
The Ultrastructure of Hepatic Stellate Cell–Macrophage Intercellular Crosstalk as a New Morphological Insight into Phenomenon of Fibrogenesis in Pediatric Autoimmune Hepatitis
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Material and Methods
2.1. Study Patients’ Profile
2.2. Liver Tissue Processing for Transmission Electron Microscopy
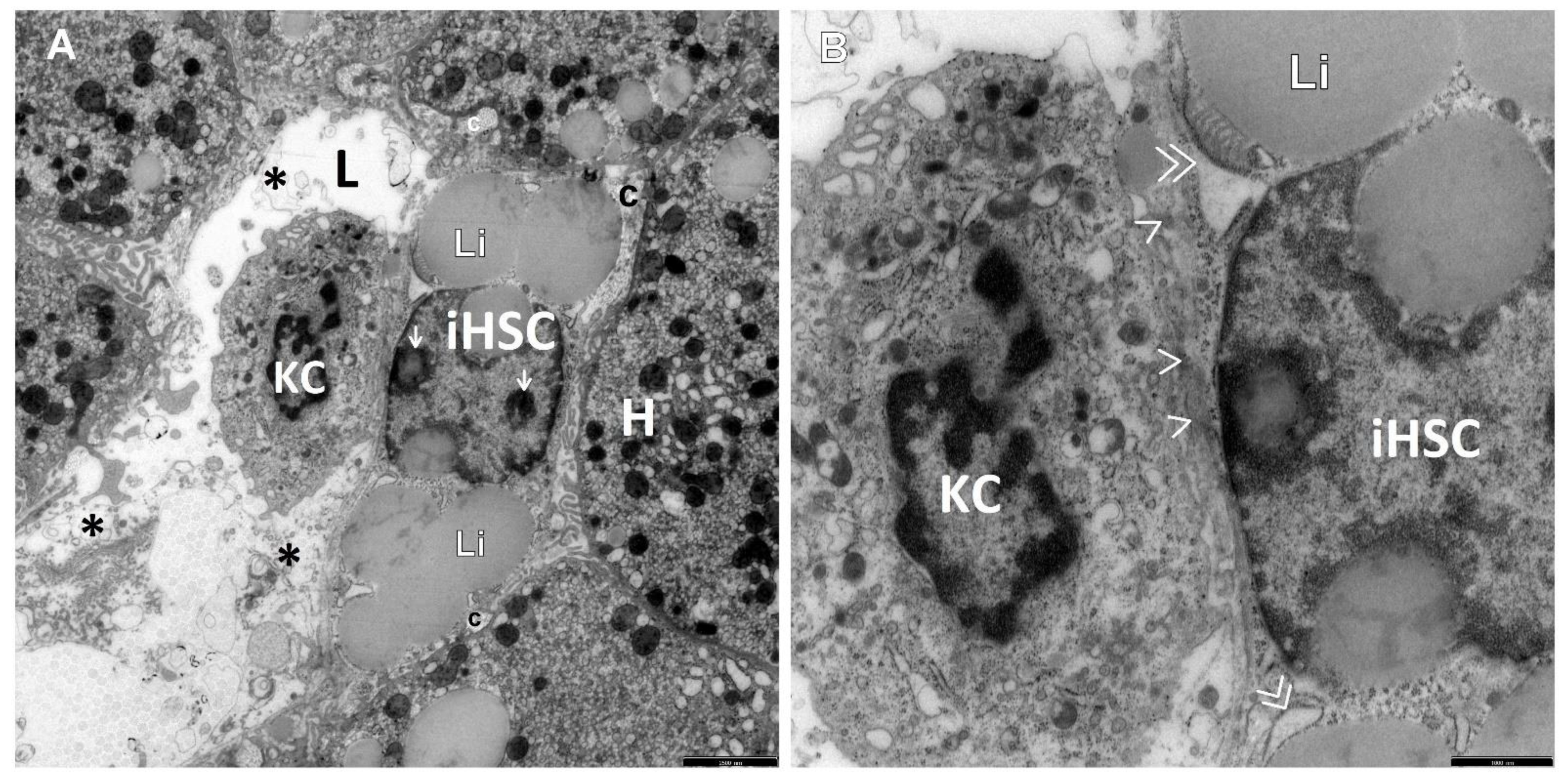
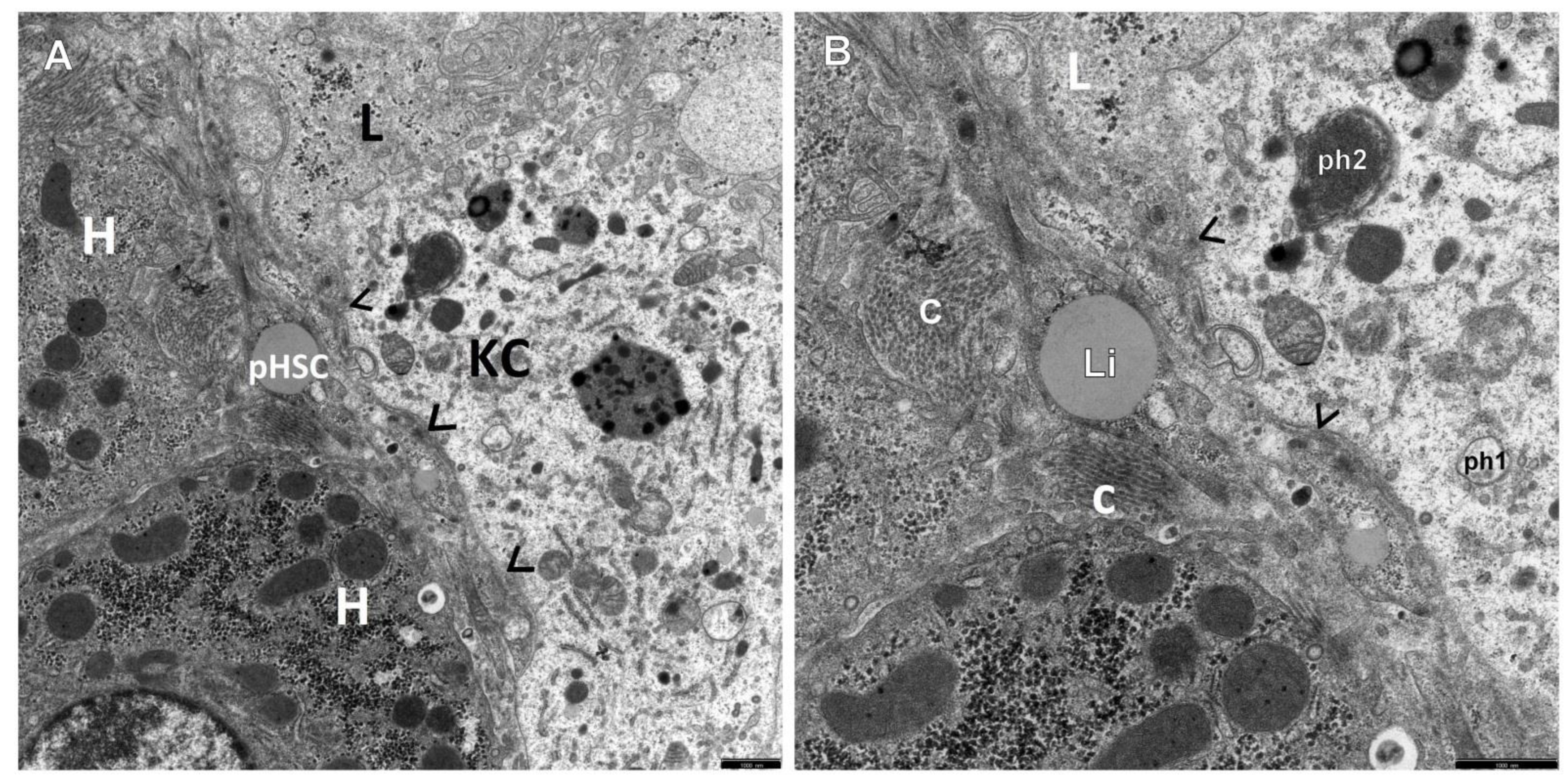
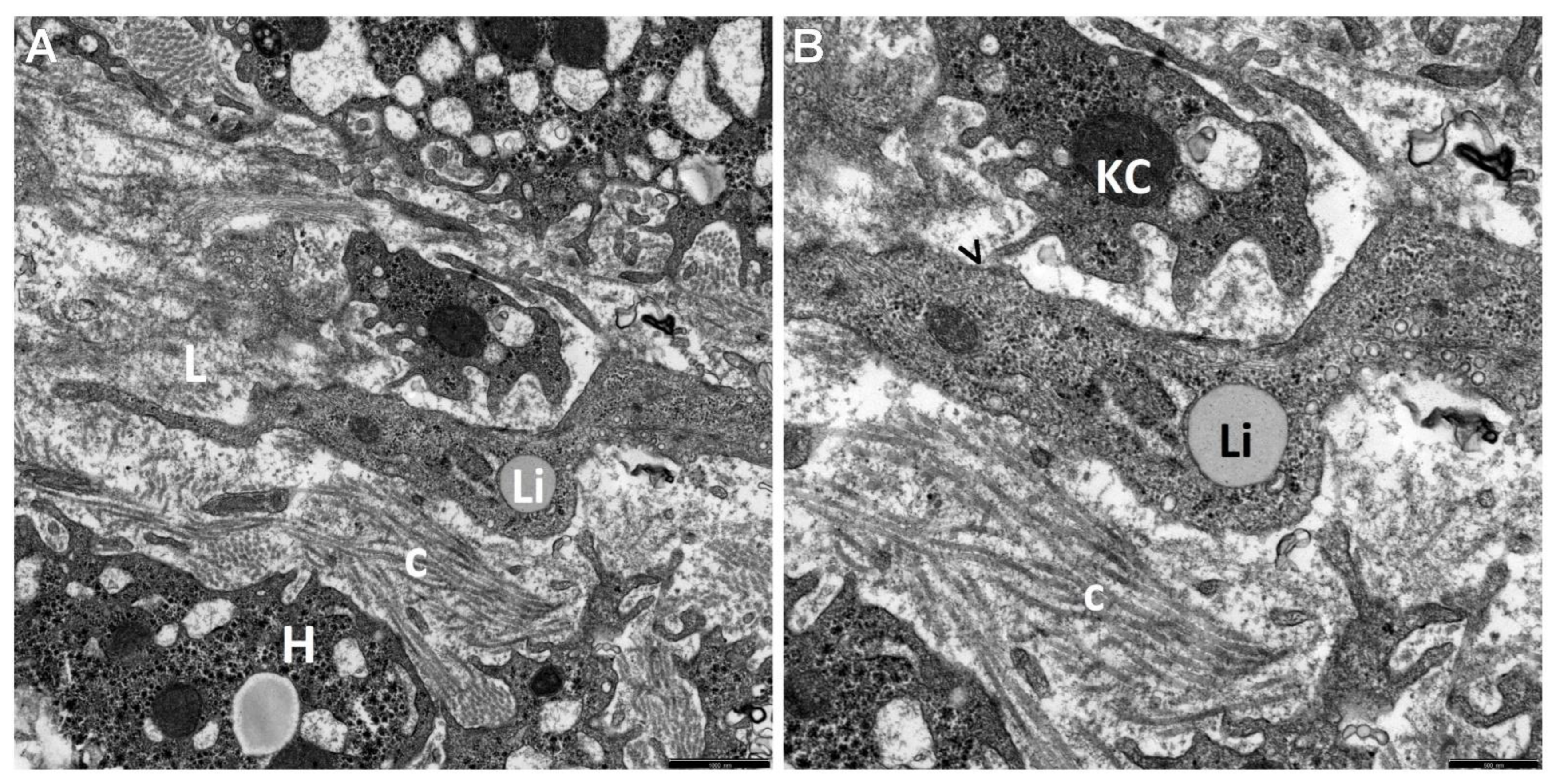
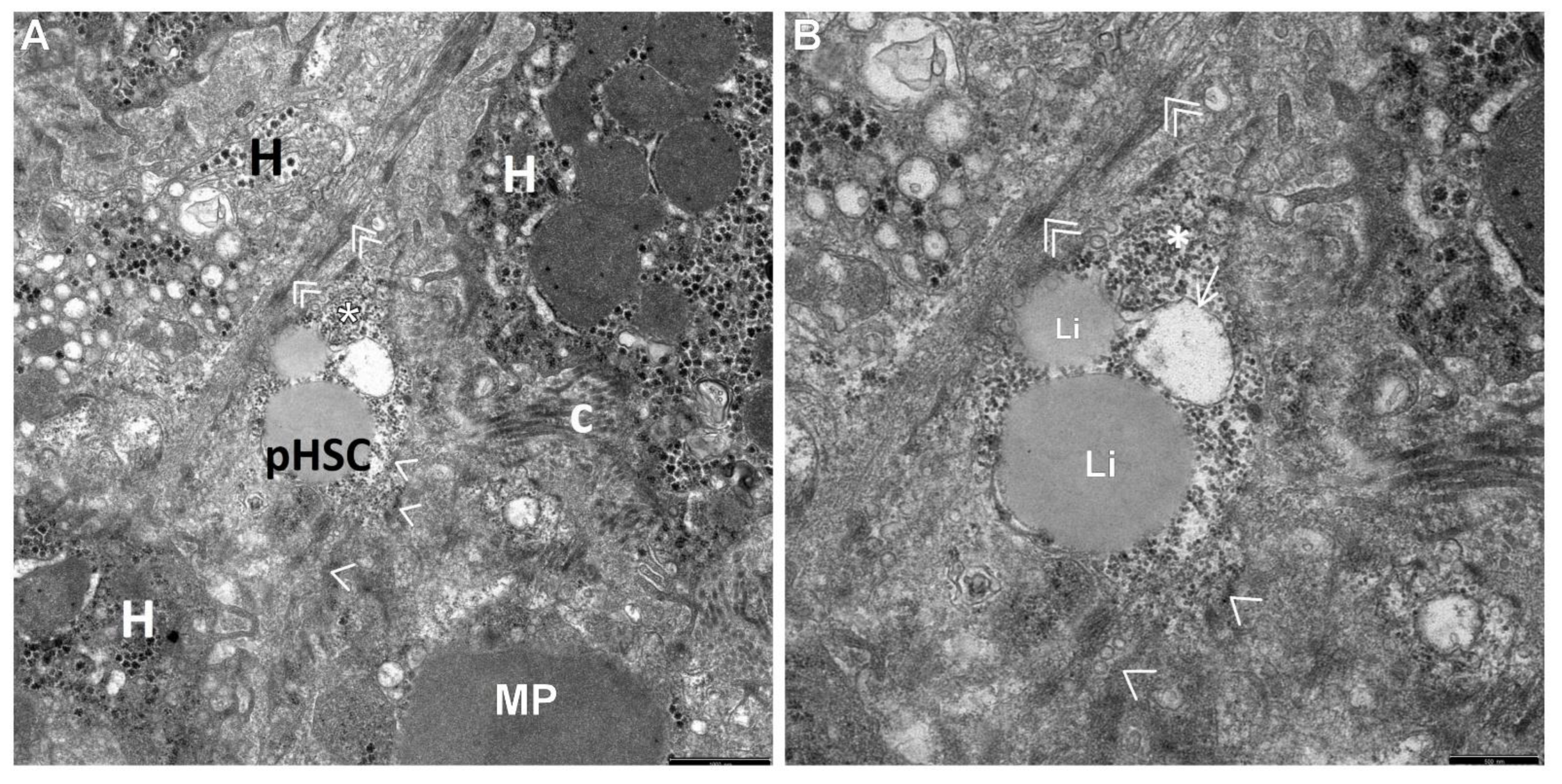
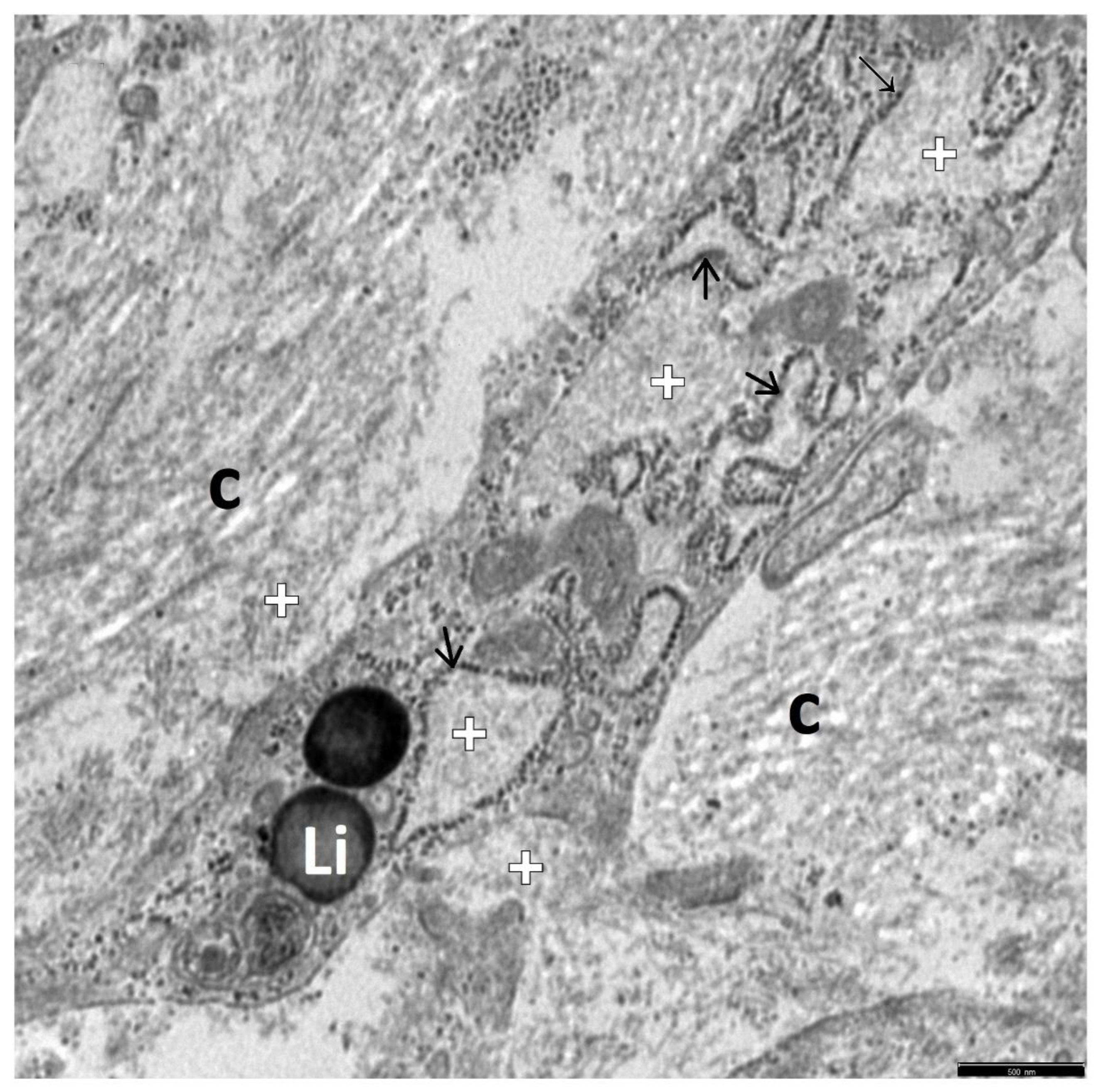
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mieli-Vergani, G.; Vergani, D.; Czaja, A.J.; Manns, M.P.; Krawitt, E.L.; Vierling, J.M.; Lohse, A.W.; Montano-Loza, A.J. Autoimmunehepatitis. Nat. Rev. Dis. Prim. 2018, 4, 18017. [Google Scholar] [CrossRef]
- Cha, H.J.; Hwang, J.; Lee, L.E.; Park, Y.; Song, J.J. The significance of cytoplasmic antinuclear antibody patterns in autoimmune liver disease. PLoS ONE 2021, 16, e0244950. [Google Scholar] [CrossRef]
- Lotowska, J.M.; Sobaniec-Lotowska, M.E.; Daniluk, U.; Lebensztejn, D.M. Glassy droplet inclusions within the cytoplasm of Kupffer cells: A novel ultrastructural feature for the diagnosis of pediatric autoimmune hepatitis. Dig. Liver Dis. 2017, 49, 929–933. [Google Scholar] [CrossRef] [PubMed]
- Dezsőfi, A.; Baumann, U.; Dhawan, A.; Durmaz, O.; Fischler, B.; Hadzic, N.; Hierro, L.; Lacaille, F.; McLin, V.A.; Nobili, V.; et al. Liver 533 biopsy in children: Position paper of the ESPGHAN Hepatology Committee. J. Pediatr. Gastroenterol. Nutr. 2015, 60, 408–420. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, A. Autoimmune Hepatitis: 2019 Update. Gut Liver 2020, 14, 430–438. [Google Scholar] [CrossRef] [PubMed]
