
Application of Electrophysiology in Non-Macular Inherited Retinal Dystrophies


Abstract
:1. Introduction
2. Application of Electrophysiology in Inherited Dystrophies
2.1. Retinitis Pigmentosa
2.2. Progressive Cone/Cone-Rod Dystrophy
2.3. Cone Dystrophy with Supernormal Rod Response
2.4. Enhanced S-Cone Syndrome
2.5. Bradyopsia
2.6. Bietti Crystalline Dystrophy
2.7. Late-Onset Retinal Degeneration (L-ORD)
2.8. Fundus Albipunctatus
2.9. Retinitis Punctata Albescens
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Liew, G.; Michaelides, M.; Bunce, C. A comparison of the causes of blindness certifications in England and Wales in working age adults (16–64 years), 1999–2000 with 2009–2010. BMJ Open 2014, 4, e004015. [Google Scholar] [CrossRef] [PubMed]
- Pontikos, N.; Arno, G.; Jurkute, N.; Schiff, E.; Ba-Abbad, R.; Malka, S.; Gimenez, A.; Georgiou, M.; Wright, G.; Armengol, M.; et al. Genetic Basis of Inherited Retinal Disease in a Molecularly Characterized Cohort of More Than 3000 Families from the United Kingdom. Ophthalmology 2020, 127, 1384–1394. [Google Scholar] [CrossRef] [PubMed]
- Chay, J.; Tang, R.W.C.; Tan, T.E.; Chan, C.M.; Mathur, R.; Lee, B.J.H.; Chan, H.H.; Sim, S.; Farooqui, S.; Teo, K.Y.C.; et al. The economic burden of inherited retinal disease in Singapore: A prevalence-based cost-of-illness study. Eye 2023. [Google Scholar] [CrossRef] [PubMed]
- Georgiou, M.; Fujinami, K.; Michaelides, M. Inherited retinal diseases: Therapeutics, clinical trials and end points—A review. Clin. Exp. Ophthalmol. 2021, 49, 270–288. [Google Scholar] [CrossRef] [PubMed]
- Robson, A.G.; Frishman, L.J.; Grigg, J.; Hamilton, R.; Jeffrey, B.G.; Kondo, M.; Li, S.; McCulloch, D.L. ISCEV Standard for full-field clinical electroretinography (2022 update). Doc. Ophthalmol. 2022, 144, 165–177. [Google Scholar] [CrossRef] [PubMed]
- McCulloch, D.L.; Marmor, M.F.; Brigell, M.G.; Hamilton, R.; Holder, G.E.; Tzekov, R.; Bach, M. ISCEV Standard for full-field clinical electroretinography (2015 update). Doc. Ophthalmol. 2015, 130, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Liao, F.; Liu, H.; Milla-Navarro, S.; Villa, P.; Germain, F. Origin of Retinal Oscillatory Potentials in the Mouse, a Tool to Specifically Locate Retinal Damage. Int. J. Mol. Sci. 2023, 24, 3126. [Google Scholar] [CrossRef]
- Hood, D.C.; Bach, M.; Brigell, M.; Keating, D.; Kondo, M.; Lyons, J.S.; Marmor, M.F.; McCulloch, D.L.; Palmowski-Wolfe, A.M. ISCEV standard for clinical multifocal electroretinography (mfERG) (2011 edition). Doc. Ophthalmol. 2012, 124, 1–13. [Google Scholar] [CrossRef]
- Hoffmann, M.B.; Bach, M.; Kondo, M.; Li, S.; Walker, S.; Holopigian, K.; Viswanathan, S.; Robson, A.G. ISCEV standard for clinical multifocal electroretinography (mfERG) (2021 update). Doc. Ophthalmol. 2021, 142, 5–16. [Google Scholar] [CrossRef]
- Parisi, V.; Ziccardi, L.; Stifano, G.; Montrone, L.; Gallinaro, G.; Falsini, B. Impact of regional retinal responses on cortical visually evoked responses: Multifocal ERGs and VEPs in the retinitis pigmentosa model. Clin. Neurophysiol. 2010, 121, 380–385. [Google Scholar] [CrossRef]
- Bach, M.; Brigell, M.G.; Hawlina, M.; Holder, G.E.; Johnson, M.A.; McCulloch, D.L.; Meigen, T.; Viswanathan, S. ISCEV standard for clinical pattern electroretinography (PERG): 2012 update. Doc. Ophthalmol. 2013, 126, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Holder, G.E. Electrophysiological assessment of optic nerve disease. Eye 2004, 18, 1133–1143. [Google Scholar] [CrossRef] [PubMed]
- Chiang, T.K.; Yu, M. Electrophysiological Evaluation of Macular Dystrophies. J. Clin. Med. 2023, 12, 1430. [Google Scholar] [CrossRef] [PubMed]
- Lyraki, R.; Megaw, R.; Hurd, T. Disease mechanisms of X-linked retinitis pigmentosa due to RP2 and RPGR mutations. Biochem. Soc. Trans. 2016, 44, 1235–1244. [Google Scholar] [CrossRef] [PubMed]
- Bravo-Gil, N.; Gonzalez-Del Pozo, M.; Martin-Sanchez, M.; Mendez-Vidal, C.; Rodriguez-de la Rua, E.; Borrego, S.; Antinolo, G. Unravelling the genetic basis of simplex Retinitis Pigmentosa cases. Sci. Rep. 2017, 7, 41937. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, S.; Di Iorio, E.; Barbaro, V.; Ponzin, D.; Sorrentino, F.S.; Parmeggiani, F. Retinitis pigmentosa: Genes and disease mechanisms. Curr. Genom. 2011, 12, 238–249. [Google Scholar] [PubMed]
- Michaelides, M.; Hardcastle, A.J.; Hunt, D.M.; Moore, A.T. Progressive cone and cone-rod dystrophies: Phenotypes and underlying molecular genetic basis. Surv. Ophthalmol. 2006, 51, 232–258. [Google Scholar] [CrossRef] [PubMed]
- Gill, J.S.; Georgiou, M.; Kalitzeos, A.; Moore, A.T.; Michaelides, M. Progressive cone and cone-rod dystrophies: Clinical features, molecular genetics and prospects for therapy. Br. J. Ophthalmol. 2019, 103, 711–720. [Google Scholar] [CrossRef]
- Wu, H.; Cowing, J.A.; Michaelides, M.; Wilkie, S.E.; Jeffery, G.; Jenkins, S.A.; Mester, V.; Bird, A.C.; Robson, A.G.; Holder, G.E.; et al. Mutations in the gene KCNV2 encoding a voltage-gated potassium channel subunit cause “cone dystrophy with supernormal rod electroretinogram” in humans. Am. J. Hum. Genet. 2006, 79, 574–579. [Google Scholar] [CrossRef]
- Thiagalingam, S.; McGee, T.L.; Weleber, R.G.; Sandberg, M.A.; Trzupek, K.M.; Berson, E.L.; Dryja, T.P. Novel mutations in the KCNV2 gene in patients with cone dystrophy and a supernormal rod electroretinogram. Ophthalmic Genet. 2007, 28, 135–142. [Google Scholar] [CrossRef]
