
Case Series on Autosomal Recessive Non-Syndromic Retinitis Pigmentosa Caused by POMGNT1 Mutations with a Report of a New Variant

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
3. Results
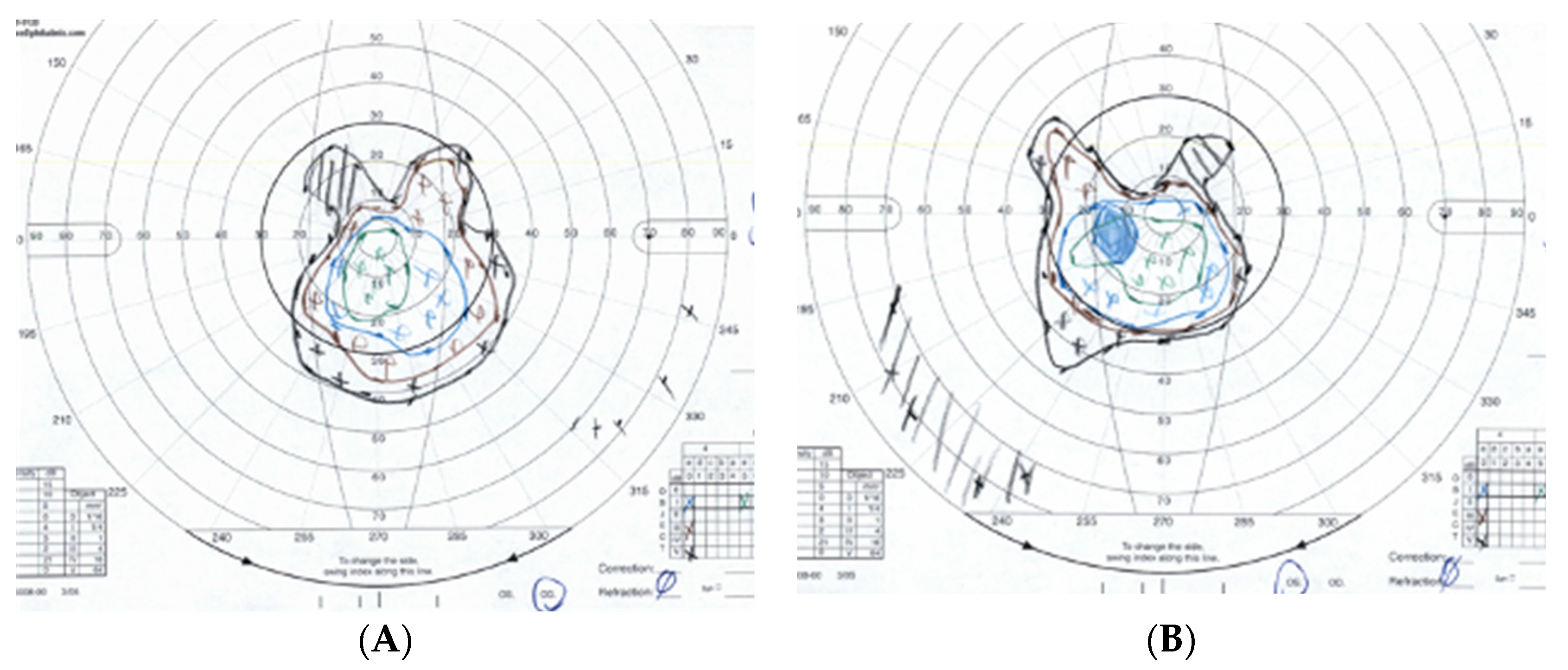
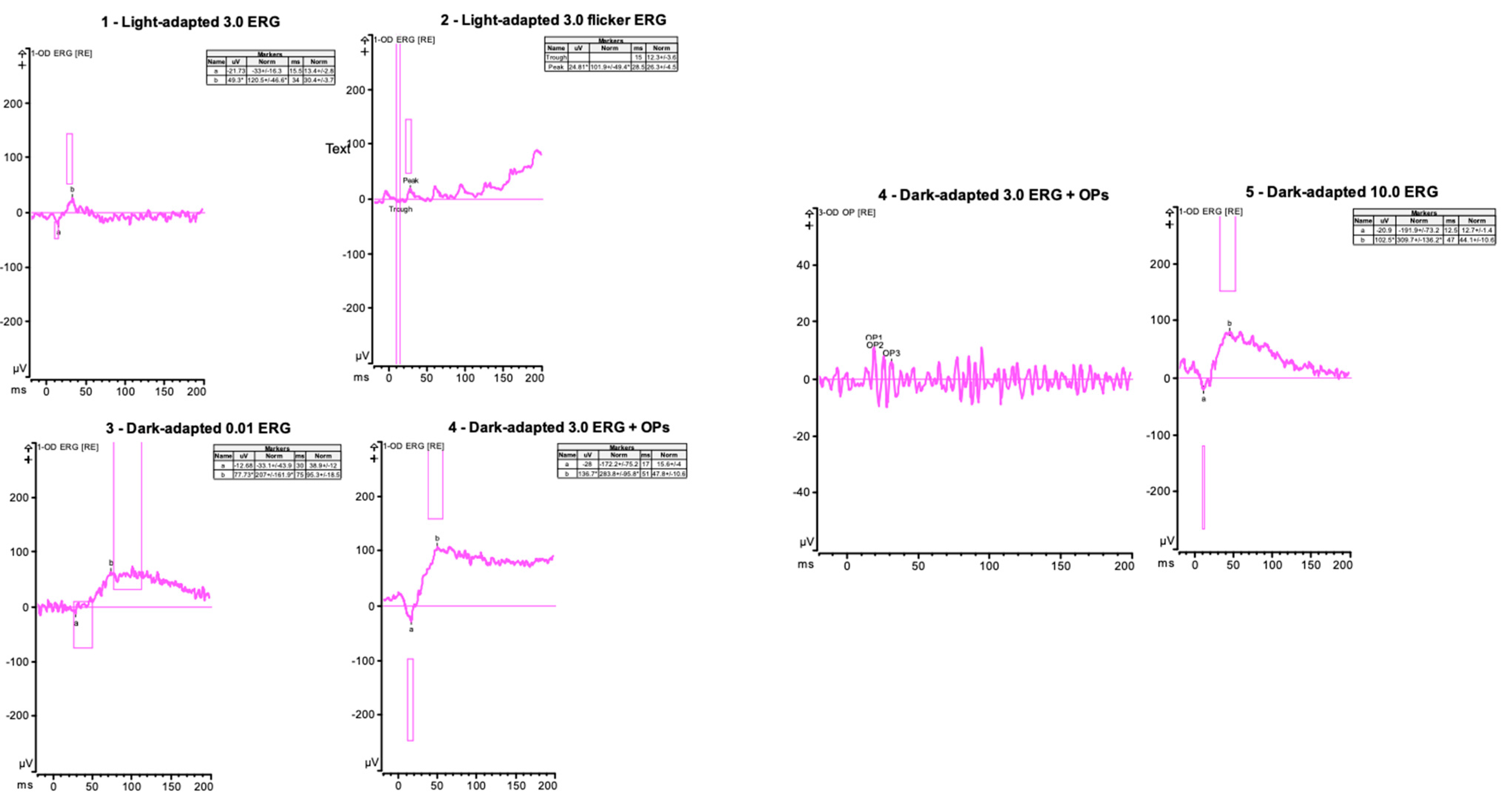
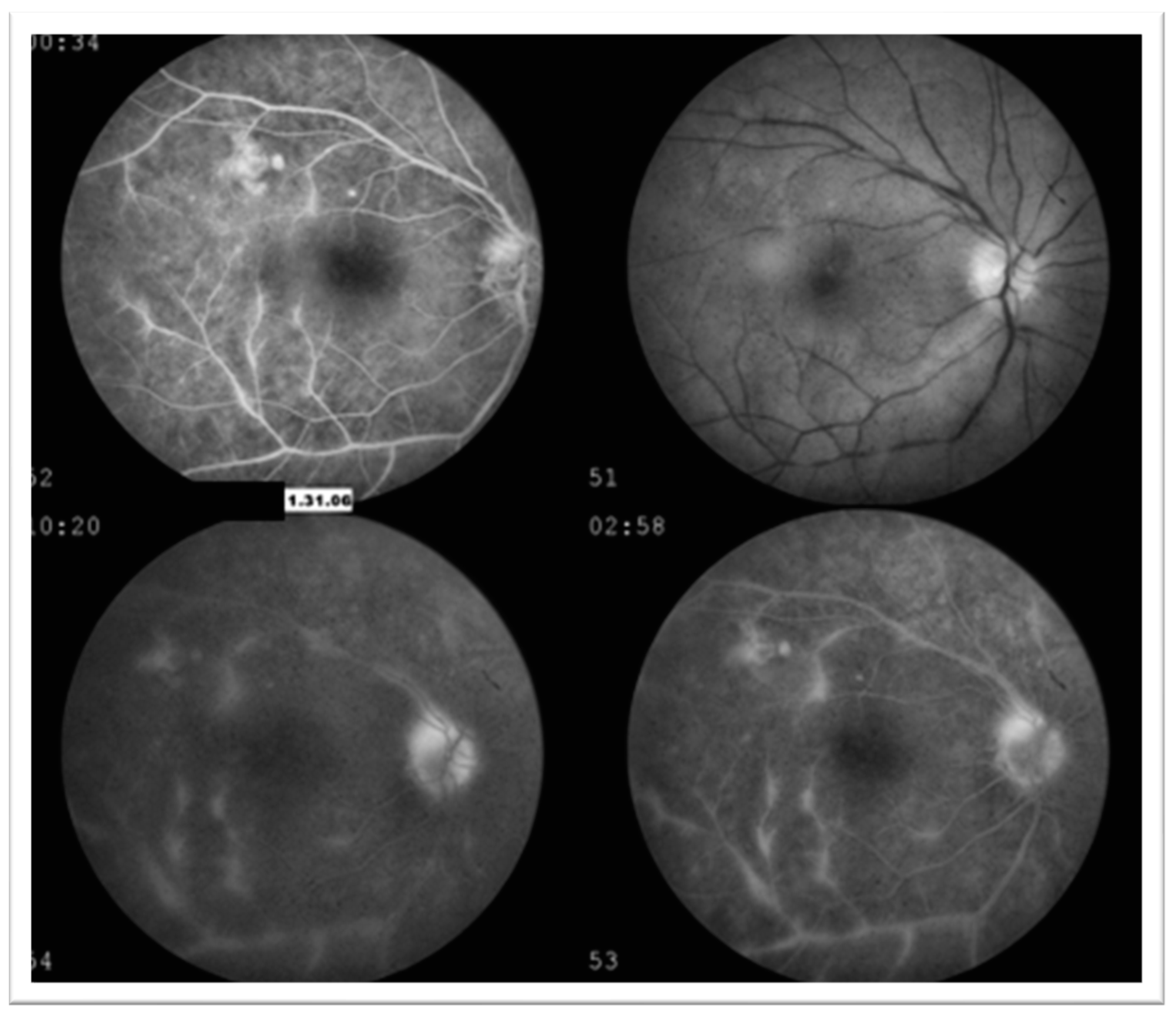
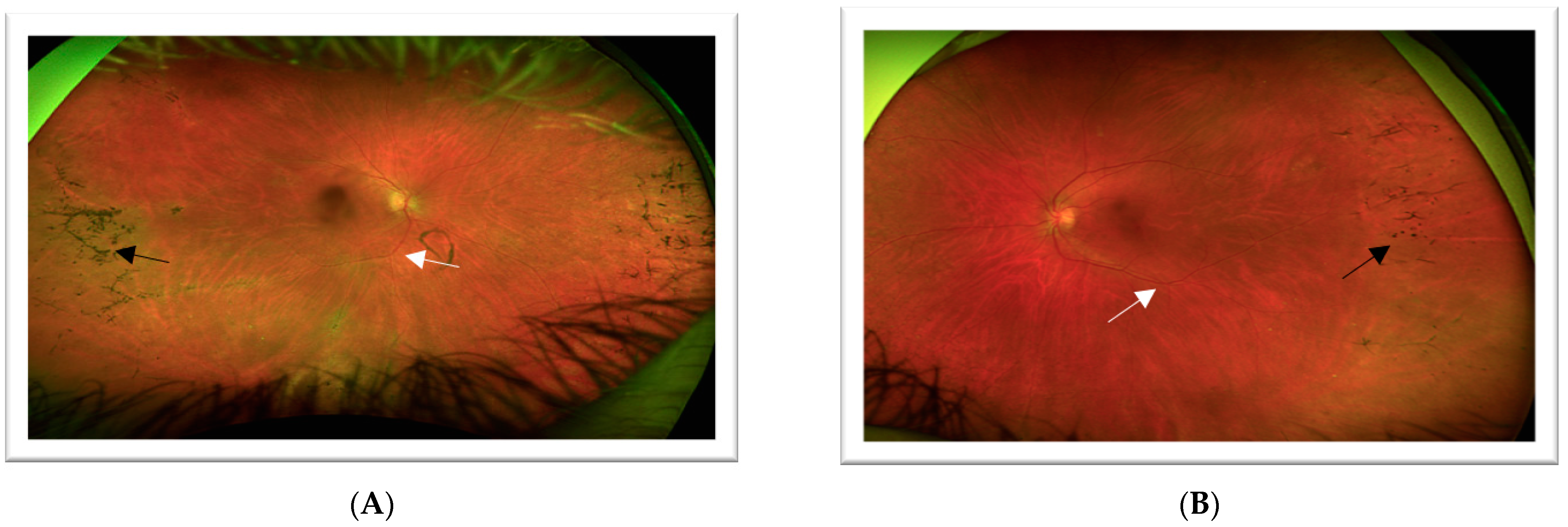
3.1. Case 1: 42-Year-Old Female for Cataract Evaluation (Proband)
3.2. Case 2: 45-Year-Old Female with Recurrent Anterior Uveitis and Retinal Vasculitis
3.3. Case 3: 54-Year-Old Female with Multiple Sclerosis
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Verbakel, S.K.; van Huet, R.A.; Boon, C.J.; den Hollander, A.I.; Collin, R.W.; Klaver, C.C.; Hoyng, C.B.; Roepman, R.; Klevering, B.J. Non-syndromic retinitis pigmentosa. Prog. Retin. Eye Res. 2018, 66, 157–186. [Google Scholar] [CrossRef] [PubMed]
- Fahim, A.T.; Daiger, S.P.; Weleber, R.G. Nonsyndromic retinitis pigmentosa overview. In GeneReviews®; University of Washington: Seattle, WA, USA, 2023. [Google Scholar]