- Arcos-Machancoses, J.V.; Busoms, C.M.; Tatis, E.J.; Bovo, M.V.; Bernabeu, J.Q.; Goñi, J.J.; Martínez, V.C.; de Carpi, J.M. Development and validation of a new simplified diagnostic scoring system for pediatric autoimmune hepatitis. Multicenter Study. Dig. Liver Dis. 2019, 51, 1308–13135. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, K.; Fukuda, Y.; Seza, K.; Saito, M.; Yasui, S.; Nakano, M.; Yokosuka, O.; Kato, N. Long-term observation of acute-onset autoimmune hepatitis presenting clinically and radiologically as acute hepatitis. Hepatol. Int. 2018, 12, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Sokollik, C.; McLin, V.A.; Vergani, D.; Beretta-Piccoli, B.T.; Mieli-Vergani, G. Juvenile autoimmune hepatitis: A comprehensive review. J. Autoimmun. 2018, 95, 69–76. [Google Scholar] [CrossRef]
- Puustinen, L.; Barner-Rasmussen, N.; Pukkala, E.; Färkkilä, M. Incidence, prevalence, and causes of death of patients with autoimmune hepatitis: A nationwide register-based cohort study in Finland. Dig. Liver Dis. 2019, 51, 1294–1299. [Google Scholar] [CrossRef] [Green Version]
- Nares-Cisneros, J.; Jaramillo-Rodríguez, Y. Autoimmune hepatitis in children: Progression of 20 cases in northern Mexico. Rev. Gastroenterol. Mex. 2014, 79, 238–243. [Google Scholar] [CrossRef]
- Floreani, A.; Liberal, R.; Vergani, D.; Mieli-Vergani, G. Autoimmune hepatitis: Contrasts and comparisons in children and adults—A comprehensive review. J. Autoimmun. 2013, 46, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Soares, J.C.; Borgonovo, A.; Maggi, D.C.; Pasinato, A.P.; Ramos, F.G.; Dantas-Corrêa, E.B.; Schiavon, L.L.; Narciso-Schiavon, J.L. Liver dysfunction and fibrosis as predictors of biochemical response to autoimmune hepatitis treatment. Minerva Gastroenterol. Dietol. 2016, 62, 138–147. [Google Scholar] [PubMed]
