A Morphological Study”. | Researching the differences in brain volume between CM-I patients and a healthy population revealed notable findings. | An increase in cerebrospinal fluid (CSF) and lateral ventricle volume was observed in CM-I patients compared to the control group, supporting ventricular enlargement and hydrocephalus in these patients. However, the volume of the brain and cerebellum was reduced. While there were no significant differences in white matter volumes, gray matter was significantly lower in CM-I patients. This reduction in gray matter might underlie certain cortical dysfunctions seen in individuals with this condition. |
| Heffez et al. [16] | 2020 | “Is there a relationship between the extent of tonsillar ectopia and the severity of the clinical Chiari syndrome?”. | Investigating the correlation between the extent of tonsillar ectopia and the prevalence and severity of symptoms associated with CM. | Regression analysis confirmed a connection between the extent of tonsillar ectopia and the likelihood of experiencing many symptoms. However, the severity of symptoms does not directly correlate with the extent of tonsillar ectopia. |
| Lázaro et al. [17] | 2018 | “Chiari Type I Malformation Associated With Verbal Fluency Impairment”. | Investigating if verbal fluency is affected in CM-I patients. | Significantly lower scores were found for the CM-I group in terms of verbal fluency compared to the control group. After controlling for tendencies of anxious and depressive symptoms, it was observed that verbal fluency could not be predicted by these variables. Therefore, it can be concluded that individuals with CM-I exhibit reduced verbal fluency, and this difference is not attributed to depression or anxiety. |
| Ciaramitaro et al. [18] | 2022 | “Migraine in Chiari 1 Malformation: a cross-sectional, single centre study”. | Examine the prevalence of migraine in CM-I patients and compare the clinical-demographic characteristics with CM-I without migraine. | Out of 230 patients, 78 exhibited migraine headaches, with 44 out of those 78 experiencing aura; 0.5387 CI 95% (0.4487–0.6287). Among these, 58 had comorbid migraines attributed to CM-I; 0.1357 CI 9% (0.0779–0.1935). The prevalence of migraine was higher in isolated CM-I patients compared to the remaining subjects. While migraine was more prevalent in females, age and gender were not risk factors for its occurrence; however, it was associated with the isolated CM-I phenotype. Thus, a high prevalence of migraine in CM-I patients is demonstrated, with a significant association between migraine and isolated CM-I. |
| Di Rocco et al. [19] | 2023 | “Surgical management of Chiari malformation type 1 associated with MCAP syndrome and study of cerebellar and adjacent tissues for PIK3CA mosaicism”. | Propose a potential relationship between the PIK3CA gene and the cerebral phenotype in patients with megalencephaly-capillary malformation polygyria (MCAP) and CM-I. | Both in CM-I and MCAP, alterations in cerebrospinal fluid (CSF) dynamics were observed, specifically syringomyelia and hydrocephalus, which necessitated surgical intervention. Targeted sequencing of PIK3CA determined the variable allelic frequency of the postzygotic variant in both the cerebellum and adjacent bone/connective tissues. |
| Eppelheimer et al. [20] | 2018 | “A Retrospective 2D Morphometric Analysis of Adult Female Chiari Type I Patients with Commonly Reported and Related Conditions”. | Identify the morphological characteristics of conditions associated with CM-I and explain the inconsistent results of comparisons between cases and controls. | A reduced McRae line length was observed in CM-I participants with syringomyelia. On the other hand, tonsillar position was decreased in CM-I participants with Ehlers–Danlos syndrome, and the basion to axial line posterior distance was greater in CM-I participants with scoliosis. Furthermore, differences were found between CM-I participants and healthy controls regardless of associated conditions, indicating that the prevalence of these conditions is not strongly linked to CM-I. The inconsistent findings in the radiographic literature are not explained by these conditions in the CM-I samples. |