- Wissinger, B.; Dangel, S.; Jagle, H.; Hansen, L.; Baumann, B.; Rudolph, G.; Wolf, C.; Bonin, M.; Koeppen, K.; Ladewig, T.; et al. Cone dystrophy with supernormal rod response is strictly associated with mutations in KCNV2. Investig. Ophthalmol. Vis. Sci. 2008, 49, 751–757. [Google Scholar] [CrossRef] [PubMed]
- Coppieters, F.; Leroy, B.P.; Beysen, D.; Hellemans, J.; De Bosscher, K.; Haegeman, G.; Robberecht, K.; Wuyts, W.; Coucke, P.J.; De Baere, E. Recurrent mutation in the first zinc finger of the orphan nuclear receptor NR2E3 causes autosomal dominant retinitis pigmentosa. Am. J. Hum. Genet. 2007, 81, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Escher, P.; Gouras, P.; Roduit, R.; Tiab, L.; Bolay, S.; Delarive, T.; Chen, S.; Tsai, C.C.; Hayashi, M.; Zernant, J.; et al. Mutations in NR2E3 can cause dominant or recessive retinal degenerations in the same family. Hum. Mutat. 2009, 30, 342–351. [Google Scholar] [CrossRef] [PubMed]
- Nishiguchi, K.M.; Sandberg, M.A.; Kooijman, A.C.; Martemyanov, K.A.; Pott, J.W.; Hagstrom, S.A.; Arshavsky, V.Y.; Berson, E.L.; Dryja, T.P. Defects in RGS9 or its anchor protein R9AP in patients with slow photoreceptor deactivation. Nature 2004, 427, 75–78. [Google Scholar] [CrossRef] [PubMed]
- Lyubarsky, A.L.; Naarendorp, F.; Zhang, X.; Wensel, T.; Simon, M.I.; Pugh, E.N., Jr. RGS9-1 is required for normal inactivation of mouse cone phototransduction. Mol. Vis. 2001, 7, 71–78. [Google Scholar] [PubMed]
- Nakano, M.; Kelly, E.J.; Wiek, C.; Hanenberg, H.; Rettie, A.E. CYP4V2 in Bietti’s crystalline dystrophy: Ocular localization, metabolism of omega-3-polyunsaturated fatty acids, and functional deficit of the p.H331P variant. Mol. Pharmacol. 2012, 82, 679–686. [Google Scholar] [CrossRef] [PubMed]
- Osman Saatci, A.; Can Doruk, H. An Overview of Rare and Unusual Clinical Features of Bietti’s Crystalline Dystrophy. Med. Hypothesis Discov. Innov. Ophthalmol. 2014, 3, 51–56. [Google Scholar]
- Saatci, A.O.; Atas, F.; Cetin, G.O.; Kayabasi, M. Diagnostic and Management Strategies of Bietti Crystalline Dystrophy: Current Perspectives. Clin. Ophthalmol. 2023, 17, 953–967. [Google Scholar] [CrossRef]
- Chekuri, A.; Zientara-Rytter, K.; Soto-Hermida, A.; Borooah, S.; Voronchikhina, M.; Biswas, P.; Kumar, V.; Goodsell, D.; Hayward, C.; Shaw, P.; et al. Late-onset retinal degeneration pathology due to mutations in CTRP5 is mediated through HTRA1. Aging Cell 2019, 18, e13011. [Google Scholar] [CrossRef]
- De Zaeytijd, J.; Coppieters, F.; De Bruyne, M.; Van Royen, J.; Roels, D.; Six, R.; Van Cauwenbergh, C.; De Baere, E.; Leroy, B.P. Longitudinal phenotypic study of late-onset retinal degeneration due to a founder variant c.562C>A p.(Pro188Thr) in the C1QTNF5 gene. Ophthalmic Genet. 2021, 42, 521–532. [Google Scholar] [CrossRef]
- Yamamoto, H.; Simon, A.; Eriksson, U.; Harris, E.; Berson, E.L.; Dryja, T.P. Mutations in the gene encoding 11-cis retinol dehydrogenase cause delayed dark adaptation and Fundus albipunctatus. Nat. Genet. 1999, 22, 188–191. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.; Hotta, Y.; Tanikawa, A.; Terasaki, H.; Miyake, Y. A high association with cone dystrophy in Fundus albipunctatus caused by mutations of the RDH5 gene. Investig. Ophthalmol. Vis. Sci. 2000, 41, 3925–3932. [Google Scholar] [PubMed]
- Scimone, C.; Donato, L.; Esposito, T.; Rinaldi, C.; D’Angelo, R.; Sidoti, A. A novel RLBP1 gene geographical area-related mutation present in a young patient with Retinitis punctata albescens. Hum. Genom. 2017, 11, 18. [Google Scholar] [CrossRef] [PubMed]
- Kajiwara, K.; Sandberg, M.A.; Berson, E.L.; Dryja, T.P. A null mutation in the human peripherin/RDS gene in a family with autosomal dominant retinitis punctata albescens. Nat. Genet. 1993, 3, 208–212. [Google Scholar] [CrossRef] [PubMed]
- Souied, E.; Soubrane, G.; Benlian, P.; Coscas, G.J.; Gerber, S.; Munnich, A.; Kaplan, J. Retinitis punctata albescens associated with the Arg135Trp mutation in the rhodopsin gene. Am. J. Ophthalmol. 1996, 121, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Hamel, C. Retinitis pigmentosa. Orphanet J. Rare Dis. 2006, 1, 40. [Google Scholar] [CrossRef] [PubMed]
- Pagon, R.A. Retinitis pigmentosa. Surv. Ophthalmol. 1988, 33, 137–177. [Google Scholar] [CrossRef] [PubMed]
- Verbakel, S.K.; van Huet, R.A.C.; Boon, C.J.F.; den Hollander, A.I.; Collin, R.W.J.; Klaver, C.C.W.; Hoyng, C.B.; Roepman, R.; Klevering, B.J. Non-syndromic retinitis pigmentosa. Prog. Retin. Eye Res. 2018, 66, 157–186. [Google Scholar] [CrossRef]
- Tsang, S.H.; Sharma, T. Retinitis Pigmentosa (Non-syndromic). Adv. Exp. Med. Biol. 2018, 1085, 125–130. [Google Scholar]
- Tsang, S.H.; Sharma, T. Autosomal Dominant Retinitis Pigmentosa. Adv. Exp. Med. Biol. 2018, 1085, 69–77. [Google Scholar]
- Diakatou, M.; Manes, G.; Bocquet, B.; Meunier, I.; Kalatzis, V. Genome Editing as a Treatment for the Most Prevalent Causative Genes of Autosomal Dominant Retinitis Pigmentosa. Int. J. Mol. Sci. 2019, 20, 2542. [Google Scholar] [CrossRef] [PubMed]
- Tsang, S.H.; Sharma, T. X-linked Retinitis Pigmentosa. Adv. Exp. Med. Biol. 2018, 1085, 31–35. [Google Scholar] [PubMed]