- Fujiwara, K.; Yasui, S.; Yokosuka, O. Autoimmune acute liver failure: An emerging etiology for intractable acute liver failure. Hepatol. Int. 2013, 7, 335–346. [Google Scholar] [CrossRef]
- Lohse, A.W.; Sebode, M.; Bhathal, P.S.; Clouston, A.D.; Dienes, H.P.; Jain, D.; Gouw, A.S.H.; Guindi, M.; Kakar, S.; Kleiner, D.E.; et al. Consensus recommendations for histological criteria of autoimmune hepatitis from the International AIH Pathology Group: Results of a workshop on AIH histology hosted by the European Reference Network on Hepatological Diseases and the European Society of Pathology. Liver Int. 2022, 42, 1058–1069. [Google Scholar] [CrossRef] [PubMed]
- Griessmair, L.; Pirringer, L.; Mountford, S.; Sendelhofert, A.; Makeschin, M.C.; Koletzko, S.; Mayr, D.; Bufler, P. Expression of IL-37 Correlates with Immune Cell Infiltrate and Fibrosis in Pediatric Autoimmune Liver Diseases. J. Pediatr. Gastroenterol. Nutr. 2022, 74, 742–749. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.C.; Zhang, Q.B.; Qiao, L. Pathogenesis of liver cirrhosis. World J. Gastroenterol. 2014, 20, 7312–7324. [Google Scholar] [CrossRef]
- Zhang, C.Y.; Yuan, W.G.; He, P.; Lei, J.H.; Wang, C.X. Liver fibrosis and hepatic stellate cells: Etiology, pathological hallmarks and therapeutic targets. World J. Gastroenterol. 2016, 22, 10512–10522. [Google Scholar] [CrossRef]
- Tsuchida, T.; Friedman, S.L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–411. [Google Scholar] [CrossRef]
- Pinzani, M. Pathophysiology of liver fibrosis. Dig. Dis. 2015, 33, 492–497. [Google Scholar] [CrossRef] [PubMed]