- Xu, M.; Yamada, T.; Sun, Z.; Eblimit, A.; Lopez, I.; Wang, F.; Manya, H.; Xu, S.; Zhao, L.; Li, Y.; et al. Mutations in POMGNT1 cause non-syndromic retinitis pigmentosa. Hum. Mol. Genet. 2016, 25, 1479–1488. [Google Scholar] [CrossRef] [PubMed]
- Sheikh, M.O.; Halmo, S.M.; Wells, L. Recent advancements in understanding mammalian O-mannosylation. Glycobiology 2017, 27, 806–819. [Google Scholar] [CrossRef] [PubMed]
- Lommel, M.; Strahl, S. Protein O-mannosylation: Conserved from bacteria to humans. Glycobiology 2009, 19, 816–828. [Google Scholar] [CrossRef] [PubMed]
- Shenoy, A.M.; Markowitz, J.A.; Bonnemann, C.G.; Krishnamoorthy, K.; Bossler, A.D.; Tseng, B.S. Muscle-Eye-Brain disease. J. Clin. Neuromuscul. Dis. 2010, 11, 124–126. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.H.H.; Chen, S.J.; Yang, C.F.; Chen, H.W.; Chuang, H.P.; Lu, Y.H.; Chen, C.H.; Wu, J.Y.; Niu, D.M.; Chen, Y.T. Homozygosity Mapping and Whole-Genome Sequencing Links a Missense Mutation in POMGNT1 to Autosomal Recessive Retinitis Pigmentosa. Investig. Ophthalmol. Vis. Sci. 2016, 57, 3601–3609. [Google Scholar] [CrossRef] [PubMed]