- Forsythe, E.; Beales, P.L. Bardet-Biedl syndrome. Eur. J. Hum. Genet. 2013, 21, 8–13. [Google Scholar] [CrossRef]
- Mathur, P.; Yang, J. Usher syndrome: Hearing loss, retinal degeneration and associated abnormalities. Biochim. Biophys. Acta 2015, 1852, 406–420. [Google Scholar] [CrossRef] [PubMed]
- Dias, M.F.; Joo, K.; Kemp, J.A.; Fialho, S.L.; da Silva Cunha, A., Jr.; Woo, S.J.; Kwon, Y.J. Molecular genetics and emerging therapies for retinitis pigmentosa: Basic research and clinical perspectives. Prog. Retin. Eye Res. 2018, 63, 107–131. [Google Scholar] [CrossRef] [PubMed]
- Mirochnik, R.M.; Pezaris, J.S. Contemporary approaches to visual prostheses. Mil. Med. Res. 2019, 6, 19. [Google Scholar] [CrossRef]
- Papaioannou, I.; Owen, J.S.; Yanez-Munoz, R.J. Clinical applications of gene therapy for rare diseases: A review. Int. J. Exp. Pathol. 2023, 104, 154–176. [Google Scholar] [CrossRef] [PubMed]
- Audo, I.; Nassisi, M.; Zeitz, C.; Sahel, J.A. The Extraordinary Phenotypic and Genetic Variability of Retinal and Macular Degenerations: The Relevance to Therapeutic Developments. Cold Spring Harb. Perspect. Med. 2023, 13, a041652. [Google Scholar] [CrossRef]
- Chan, H.W.; Oh, J.; Leroy, B. Therapeutic landscape for inherited ocular diseases: Current and emerging therapies. Singap. Med. J. 2023, 64, 17–26. [Google Scholar] [CrossRef]
- Farris, M.; Goodall, S.; De Abreu Lourenco, R. A systematic review of economic evaluations for RPE65-mediated inherited retinal disease including HTA assessment of broader value. Int. J. Technol. Assess. Health Care 2023, 39, e38. [Google Scholar] [CrossRef]
- Han, J.; Joo, K.; Kim, U.S.; Woo, S.J.; Lee, E.K.; Lee, J.Y.; Park, T.K.; Kim, S.J.; Byeon, S.H. Voretigene Neparvovec for the Treatment of RPE65-associated Retinal Dystrophy: Consensus and Recommendations from the Korea RPE65-IRD Consensus Paper Committee. Korean J. Ophthalmol. 2023, 37, 166–186. [Google Scholar] [CrossRef] [PubMed]
- Janossy, A.; Vizvari, E.; Lorincz, M.; Pal, S.; Nagy, D.; Benedek, G.; Toth-Molnar, E.; Janaky, M. Long-Term Follow-Up of a Family with Retinal Dystrophy Caused by RPE65 Mutation. Case Rep. Ophthalmol. 2023, 14, 454–461. [Google Scholar] [CrossRef] [PubMed]
- Jung, R.; Kempf, M.; Holocher, S.; Kortum, F.C.; Stingl, K.; Stingl, K. Multi-luminance mobility testing after gene therapy in the context of retinal functional diagnostics. Graefes Arch. Clin. Exp. Ophthalmol. 2023. online ahead of print. [Google Scholar]
- Kiraly, P.; Cottriall, C.L.; Taylor, L.J.; Jolly, J.K.; Cehajic-Kapetanovic, J.; Yusuf, I.H.; Martinez-Fernandez de la Camara, C.; Shanks, M.; Downes, S.M.; MacLaren, R.E.; et al. Outcomes and Adverse Effects of Voretigene Neparvovec Treatment for Biallelic RPE65-Mediated Inherited Retinal Dystrophies in a Cohort of Patients from a Single Center. Biomolecules 2023, 13, 1484. [Google Scholar] [CrossRef] [PubMed]
- Ku, C.A.; Igelman, A.D.; Huang, S.J.; Vasconcelos, H.; da Palma, M.M.; Bailey, S.T.; Lauer, A.K.; Weleber, R.G.; Yang, P.; Pennesi, M.E. Improved Rod Sensitivity as Assessed by Two-Color Dark-Adapted Perimetry in Patients with RPE65-Related Retinopathy Treated with Voretigene Neparvovec-rzyl. Transl. Vis. Sci. Technol. 2023, 12, 17. [Google Scholar] [CrossRef]
- Lorenz, B.; Kunzel, S.H.; Preising, M.N.; Scholz, J.P.; Chang, P.; Holz, F.G.; Herrmann, P. Real-world experience with Voretigene Neparvovec gene augmentation therapy in RPE65-mutation associated inherited retinal degeneration. Ophthalmology 2023. online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Malvasi, M.; Casillo, L.; Avogaro, F.; Abbouda, A.; Vingolo, E.M. Gene Therapy in Hereditary Retinal Dystrophies: The Usefulness of Diagnostic Tools in Candidate Patient Selections. Int. J. Mol. Sci. 2023, 24, 13756. [Google Scholar] [CrossRef] [PubMed]
- Stingl, K.; Stingl, K.; Schwartz, H.; Reid, M.W.; Kempf, M.; Dimopoulos, S.; Kortuem, F.; Borchert, M.S.; Lee, T.C.; Nagiel, A. Full-field Scotopic Threshold Improvement after Voretigene Neparvovec-rzyl Treatment Correlates with Chorioretinal Atrophy. Ophthalmology 2023, 130, 764–770. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.Y.; Kulbay, M.; Toameh, D.; Xu, A.Q.; Kalevar, A.; Tran, S.D. Retinitis Pigmentosa: Novel Therapeutic Targets and Drug Development. Pharmaceutics 2023, 15, 685. [Google Scholar] [CrossRef]
- Bhatti, M.T. Retinitis pigmentosa, pigmentary retinopathies, and neurologic diseases. Curr. Neurol. Neurosci. Rep. 2006, 6, 403–413. [Google Scholar] [CrossRef]
- Newton, F.; Megaw, R. Mechanisms of Photoreceptor Death in Retinitis Pigmentosa. Genes 2020, 11, 1120. [Google Scholar] [CrossRef]
- Konieczka, K.; Bojinova, R.I.; Valmaggia, C.; Schorderet, D.F.; Todorova, M.G. Preserved functional and structural integrity of the papillomacular area correlates with better visual acuity in retinitis pigmentosa. Eye 2016, 30, 1310–1323. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.M.; Oh, J.K.; Tsang, S.H. The Use of Optical Coherence Tomography in Evaluation of Retinitis Pigmentosa. Methods Mol. Biol. 2023, 2560, 91–100. [Google Scholar] [PubMed]
- Sandberg, M.A.; Brockhurst, R.J.; Gaudio, A.R.; Berson, E.L. The association between visual acuity and central retinal thickness in retinitis pigmentosa. Investig. Ophthalmol. Vis. Sci. 2005, 46, 3349–3354. [Google Scholar] [CrossRef] [PubMed]