- Helal, T.E.S.A.; Ehsan, N.A.; Radwan, N.A.; Abdelsameea, E. Relationship between hepatic progenitor cells and stellate cells in chronic hepatitis C genotype 4. APMIS 2018, 126, 14–20. [Google Scholar] [CrossRef]
- Yang, F.; Li, H.; Li, Y.; Hao, Y.; Wang, Ch.; Jia, P.; Chen, X.; Ma, S.; Xiao, Z. Crosstalk between hepatic stellate cells and surrounding cells in hepatic fibrosis. Int. Immunopharmacol. 2021, 99, 108051. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, M.; Seki, E. Hepatic Stellate Cell-Macrophage Crosstalk in Liver Fibrosis and Carcinogenesis. Semin. Liver Dis. 2020, 40, 307–320. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L. Hepatic stellate cells: Protean, multifunctional, and enigmatic cells of the liver. Physiol Rev. 2008, 88, 125–178. [Google Scholar] [CrossRef]
- Park, S.Y.; Shin, H.W.; Lee, K.B.; Lee, M.J.; Jang, J.J. Differential expression of matrix metalloproteinases and tissue inhibitors of metalloproteinases in thioacetamide-induced chronic liver injury. J. Korean Med. Sci. 2010, 25, 570–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roeb, E. Matrix metalloproteinases and liver fibrosis (translational aspects). Matrix Biol. 2018, 68–69, 463–473. [Google Scholar] [CrossRef]
- Geerts, A. History, heterogeneity, developmental biology, and functions of quiescent hepatic stellate cells. Semin. Liver Dis. 2001, 21, 311–335. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Wang, J.; Wang, J.; Zhou, Q.; Yang, B.; He, Q.; Weng, Q. Intercellular crosstalk of hepatic stellate cells in liver fibrosis: New insights into therapy. Pharmacol. Res. 2020, 155, 104720. [Google Scholar] [CrossRef]