| Pan et al. [21] | 2018 | “Chiari I Malformation and Basilar Invagination in Fibrous Dysplasia: Prevalence, Mechanisms, and Clinical Implications”. | Determine the prevalence and risk factors of CM-I and basilar invagination (BI) in patients with McCune Albright syndrome (MAS). | Craniomorphometric and volumetric analyses in patients with MAS identified cranial constriction and settling as the primary mechanisms for anomalies at the skull base, with intracranial hypertension playing a lesser role. Furthermore, there was a noted progression of odontoid changes with age, but not in the tonsillar position. Endocrinopathies associated with BI in MAS included precocious puberty, hyperthyroidism, and hypophosphatemia, but they were not linked to CM-I. Scoliosis was correlated with both CM-I and BI. |
| Turgut et al. [22] | 2020 | “Chiari I Malformation and Craniosynostosis”. | Highlight the existing association between CM-I and craniosynostosis. | CM-I is commonly associated with syndromic multisutural craniosynostosis, characterized by early fusion of the lambdoid sutures and synchondrosis of the skull base. The standard treatment for these conditions involves simultaneous or sequential remodeling of the cranial vault and suboccipital decompression. However, it is widely accepted to perform cranial vault expansion or remodeling procedures before the decompression. |
| García et al. [23] | 2018 | “Comparison between decompressed and non-decompressed Chiari T Malformation type I patients: A neuropsychological study”. | Examine whether posterior fossa decompression leads to an improvement in the cognitive profile deficit in CM-I patients. | CM-I patients exhibit lower cognitive performance in executive function, verbal fluency, spatial cognition, language, processing speed, verbal memory, facial recognition, and theory of mind compared to patients in the control group. These results persist even after statistically controlling for physical pain and anxious–depressive symptoms. These findings underscore a cognitive deficit associated with CM-I, independent of posterior fossa decompression surgery. |
| Menezes et al. [24] | 2018 | “Syringobulbia in pediatric patients with Chiari malformation type I”. | Enhancing the understanding of the presentation, treatment, and surgical outcomes of CM-I. | The incidence of syringobulbia in CM-I patients was 4%, and all of them had concurrent syringomyelia. The syringobulbia involved the medulla in all cases and communicated with the fourth ventricle in 54% of cases. Posterior fossa decompression with intradural exploration and duraplasty has proven effective for these patients. |
| Rangari et al. [25] | 2021 | “Type I Chiari Malformation Without Concomitant Bony Instability: Assessment of Different Surgical Procedures and Outcomes in 73 patients”. | Compare different treatment strategies for CM-I using objective outcome measures. | Patients with minimal symptoms underwent posterior fossa bony decompression (PFBD), while those with more severe symptoms received both bony and dural decompression (PFBDD) of the posterior fossa. The PFBDD group predicted a poor short-term postoperative outcome. Findings indicate that PFBD appears to be a durable procedure, whereas the PFBDD group experiences complications and late deterioration. Posterior fossa fixation does not yield better long-term outcomes compared to decompression alone. |
| Spena et al. [26] | 2020 | “Management of Chiari Malformation”. | Provide detailed information about the management of CM-I. | Surgical management is the preferred treatment for this condition. However, there are still many patients who do not experience symptom relief after surgery. There are surgical nuances that are still under debate, but it appears that the issue lies in understanding the subtle differences between patients. One possible solution would be to adopt a more comprehensive approach that does not solely focus on the skull base and posterior fossa. |
| Goel et al. [27] | 2020 | “Tethered cord and Chiari formation: Analysis of treatment in a relatively rare clinical situation”. | Contributing additional information to the literature on Chiari Malformation by explaining a real-life case. | Following surgery for a tethered spinal cord, the patient’s neurological symptoms continued to worsen, leading to the development of spastic tetraparesis, urinary retention, and constipation. An atlantoaxial fixation procedure was performed, resulting in improved limb function and urinary and bowel control. The presence of symptomatic Chiari in conjunction with a tethered cord is a rare clinical event. Surgical treatment of Chiari formation can lead to a rewarding clinical recovery. |