- Ebdali, S.; Hashemi, B.; Hashemi, H.; Jafarzadehpur, E.; Asgari, S. Time and frequency components of ERG responses in retinitis pigmentosa. Int. Ophthalmol. 2018, 38, 2435–2444. [Google Scholar] [CrossRef] [PubMed]
- Hassan-Karimi, H.; Jafarzadehpur, E.; Blouri, B.; Hashemi, H.; Sadeghi, A.Z.; Mirzajani, A. Frequency Domain Electroretinography in Retinitis Pigmentosa versus Normal Eyes. J. Ophthalmic Vis. Res. 2012, 7, 34–38. [Google Scholar] [PubMed]
- Arsiwalla, T.A.; Cornish, E.E.; Nguyen, P.V.; Korsakova, M.; Ali, H.; Saakova, N.; Fraser, C.L.; Jamieson, R.V.; Grigg, J.R. Assessing Residual Cone Function in Retinitis Pigmentosa Patients. Transl. Vis. Sci. Technol. 2020, 9, 29. [Google Scholar] [CrossRef] [PubMed]
- Karali, M.; Testa, F.; Brunetti-Pierri, R.; Di Iorio, V.; Pizzo, M.; Melillo, P.; Barillari, M.R.; Torella, A.; Musacchia, F.; D’Angelo, L.; et al. Clinical and Genetic Analysis of a European Cohort with Pericentral Retinitis Pigmentosa. Int. J. Mol. Sci. 2019, 21, 86. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.W.; Yang, J.J.; Yang, C.H.; Yang, C.M.; Hu, F.R.; Ho, T.C.; Chen, T.C. The structure-function correlation analysed by OCT and full field ERG in typical and pericentral subtypes of retinitis pigmentosa. Sci. Rep. 2021, 11, 16883. [Google Scholar] [CrossRef]
- Todorova, M.G.; Turksever, C.; Schotzau, A.; Schorderet, D.F.; Valmaggia, C. Metabolic and functional changes in retinitis pigmentosa: Comparing retinal vessel oximetry to full-field electroretinography, electrooculogram and multifocal electroretinography. Acta Ophthalmol. 2016, 94, e231–e241. [Google Scholar] [CrossRef]
- Hood, D.C.; Holopigian, K.; Greenstein, V.; Seiple, W.; Li, J.; Sutter, E.E.; Carr, R.E. Assessment of local retinal function in patients with retinitis pigmentosa using the multi-focal ERG technique. Vis. Res. 1998, 38, 163–179. [Google Scholar] [CrossRef]
- Moschos, M.M.; Chatziralli, I.P.; Verriopoulos, G.; Triglianos, A.; Ladas, D.S.; Brouzas, D. Correlation between optical coherence tomography and multifocal electroretinogram findings with visual acuity in retinitis pigmentosa. Clin. Ophthalmol. 2013, 7, 2073–2078. [Google Scholar] [CrossRef] [PubMed]
- Okado, S.; Koyanagi, Y.; Inooka, T.; Kominami, T.; Terasaki, H.; Nishiguchi, K.M.; Ueno, S. Assessments of Macular Function by Focal Macular Electroretinography and Static Perimetry in Eyes with Retinitis Pigmentosa. Retina 2022, 42, 2184–2193. [Google Scholar] [CrossRef] [PubMed]
- Gerth, C.; Wright, T.; Heon, E.; Westall, C.A. Assessment of central retinal function in patients with advanced retinitis pigmentosa. Investig. Ophthalmol. Vis. Sci. 2007, 48, 1312–1318. [Google Scholar] [CrossRef] [PubMed]
- Arden, G.B.; Wolf, J.E. The electro-oculographic responses to alcohol and light in a series of patients with retinitis pigmentosa. Investig. Ophthalmol. Vis. Sci. 2000, 41, 2730–2734. [Google Scholar] [PubMed]
- Vingolo, E.M.; Livani, M.L.; Domanico, D.; Mendonca, R.H.; Rispoli, E. Optical coherence tomography and electro-oculogram abnormalities in X-linked retinitis pigmentosa. Doc. Ophthalmol. 2006, 113, 5–10. [Google Scholar] [CrossRef] [PubMed]
- Pinckers, A.; van Aarem, A.; Brink, H. The electrooculogram in heterozygote carriers of Usher syndrome, retinitis pigmentosa, neuronal ceroid lipofuscinosis, senior syndrome and choroideremia. Ophthalmic Genet. 1994, 15, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Asanad, S.; Karanjia, R. Multifocal Electroretinogram; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Granse, L.; Ponjavic, V.; Andreasson, S. Full-field ERG, multifocal ERG and multifocal VEP in patients with retinitis pigmentosa and residual central visual fields. Acta Ophthalmol. Scand. 2004, 82, 701–706. [Google Scholar] [CrossRef] [PubMed]
- Holder, G.E. Pattern electroretinography (PERG) and an integrated approach to visual pathway diagnosis. Prog. Retin. Eye Res. 2001, 20, 531–561. [Google Scholar] [CrossRef]
- Janaky, M.; Palffy, A.; Horvath, G.; Tuboly, G.; Benedek, G. Pattern-reversal electroretinograms and visual evoked potentials in retinitis pigmentosa. Doc. Ophthalmol. 2008, 117, 27–36. [Google Scholar] [CrossRef]
- Popovic, P.; Jarc-Vidmar, M.; Hawlina, M. Abnormal fundus autofluorescence in relation to retinal function in patients with retinitis pigmentosa. Graefes Arch. Clin. Exp. Ophthalmol. 2005, 243, 1018–1027. [Google Scholar] [CrossRef]
- Santos, A.; Humayun, M.S.; de Juan, E., Jr.; Greenburg, R.J.; Marsh, M.J.; Klock, I.B.; Milam, A.H. Preservation of the inner retina in retinitis pigmentosa. A morphometric analysis. Arch. Ophthalmol. 1997, 115, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Stone, J.L.; Barlow, W.E.; Humayun, M.S.; de Juan, E., Jr.; Milam, A.H. Morphometric analysis of macular photoreceptors and ganglion cells in retinas with retinitis pigmentosa. Arch. Ophthalmol. 1992, 110, 1634–1639. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Ouyang, W.; Wang, H.; Meng, X.; Li, S.; Yin, Z.Q. Quantitative assessment of visual pathway function in blind retinitis pigmentosa patients. Clin. Neurophysiol. 2021, 132, 392–403. [Google Scholar] [CrossRef] [PubMed]