- Lotowska, J.M.; Lebensztejn, D.M. Immunoreactive hepatic stellate cells in biopsy material in children with hepatitis B: The first report in children. Pol. J. Pathol. 2015, 66, 224–230. [Google Scholar] [CrossRef] [Green Version]
- Lotowska, J.M.; Sobaniec-Lotowska, M.E.; Lebensztejn, D. M. Ultrastructural characteristics of the respective forms of hepatic stellate cells in chronic hepatitis B as an example of high fibroblastic cell plasticity. The first assessment in children. Adv. Med. Sci. 2018, 63, 127–133. [Google Scholar] [CrossRef]
- Mehal, W.Z.; Azzaroli, F.; Crispe, I.N. Immunology of the healthy liver: Old questions and new insights. Gastroenterology 2001, 120, 250–260. [Google Scholar] [CrossRef]
- Kmieć, Z. Cooperation of Liver Cells in Health and Disease. In Advances in Anatomy Embryology and Cell Biology Book Series; Springer: Berlin/Heidelberg, Germany, 2001; Volume 161, Chapter III–XIII; pp. 1–151. [Google Scholar]
- Puche, J.E.; Saiman, Y.; Friedman, S.L. Hepatic stellate cells and liver fibrosis. Compr. Physiol. 2013, 3, 1473–1492. [Google Scholar]
- Elzamly, S.; Agina, H.A.; Elbalshy, Abd El-Latif; Abuhashim, M.; Saad, E.; Abd Elmageed, Z.Y. Integration of VEGF and α-SMA Expression Improves the Prediction Accuracy of Fibrosis in Chronic Hepatitis C Liver Biopsy. Appl. Immunohistochem. Mol. Morphol. 2017, 25, 261–270. [Google Scholar] [CrossRef]
- Senoo, H.; Yoshikawa, K.; Morii, M.; Miura, M.; Imai, K.; Mezaki, Y. Hepatic stellate cell (vitamin A-storing cell) and its relative--past, present and future. Cell. Biol. Int. 2010, 34, 1247–1272. [Google Scholar] [CrossRef] [PubMed]