| Chrysoula et al. [28] | 2020 | “Acute Visual Loss Secondary to Arnold Chiari Type I Malformation Completely Resolving After Decompressive Posterior Fossa Surgery”. | Illustrating a real case of CM-I. | Consider the case of a 22-year-old young woman who presented with sudden unilateral visual loss following hearing loss. MRI revealed a small, almost subclinical herniation of the cerebellar tonsils. The patient underwent occipital craniotomy with posterior fossa decompression, resulting in a favorable outcome with papilledema regression and full recovery of visual acuity. The occurrence of papilledema associated with CM-I is rare. |
| Florefice et al. [29] | 2022 | “Pseudo Chiari with holocord syringomyelia secondary to cerebrospinal fluid hypotension. Case report”. | Contributing novel information about Pseudo Chiari with the presentation of a clinical case. | After surgically decompressing the posterior fossa using an epidural blood patch and a dural patch, it was observed that the patient achieved favorable clinical outcomes. |
| Gabr et al. [30] | 2022 | “Chiari Type III: Experience of Outcome for 15 Cases”. | Highlight the key findings from 15 cases of CM-III. | Initially, eight patients required ventriculoperitoneal shunting, while the other seven patients began with a normal delivery. Subsequently, six more patients needed such shunting due to cerebrospinal fluid leakage and increased intracranial pressure. Only four patients required a blood transfusion. Therefore, it is affirmed that there are variations in outcomes, and not all CM-III cases will result in death or severe developmental delay. Proper management leads to a favorable prognosis for the condition. |
| Deleu et al. [31] | 2022 | “Brain overgrowth associated with megalencephaly-capillary malformation syndrome causing progressive Chiari and syringomyelia”. | Gather information on a case involving the association of megalencephaly-capillary malformation (M-CM) with CM-I and syringomyelia. | The case involves an infant with M-CM who developed progressive CM-I and syringomyelia, reflecting disproportionate growth of the cerebellum/posterior fossa; this justified a suboccipital craniectomy and C1 laminectomy with duraplasty. At three, six-, and nine-months post-surgery, the only residual deficit in the patient was mild gait disturbance. |
| Chu et al. [32] | 2022 | “Neck pain and Headache Complicated by Persistent Syringomyelia After Foramen Magnum Decompression for Chiari I Malformation: Improvement with Multimodal Chiropractic Therapies”. | Present information about a real case of CM-I treated with foramen magnum decompression. | This case features a patient with neck pain, headache, and persistent syringomyelia following CM-I decompression. The symptoms improved after multimodal rehabilitation and chiropractic techniques. Due to limited and low-level evidence for these interventions in patients with syringomyelia and persistent symptoms post-decompression, these therapies cannot be broadly recommended. However, they might be considered on a case-by-case basis. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodríguez-Blanque, R.; Almazán-Soto, C.; Piqueras-Sola, B.; Sánchez-García, J.C.; Reinoso-Cobo, A.; Menor-Rodríguez, M.J.; Cortés-Martín, J. Chiari Syndrome: Advances in Epidemiology and Pathogenesis: A Systematic Review. J. Clin. Med. 2023, 12, 6694. https://doi.org/10.3390/jcm12206694
Rodríguez-Blanque R, Almazán-Soto C, Piqueras-Sola B, Sánchez-García JC, Reinoso-Cobo A, Menor-Rodríguez MJ, Cortés-Martín J. Chiari Syndrome: Advances in Epidemiology and Pathogenesis: A Systematic Review. Journal of Clinical Medicine. 2023; 12(20):6694. https://doi.org/10.3390/jcm12206694
Chicago/Turabian StyleRodríguez-Blanque, Raquel, Cristina Almazán-Soto, Beatriz Piqueras-Sola, Juan Carlos Sánchez-García, Andrés Reinoso-Cobo, María José Menor-Rodríguez, and Jonathan Cortés-Martín. 2023. "Chiari Syndrome: Advances in Epidemiology and Pathogenesis: A Systematic Review" Journal of Clinical Medicine 12, no. 20: 6694. https://doi.org/10.3390/jcm12206694
APA StyleRodríguez-Blanque, R., Almazán-Soto, C., Piqueras-Sola, B., Sánchez-García, J. C., Reinoso-Cobo, A., Menor-Rodríguez, M. J., & Cortés-Martín, J. (2023). Chiari Syndrome: Advances in Epidemiology and Pathogenesis: A Systematic Review. Journal of Clinical Medicine, 12(20), 6694. https://doi.org/10.3390/jcm12206694