- Klein, M.; Birch, D.G. Psychophysical assessment of low visual function in patients with retinal degenerative diseases (RDDs) with the Diagnosys full-field stimulus threshold (D-FST). Doc. Ophthalmol. 2009, 119, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Roman, A.J.; Schwartz, S.B.; Aleman, T.S.; Cideciyan, A.V.; Chico, J.D.; Windsor, E.A.; Gardner, L.M.; Ying, G.S.; Smilko, E.E.; Maguire, M.G.; et al. Quantifying rod photoreceptor-mediated vision in retinal degenerations: Dark-adapted thresholds as outcome measures. Exp. Eye Res. 2005, 80, 259–272. [Google Scholar] [CrossRef] [PubMed]
- Messias, K.; Jagle, H.; Saran, R.; Ruppert, A.D.; Siqueira, R.; Jorge, R.; Messias, A. Psychophysically determined full-field stimulus thresholds (FST) in retinitis pigmentosa: Relationships with electroretinography and visual field outcomes. Doc. Ophthalmol. 2013, 127, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Hamel, C.P.; Griffoin, J.M.; Bazalgette, C.; Lasquellec, L.; Duval, P.A.; Bareil, C.; Beaufrere, L.; Bonnet, S.; Eliaou, C.; Marlhens, F.; et al. Molecular genetics of pigmentary retinopathies: Identification of mutations in CHM, RDS, RHO, RPE65, USH2A and XLRS1 genes. J. Fr. Ophtalmol. 2000, 23, 985–995. [Google Scholar]
- Hamel, C.P. Cone rod dystrophies. Orphanet J. Rare Dis. 2007, 2, 7. [Google Scholar] [CrossRef]
- Tsang, S.H.; Sharma, T. Progressive Cone Dystrophy and Cone-Rod Dystrophy (XL, AD, and AR). Adv. Exp. Med. Biol. 2018, 1085, 53–60. [Google Scholar]
- Sadowski, B.; Zrenner, E. Cone and rod function in cone degenerations. Vis. Res. 1997, 37, 2303–2314. [Google Scholar] [CrossRef]
- Kamenarova, K.; Corton, M.; Garcia-Sandoval, B.; Fernandez-San Jose, P.; Panchev, V.; Avila-Fernandez, A.; Lopez-Molina, M.I.; Chakarova, C.; Ayuso, C.; Bhattacharya, S.S. Novel GUCA1A mutations suggesting possible mechanisms of pathogenesis in cone, cone-rod, and macular dystrophy patients. BioMed Res. Int. 2013, 2013, 517570. [Google Scholar] [CrossRef] [PubMed]
- Sokal, I.; Dupps, W.J.; Grassi, M.A.; Brown, J., Jr.; Affatigato, L.M.; Roychowdhury, N.; Yang, L.; Filipek, S.; Palczewski, K.; Stone, E.M.; et al. A novel GCAP1 missense mutation (L151F) in a large family with autosomal dominant cone-rod dystrophy (adCORD). Investig. Ophthalmol. Vis. Sci. 2005, 46, 1124–1132. [Google Scholar] [CrossRef] [PubMed]
- Payne, A.M.; Downes, S.M.; Bessant, D.A.; Taylor, R.; Holder, G.E.; Warren, M.J.; Bird, A.C.; Bhattacharya, S.S. A mutation in guanylate cyclase activator 1A (GUCA1A) in an autosomal dominant cone dystrophy pedigree mapping to a new locus on chromosome 6p21.1. Hum. Mol. Genet. 1998, 7, 273–277. [Google Scholar] [CrossRef] [PubMed]
- Zahlava, J.; Lestak, J.; Karel, I. Optical coherence tomography in progressive cone dystrophy. Biomed. Pap. Med. Fac. Univ. Palacky Olomouc. Czech Repub. 2014, 158, 628–634. [Google Scholar] [CrossRef]
- Simunovic, M.P.; Moore, A.T. The cone dystrophies. Eye 1998, 12, 553–565. [Google Scholar] [CrossRef] [PubMed]
- Szlyk, J.P.; Fishman, G.A.; Alexander, K.R.; Peachey, N.S.; Derlacki, D.J. Clinical subtypes of cone-rod dystrophy. Arch. Ophthalmol. 1993, 111, 781–788. [Google Scholar] [CrossRef] [PubMed]
- Downes, S.M.; Holder, G.E.; Fitzke, F.W.; Payne, A.M.; Warren, M.J.; Bhattacharya, S.S.; Bird, A.C. Autosomal dominant cone and cone-rod dystrophy with mutations in the guanylate cyclase activator 1A gene-encoding guanylate cyclase activating protein-1. Arch. Ophthalmol. 2001, 119, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Garafalo, A.V.; Sheplock, R.; Sumaroka, A.; Roman, A.J.; Cideciyan, A.V.; Jacobson, S.G. Childhood-onset genetic cone-rod photoreceptor diseases and underlying pathobiology. eBioMedicine 2021, 63, 103200. [Google Scholar] [CrossRef]
- Wang, I.; Khan, N.W.; Branham, K.; Wissinger, B.; Kohl, S.; Heckenlively, J.R. Establishing baseline rod electroretinogram values in achromatopsia and cone dystrophy. Doc. Ophthalmol. 2012, 125, 229–233. [Google Scholar] [CrossRef]
- Gouras, P.; Eggers, H.M.; MacKay, C.J. Cone dystrophy, nyctalopia, and supernormal rod responses. A new retinal degeneration. Arch. Ophthalmol. 1983, 101, 718–724. [Google Scholar] [CrossRef]
- Michaelides, M.; Holder, G.E.; Webster, A.R.; Hunt, D.M.; Bird, A.C.; Fitzke, F.W.; Mollon, J.D.; Moore, A.T. A detailed phenotypic study of “cone dystrophy with supernormal rod ERG”. Br. J. Ophthalmol. 2005, 89, 332–339. [Google Scholar] [CrossRef] [PubMed]
- Piri, N.; Gao, Y.Q.; Danciger, M.; Mendoza, E.; Fishman, G.A.; Farber, D.B. A substitution of G to C in the cone cGMP-phosphodiesterase gamma subunit gene found in a distinctive form of cone dystrophy. Ophthalmology 2005, 112, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Sergouniotis, P.I.; Holder, G.E.; Robson, A.G.; Michaelides, M.; Webster, A.R.; Moore, A.T. High-resolution optical coherence tomography imaging in KCNV2 retinopathy. Br. J. Ophthalmol. 2012, 96, 213–217. [Google Scholar] [CrossRef] [PubMed]
- Robson, A.G.; Webster, A.R.; Michaelides, M.; Downes, S.M.; Cowing, J.A.; Hunt, D.M.; Moore, A.T.; Holder, G.E. “Cone dystrophy with supernormal rod electroretinogram”: A comprehensive genotype/phenotype study including fundus autofluorescence and extensive electrophysiology. Retina 2010, 30, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Robson, A.G.; Michaelides, M.; Saihan, Z.; Bird, A.C.; Webster, A.R.; Moore, A.T.; Fitzke, F.W.; Holder, G.E. Functional characteristics of patients with retinal dystrophy that manifest abnormal parafoveal annuli of high density fundus autofluorescence; a review and update. Doc. Ophthalmol. 2008, 116, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Vincent, A.; Robson, A.G.; Holder, G.E. Pathognomonic (diagnostic) ERGs. A review and update. Retina 2013, 33, 5–12. [Google Scholar] [CrossRef]