- Higashi, T.; Friedman, S.L.; Hoshida, Y. Hepatic stellate cells as key target in liver fibrosis. Adv. Drug Deliver Rev. 2017, 121, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L.; Sheppard, D.; Duffield, J.S.; Violette, S. Therapy for fibrotic diseases: Nearing the starting line. Sci. Transl. Med. 2013, 5, 167sr1. [Google Scholar] [CrossRef] [Green Version]
- Campana, L.; Iredale, J.P. Regression of liver fibrosis. Semin. Liver Dis. 2017, 37, 1–10. [Google Scholar]
- Schuppan, D.; Kim, Y.O. Evolving therapies for liver fibrosis. J. Clin. Invest. 2013, 123, 1887–1901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czaja, A.J. Review article: The prevention and reversal of hepatic fibrosis in autoimmune hepatitis. Aliment. Pharmacol. Ther. 2014, 39, 385–406. [Google Scholar] [CrossRef]
- Pellicoro, A.; Ramachandran, P.; Iredale, J.P.; Fallowfield, J.A. Liver fibrosis and repair: Immune regulation of wound healing in a solid organ. Nat. Rev. Immunol. 2014, 14, 181–194. [Google Scholar] [CrossRef]
- Zong, Z.; Liu, J.; Wang, N.; Yang, C.; Wang, Q.; Zhang, W.; Chen, Y.; Liu, X.; Deng, H. Nicotinamide mononucleotide inhibits hepatic stellate cell activation to prevent liver fibrosis via promoting PGE(2) degradation. Free Radic. Biol. Med. 2021, 162, 571–581. [Google Scholar] [CrossRef]
- Weiskirchen, R.; Tacke, F. Liver Fibrosis: From Pathogenesis to Novel Therapies. Dig. Dis. 2016, 34, 410–422. [Google Scholar] [CrossRef]
- Marrone, G.; Shah, V.H.; Gracia-Sancho, J. Sinusoidal communication in liver fibrosis and regeneration. J. Hepatol. 2016, 65, 608–617. [Google Scholar] [CrossRef] [PubMed]
- Lotowska, J.M.; Sobaniec-Lotowska, M.E.; Lebensztejn, D.M.; Daniluk, U.; Sobaniec, P.; Sendrowski, K.; Daniluk, J.; Reszec, J.; Debek, W. Ultrastructural characteristics of rat hepatic oval cells and their intercellular contacts in the model of biliary fibrosis: New insights into experimental liver fibrogenesis. Gastroenterol. Res. Pract. 