- National Center for Biotechnology Information. ClinVar. [VCV000632108.5]. Available online: https://www.ncbi.nlm.nih.gov/clinvar/variation/VCV000632108.5 (accessed on 26 November 2023).
- Raskin, E.; Achiron, A.; Zloto, O.; Neuman, R.; Vishnevskia-Dai, V. Uveitis prior to clinical presentation of Multiple Sclerosis (MS) is associated with better MS prognosis. PLoS ONE 2022, 17, e0264918. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, N.; Ikeda, Y.; Notomi, S.; Ishikawa, K.; Murakami, Y.; Hisatomi, T.; Enaida, H.; Ishibashi, T. Clinical evidence of sustained chronic inflammatory reaction in retinitis pigmentosa. Ophthalmology 2013, 120, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Dutta Majumder, P.; Menia, N.; Roy, R.; Sen, P.; George, A.E.; Ganesh, S.K.; Biswas, J. Uveitis in Patients with Retinitis Pigmentosa: 30 Years’ Consecutive Data. Ocul. Immunol. Inflamm. 2018, 26, 1283–1288. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Patel, A.; Cui, R.; Odom, J.V.; Leys, M. Case Series on Autosomal Recessive Non-Syndromic Retinitis Pigmentosa Caused by POMGNT1 Mutations with a Report of a New Variant. J. Clin. Med. 2023, 12, 7549. https://doi.org/10.3390/jcm12247549
Patel A, Cui R, Odom JV, Leys M. Case Series on Autosomal Recessive Non-Syndromic Retinitis Pigmentosa Caused by POMGNT1 Mutations with a Report of a New Variant. Journal of Clinical Medicine. 2023; 12(24):7549. https://doi.org/10.3390/jcm12247549
Chicago/Turabian StylePatel, Ami, Ruifeng Cui, James Vernon Odom, and Monique Leys. 2023. "Case Series on Autosomal Recessive Non-Syndromic Retinitis Pigmentosa Caused by POMGNT1 Mutations with a Report of a New Variant" Journal of Clinical Medicine 12, no. 24: 7549. https://doi.org/10.3390/jcm12247549
APA StylePatel, A., Cui, R., Odom, J. V., & Leys, M. (2023). Case Series on Autosomal Recessive Non-Syndromic Retinitis Pigmentosa Caused by POMGNT1 Mutations with a Report of a New Variant. Journal of Clinical Medicine, 12(24), 7549. https://doi.org/10.3390/jcm12247549