- Hull, S.; Arno, G.; Sergouniotis, P.I.; Tiffin, P.; Borman, A.D.; Chandra, A.; Robson, A.G.; Holder, G.E.; Webster, A.R.; Moore, A.T. Clinical and molecular characterization of enhanced S-cone syndrome in children. JAMA Ophthalmol. 2014, 132, 1341–1349. [Google Scholar] [CrossRef]
- Marmor, M.F.; Jacobson, S.G.; Foerster, M.H.; Kellner, U.; Weleber, R.G. Diagnostic clinical findings of a new syndrome with night blindness, maculopathy, and enhanced S cone sensitivity. Am. J. Ophthalmol. 1990, 110, 124–134. [Google Scholar] [CrossRef]
- Pachydaki, S.I.; Klaver, C.C.; Barbazetto, I.A.; Roy, M.S.; Gouras, P.; Allikmets, R.; Yannuzzi, L.A. Phenotypic features of patients with NR2E3 mutations. Arch. Ophthalmol. 2009, 127, 71–75. [Google Scholar] [CrossRef]
- Milam, A.H.; Rose, L.; Cideciyan, A.V.; Barakat, M.R.; Tang, W.X.; Gupta, N.; Aleman, T.S.; Wright, A.F.; Stone, E.M.; Sheffield, V.C.; et al. The nuclear receptor NR2E3 plays a role in human retinal photoreceptor differentiation and degeneration. Proc. Natl. Acad. Sci. USA 2002, 99, 473–478. [Google Scholar] [CrossRef]
- Alsalamah, A.K.; Khan, A.O.; Bakar, A.A.; Schatz, P.; Nowilaty, S.R. Recognizable Patterns of Submacular Fibrosis in Enhanced S-Cone Syndrome. Ophthalmol. Retina 2021, 5, 918–927. [Google Scholar] [CrossRef] [PubMed]
- Sustar, M.; Perovsek, D.; Cima, I.; Stirn-Kranjc, B.; Hawlina, M.; Brecelj, J. Electroretinography and optical coherence tomography reveal abnormal post-photoreceptoral activity and altered retinal lamination in patients with enhanced S-cone syndrome. Doc. Ophthalmol. 2015, 130, 165–177. [Google Scholar] [CrossRef] [PubMed]
- Lam, B.L.; Goldberg, J.L.; Hartley, K.L.; Stone, E.M.; Liu, M. Atypical mild enhanced S-cone syndrome with novel compound heterozygosity of the NR2E3 gene. Am. J. Ophthalmol. 2007, 144, 157–159. [Google Scholar] [CrossRef] [PubMed]
- Naik, A.; Ratra, D.; Banerjee, A.; Dalan, D.; Jandyal, S.; Rao, G.; Sen, P.; Bhende, M.; Jayaprakash, V.; Susvar, P.; et al. Enhanced S-cone syndrome: Clinical spectrum in Indian population. Indian J. Ophthalmol. 2019, 67, 523–529. [Google Scholar] [PubMed]
- Audo, I.; Michaelides, M.; Robson, A.G.; Hawlina, M.; Vaclavik, V.; Sandbach, J.M.; Neveu, M.M.; Hogg, C.R.; Hunt, D.M.; Moore, A.T.; et al. Phenotypic variation in enhanced S-cone syndrome. Investig. Ophthalmol. Vis. Sci. 2008, 49, 2082–2093. [Google Scholar] [CrossRef] [PubMed]
- De Carvalho, E.R.; Robson, A.G.; Arno, G.; Boon, C.J.F.; Webster, A.A.; Michaelides, M. Enhanced S-Cone Syndrome: Spectrum of Clinical, Imaging, Electrophysiologic, and Genetic Findings in a Retrospective Case Series of 56 Patients. Ophthalmol. Retina 2021, 5, 195–214. [Google Scholar] [CrossRef] [PubMed]
- Park, S.P.; Hong, I.H.; Tsang, S.H.; Lee, W.; Horowitz, J.; Yzer, S.; Allikmets, R.; Chang, S. Disruption of the human cone photoreceptor mosaic from a defect in NR2E3 transcription factor function in young adults. Graefes Arch. Clin. Exp. Ophthalmol. 2013, 251, 2299–2309. [Google Scholar] [CrossRef]
- Michaelides, M.; Li, Z.; Rana, N.A.; Richardson, E.C.; Hykin, P.G.; Moore, A.T.; Holder, G.E.; Webster, A.R. Novel mutations and electrophysiologic findings in RGS9- and R9AP-associated retinal dysfunction (Bradyopsia). Ophthalmology 2010, 117, 120–127.e1. [Google Scholar] [CrossRef]
- Hartong, D.T.; Pott, J.W.; Kooijman, A.C. Six patients with bradyopsia (slow vision): Clinical features and course of the disease. Ophthalmology 2007, 114, 2323–2331. [Google Scholar] [CrossRef]
- Chen, C.K.; Burns, M.E.; He, W.; Wensel, T.G.; Baylor, D.A.; Simon, M.I. Slowed recovery of rod photoresponse in mice lacking the GTPase accelerating protein RGS9-1. Nature 2000, 403, 557–560. [Google Scholar] [CrossRef]
- Welch, R.B. Bietti’s tapetoretinal degeneration with marginal corneal dystrophy crystalline retinopathy. Trans. Am. Ophthalmol. Soc. 1977, 75, 164–179. [Google Scholar] [PubMed]
- Tian, R.; Wang, S.R.; Wang, J.; Chen, Y.X. Novel CYP4V2 mutations associated with Bietti crystalline corneoretinal dystrophy in Chinese patients. Int. J. Ophthalmol. 2015, 8, 465–469. [Google Scholar] [PubMed]
- Kojima, H.; Otani, A.; Ogino, K.; Nakagawa, S.; Makiyama, Y.; Kurimoto, M.; Guo, C.; Yoshimura, N. Outer retinal circular structures in patients with Bietti crystalline retinopathy. Br. J. Ophthalmol. 2012, 96, 390–393. [Google Scholar] [CrossRef] [PubMed]
- Gocho, K.; Kameya, S.; Akeo, K.; Kikuchi, S.; Usui, A.; Yamaki, K.; Hayashi, T.; Tsuneoka, H.; Mizota, A.; Takahashi, H. High-Resolution Imaging of Patients with Bietti Crystalline Dystrophy with CYP4V2 Mutation. J. Ophthalmol. 2014, 2014, 283603. [Google Scholar] [CrossRef]
- Iriyama, A.; Aihara, Y.; Yanagi, Y. Outer retinal tubulation in inherited retinal degenerative disease. Retina 2013, 33, 1462–1465. [Google Scholar] [CrossRef] [PubMed]