2017, 2017, 2721547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sobaniec-Lotowska, M.E.; Lotowska, J.M.; Lebensztejn, D.M. Ultrastructure of oval cells in children with chronic hepatitis B, with special emphasis on the stage of liver fibrosis: The first pediatric study. World J. Gastroenterol. 2007, 13, 2918–2922. [Google Scholar] [CrossRef] [PubMed]
- Sobaniec-Lotowska, M.E.; Lebensztejn, D.M.; Lotowska, J.M.; Kanczuga-Koda, L.; Sulkowski, S. Ultrastructure of liver progenitor/oval cells in children with nonalcoholic steatohepatitis. Adv. Med. Sci. 2011, 56, 172–179. [Google Scholar] [CrossRef]
- Lotowska, J.M.; Sobaniec-Lotowska, M.E.; Lebensztejn, D.M. Electron microscopic alterations in intermediate hepatocyte-like cells in children with chronic hepatitis B. The first report in pediatric patients. Eur. J. Gastroenterol. Hepatol. 2010, 22, 741–747. [Google Scholar] [CrossRef] [PubMed]
- Lotowska, J.M.; Sobaniec-Lotowska, M.E.; Lebensztejn, D.M. The role of Kupffer cells in the morphogenesis of nonalcoholic steatohepatitis—ultrastructural findings. The first report in pediatric patients. Scand. J. Gastroenterol. 2013, 48, 352–357. [Google Scholar] [CrossRef]
- Lotowska, J.M.; Sobaniec-Lotowska, M.E.; Bockowska, S.B.; Lebensztejn, D.M. Pediatric non-alcoholic steatohepatitis: The first report on the ultrastructure of hepatocyte mitochondria. World J. Gastroenterol. 2014, 20, 4335–4340. [Google Scholar] [CrossRef]
- Lotowska, J.M.; Sobaniec-Lotowska, M.E.; Sobaniec, P.; Lebensztejn, D.M. Liver sinusoidal endothelial cells in morphogenesis of pediatric autoimmune hepatitis. Ultrastructural characteristics—A novel report. Pol. J. Pathol. 2018, 69, 327–334. [Google Scholar] [CrossRef]