- Gaucher, D.; Saleh, M.; Sauer, A.; Bourcier, T.; Speeg-Schatz, C. Spectral OCT analysis in Bietti crystalline dystrophy. Eur. J. Ophthalmol. 2010, 20, 612–614. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.Y.; Koh, A.H.; Aung, T.; Yong, V.H.; Yeung, K.; Ang, C.L.; Vithana, E.N. Characterization of Bietti crystalline dystrophy patients with CYP4V2 mutations. Investig. Ophthalmol. Vis. Sci. 2005, 46, 3812–3816. [Google Scholar] [CrossRef]
- Mansour, A.M.; Uwaydat, S.H.; Chan, C.C. Long-term follow-up in Bietti crystalline dystrophy. Eur. J. Ophthalmol. 2007, 17, 680–682. [Google Scholar] [CrossRef]
- Usui, T.; Tanimoto, N.; Takagi, M.; Hasegawa, S.; Abe, H. Rod and cone a-waves in three cases of Bietti crystalline chorioretinal dystrophy. Am. J. Ophthalmol. 2001, 132, 395–402. [Google Scholar] [CrossRef]
- Sen, P.; Ray, R.; Ravi, P. Electrophysiological findings in Bietti’s crystalline dystrophy. Clin. Exp. Optom. 2011, 94, 302–308. [Google Scholar] [CrossRef]
- Rossi, S.; Testa, F.; Li, A.; Iorio, V.D.; Zhang, J.; Gesualdo, C.; Corte, M.D.; Chan, C.C.; Fielding Hejtmancik, J.; Simonelli, F. An atypical form of Bietti crystalline dystrophy. Ophthalmic Genet. 2011, 32, 118–121. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Garcia, G.P.; Martinez-Rubio, M.; Moya-Moya, M.A.; Perez-Santonja, J.J.; Escribano, J. Current perspectives in Bietti crystalline dystrophy. Clin. Ophthalmol. 2019, 13, 1379–1399. [Google Scholar] [CrossRef] [PubMed]
- Lai, T.Y.; Ng, T.K.; Tam, P.O.; Yam, G.H.; Ngai, J.W.; Chan, W.M.; Liu, D.T.; Lam, D.S.; Pang, C.P. Genotype phenotype analysis of Bietti’s crystalline dystrophy in patients with CYP4V2 mutations. Investig. Ophthalmol. Vis. Sci. 2007, 48, 5212–5220. [Google Scholar] [CrossRef] [PubMed]
- Lockhart, C.M.; Smith, T.B.; Yang, P.; Naidu, M.; Rettie, A.E.; Nath, A.; Weleber, R.; Kelly, E.J. Longitudinal characterisation of function and structure of Bietti crystalline dystrophy: Report on a novel homozygous mutation in CYP4V2. Br. J. Ophthalmol. 2018, 102, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Borooah, S.; Collins, C.; Wright, A.; Dhillon, B. Late-onset retinal macular degeneration: Clinical insights into an inherited retinal degeneration. Br. J. Ophthalmol. 2009, 93, 284–289. [Google Scholar] [CrossRef] [PubMed]
- Ayyagari, R.; Mandal, M.N.; Karoukis, A.J.; Chen, L.; McLaren, N.C.; Lichter, M.; Wong, D.T.; Hitchcock, P.F.; Caruso, R.C.; Moroi, S.E.; et al. Late-onset macular degeneration and long anterior lens zonules result from a CTRP5 gene mutation. Investig. Ophthalmol. Vis. Sci. 2005, 46, 3363–3371. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, S.G.; Cideciyan, A.V.; Wright, E.; Wright, A.F. Phenotypic marker for early disease detection in dominant late-onset retinal degeneration. Investig. Ophthalmol. Vis. Sci. 2001, 42, 1882–1890. [Google Scholar]
- Milam, A.H.; Curcio, C.A.; Cideciyan, A.V.; Saxena, S.; John, S.K.; Kruth, H.S.; Malek, G.; Heckenlively, J.R.; Weleber, R.G.; Jacobson, S.G. Dominant late-onset retinal degeneration with regional variation of sub-retinal pigment epithelium deposits, retinal function, and photoreceptor degeneration. Ophthalmology 2000, 107, 2256–2266. [Google Scholar] [CrossRef]
- Cukras, C.; Flamendorf, J.; Wong, W.T.; Ayyagari, R.; Cunningham, D.; Sieving, P.A. Longitudinal Structural Changes in Late-Onset Retinal Degeneration. Retina 2016, 36, 2348–2356. [Google Scholar] [CrossRef]
- Soumplis, V.; Sergouniotis, P.I.; Robson, A.G.; Michaelides, M.; Moore, A.T.; Holder, G.E.; Webster, A.R. Phenotypic findings in C1QTNF5 retinopathy (late-onset retinal degeneration). Acta Ophthalmol. 2013, 91, e191–e195. [Google Scholar] [CrossRef]
- Vincent, A.; Munier, F.L.; Vandenhoven, C.C.; Wright, T.; Westall, C.A.; Heon, E. The characterization of retinal phenotype in a family with C1QTNF5-related late-onset retinal degeneration. Retina 2012, 32, 1643–1651. [Google Scholar] [CrossRef] [PubMed]
- Kuntz, C.A.; Jacobson, S.G.; Cideciyan, A.V.; Li, Z.Y.; Stone, E.M.; Possin, D.; Milam, A.H. Sub-retinal pigment epithelial deposits in a dominant late-onset retinal degeneration. Investig. Ophthalmol. Vis. Sci. 1996, 37, 1772–1782. [Google Scholar] [PubMed]
- Papastavrou, V.T.; Bradshaw, K.R.; Aye, K.H.; Turney, C.; Browning, A.C. Improvement of retinal function in L-ORD after prolonged dark adaptation. Can. J. Ophthalmol. 2015, 50, 112–118. [Google Scholar] [CrossRef] [PubMed]
- Borooah, S.; Papastavrou, V.; Lando, L.; Han, J.; Lin, J.H.; Ayyagari, R.; Dhillon, B.; Browning, A.C. Reticular Pseudodrusen in Late-Onset Retinal Degeneration. Ophthalmol. Retina 2021, 5, 1043–1051. [Google Scholar] [CrossRef] [PubMed]
- Lando, L.; Borooah, S. Late-Onset Retinal Degeneration: Clinical Perspectives. Clin. Ophthalmol. 2022, 16, 3225–3246. [Google Scholar] [CrossRef] [PubMed]
- Makiyama, Y.; Ooto, S.; Hangai, M.; Ogino, K.; Gotoh, N.; Oishi, A.; Yoshimura, N. Cone abnormalities in fundus albipunctatus associated with RDH5 mutations assessed using adaptive optics scanning laser ophthalmoscopy. Am. J. Ophthalmol. 2014, 157, 558–570.e4. [Google Scholar] [CrossRef] [PubMed]
- Genead, M.A.; Fishman, G.A.; Lindeman, M. Spectral-domain optical coherence tomography and fundus autofluorescence characteristics in patients with fundus albipunctatus and retinitis punctata albescens. Ophthalmic Genet. 2010, 31, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Schatz, P.; Preising, M.; Lorenz, B.; Sander, B.; Larsen, M.; Eckstein, C.; Rosenberg, T. Lack of autofluorescence in fundus albipunctatus associated with mutations in RDH5. Retina 2010, 30, 1704–1713. [Google Scholar] [CrossRef]
- Sergouniotis, P.I.; Sohn, E.H.; Li, Z.; McBain, V.A.; Wright, G.A.; Moore, A.T.; Robson, A.G.; Holder, G.E.; Webster, A.R. Phenotypic variability in RDH5 retinopathy (Fundus Albipunctatus). Ophthalmology 2011, 118, 1661–1670. [Google Scholar] [CrossRef]
- Nakamura, M.; Skalet, J.; Miyake, Y. RDH5 gene mutations and electroretinogram in fundus albipunctatus with or without macular dystrophy: RDH5 mutations and ERG in fundus albipunctatus. Doc. Ophthalmol. 2003, 107, 3–11. [Google Scholar] [CrossRef]
- Hajali, M.; Fishman, G.A.; Dryja, T.P.; Sweeney, M.O.; Lindeman, M. Diagnosis in a patient with fundus albipunctatus and atypical fundus changes. Doc. Ophthalmol. 2009, 118, 233–238. [Google Scholar] [CrossRef] [PubMed]
- Burstedt, M.S.; Forsman-Semb, K.; Golovleva, I.; Janunger, T.; Wachtmeister, L.; Sandgren, O. Ocular phenotype of bothnia dystrophy, an autosomal recessive retinitis pigmentosa associated with an R234W mutation in the RLBP1 gene. Arch. Ophthalmol. 2001, 119, 260–267. [Google Scholar] [PubMed]
- Morimura, H.; Berson, E.L.; Dryja, T.P. Recessive mutations in the RLBP1 gene encoding cellular retinaldehyde-binding protein in a form of retinitis punctata albescens. Investig. Ophthalmol. Vis. Sci. 1999, 40, 1000–1004. [Google Scholar] [PubMed]
- Eichers, E.R.; Green, J.S.; Stockton, D.W.; Jackman, C.S.; Whelan, J.; McNamara, J.A.; Johnson, G.J.; Lupski, J.R.; Katsanis, N. Newfoundland rod-cone dystrophy, an early-onset retinal dystrophy, is caused by splice-junction mutations in RLBP1. Am. J. Hum. Genet. 2002, 70, 955–964. [Google Scholar] [CrossRef] [PubMed]
- Fishman, G.A.; Roberts, M.F.; Derlacki, D.J.; Grimsby, J.L.; Yamamoto, H.; Sharon, D.; Nishiguchi, K.M.; Dryja, T.P. Novel mutations in the cellular retinaldehyde-binding protein gene (RLBP1) associated with retinitis punctata albescens: Evidence of interfamilial genetic heterogeneity and fundus changes in heterozygotes. Arch. Ophthalmol. 2004, 122, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Granse, L.; Abrahamson, M.; Ponjavic, V.; Andreasson, S. Electrophysiological findings in two young patients with Bothnia dystrophy and a mutation in the RLBP1 gene. Ophthalmic Genet. 2001, 22, 97–105. [Google Scholar] [CrossRef]
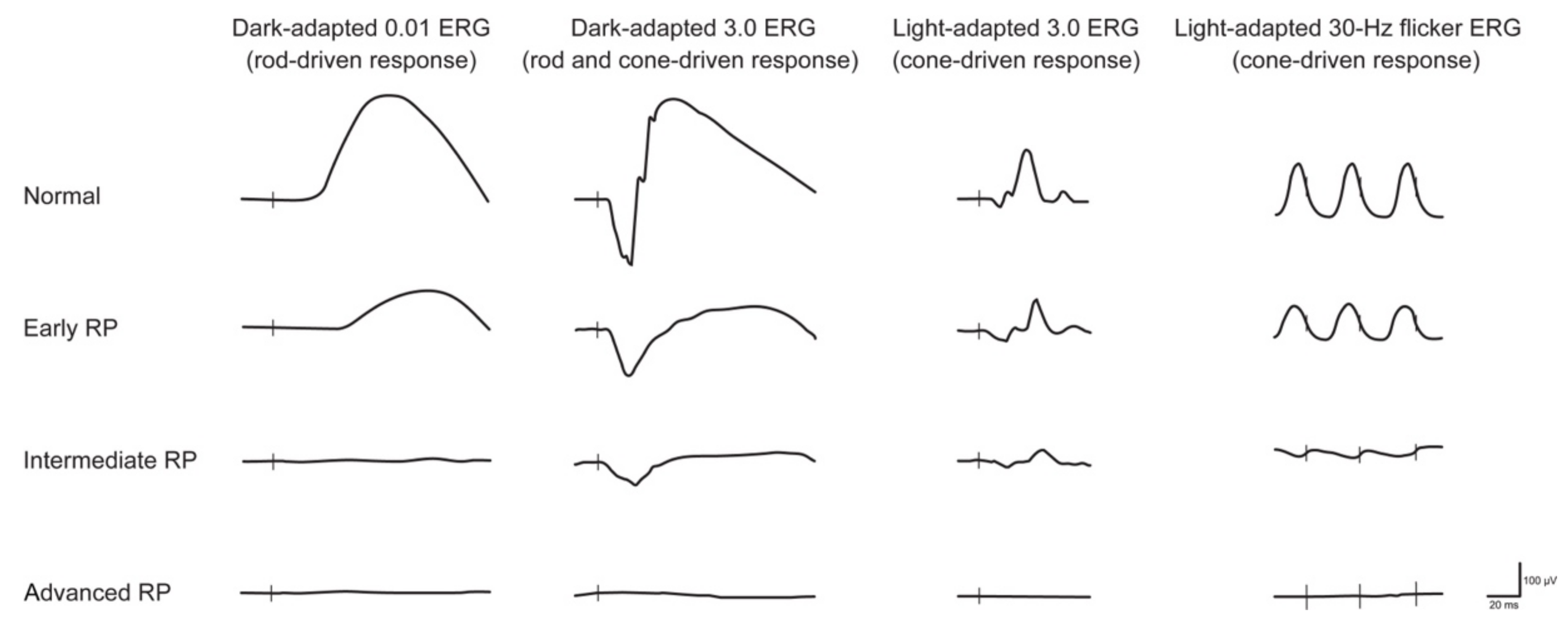
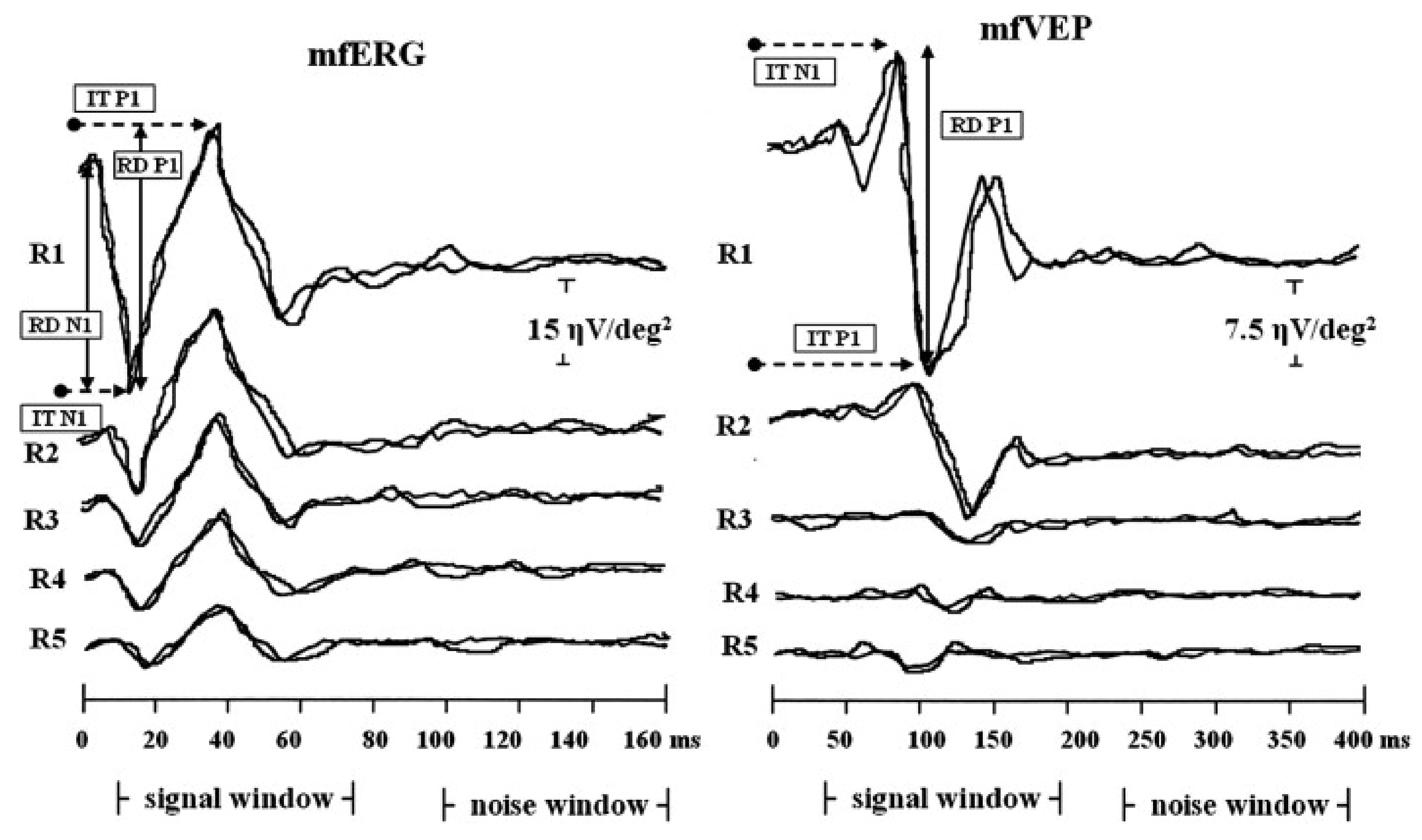
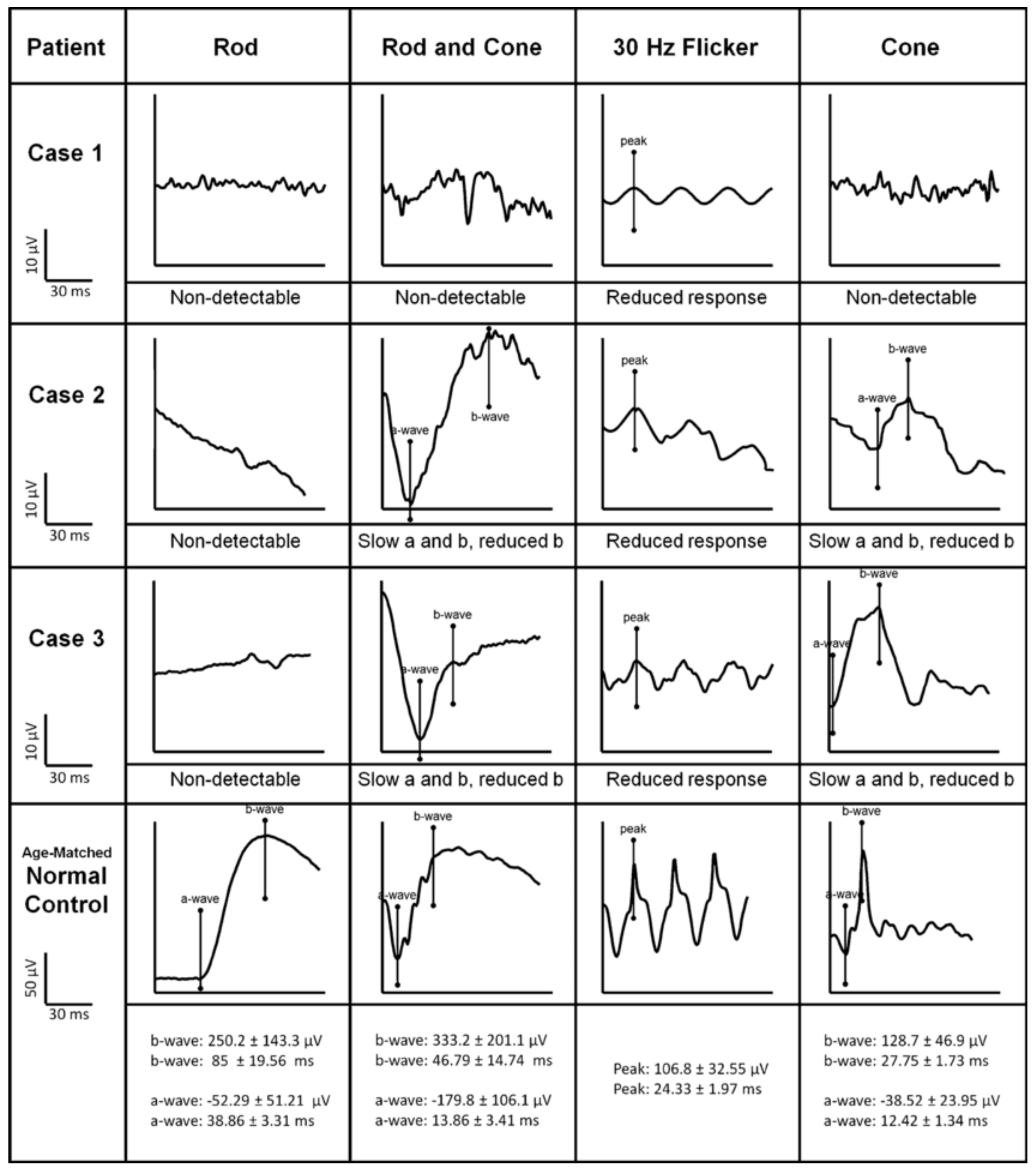

{kind=link}
{kind=link}
{kind=link}
| Name | OMIM | Gene | Inheritance | References |
|---|---|---|---|---|
| Retinitis Pigmentosa | 268000 (heterogeneous) | RHO, PRPF31, RP1, RPGR, RP2, PDE6, etc. | Autosomal dominant, X-linked, autosomal recessive | [14,15,16] |
| Cone Dystrophy | 120970, 602093, etc. (heterogeneous) | AIPL1, CRX, GUCA1A, GUCY2D, PITPNM3, PROM1, PRPH2/RDS, RIMS1, SEMA4A, and UNC119 | Autosomal dominant, X-linked, autosomal recessive | [17,18] |
| Cone Dystrophy with Supernormal Rod ERG | 610356 | KCNV2 | Autosomal recessive | [19,20,21] |
| Enhanced S-Cone Syndrome | 268100 | NR2E3 | Autosomal recessive | [22,23] |
| Bradyopsia | 608415 or 620344 | RGS9 or R9AP | Autosomal recessive | [24,25] |
| Bietti Crystalline Dystrophy | 210370 | CYP4V2 | Autosomal recessive | [26,27,28] |
| Late-Onset Retinal Degeneration | 605670 | C1QTNF5 | Autosomal dominant | [29,30] |
| Fundus Albipunctatus | 601617 | RDH5 | Autosomal recessive or dominant | [31,32] |
| Retinitis Punctata Albescens | 136880 | RLBP1, RHO, PRPH2 | Autosomal recessive or dominant | [33,34,35] |
| Name | ffERG | mfERG | Pathognomonic Findings | References |
|---|---|---|---|---|
| Retinitis Pigmentosa | A−, I− | A−, I− | No | [65,66,68,69,70,71] |
| Cone/Cone-Rod Dystrophy | A−, I− | No | [97,98] | |
| Cone Dystrophy with Supernormal Rod ERG | A− (except supernormal S-cone ERG), I− | Yes (see Section 2.3) | [106,107,108] | |
| Enhanced S-Cone Syndrome | A−, I− | Yes (see Section 2.4) | [115,116,117,118] | |
| Bradyopsia | A− | Yes (see Section 2.5) | [24,25,120,122] | |
| Bietti Crystalline Dystrophy | A−, I− | A−, I− | No | [126,127,128] |
| Late-Onset Retinal Degeneration | A− | A− | No | [30,138,140,143,145] |
| Fundus Albipunctatus | A− | No | [151,152,153] | |
| Retinitis Punctata Albescens | A− | A− | No | [157,158] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Haraguchi, Y.; Chiang, T.-K.; Yu, M. Application of Electrophysiology in Non-Macular Inherited Retinal Dystrophies. J. Clin. Med. 2023, 12, 6953. https://doi.org/10.3390/jcm12216953
Haraguchi Y, Chiang T-K, Yu M. Application of Electrophysiology in Non-Macular Inherited Retinal Dystrophies. Journal of Clinical Medicine. 2023; 12(21):6953. https://doi.org/10.3390/jcm12216953
Chicago/Turabian StyleHaraguchi, Yulia, Tsun-Kang Chiang, and Minzhong Yu. 2023. "Application of Electrophysiology in Non-Macular Inherited Retinal Dystrophies" Journal of Clinical Medicine 12, no. 21: 6953. https://doi.org/10.3390/jcm12216953
APA StyleHaraguchi, Y., Chiang, T.-K., & Yu, M. (2023). Application of Electrophysiology in Non-Macular Inherited Retinal Dystrophies. Journal of Clinical Medicine, 12(21), 6953. https://doi.org/10.3390/jcm12216953