- Lotowska, J.M.; Sobaniec-Lotowska, M.E.; Sobaniec, P. Ultrastructural Profile Combined with Immunohistochemistry of a Hepatic Progenitor Cell Line in Pediatric Autoimmune Hepatitis: New Insights into the Morphological Pattern of the Disease. Cells 2021, 10, 1899. [Google Scholar] [CrossRef]
- Batts, K.P.; Ludwig, J. Chronic hepatitis. An update on terminology and reporting. Am. J. Surg. Pathol. 1995, 19, 1409–1417. [Google Scholar] [CrossRef] [PubMed]
- Ishak, K. Histological grading and staging of chronic hepatitis. J. Hepatol. 1995, 22, 696–699. [Google Scholar] [CrossRef] [PubMed]
- Gressner, O.A.; Lahme, B.; Demirci, I.; Gressner, A.M.; Weiskirchen, R. Differential effects of TGF-beta on connective tissue growth factor (CTGF/CCN2) expression in hepatic stellate cells and hepatocytes. J. Hepatol. 2007, 47, 699–710. [Google Scholar] [CrossRef] [PubMed]
- Beljaars, L.; Daliri, S.; Dijkhuizen, C.; Poelstra, K.; Gosens, R. WNT-5A regulates TGF-β-related activities in liver fibrosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2017, 312, G219–G227. [Google Scholar] [CrossRef] [Green Version]
- Tacke, F.; Zimmermann, H.W. Macrophage heterogeneity in liver injury and fibrosis. J. Hepatol. 2014, 60, 1090–1096. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, R.; Devhare, P.B.; Steele, R.; Ray, R.; Ray, R.B. Hepatitis C virus-induced CCL5 secretion from macrophages activates hepatic stellate cells. Hepatology 2017, 66, 746–757. [Google Scholar] [CrossRef] [Green Version]
- Tacke, F. Targeting hepatic macrophages to treat liver diseases. J. Hepatol. 2017, 66, 1300–1312. [Google Scholar] [CrossRef]
- Koyama, Y.; Brenner, D.A. Liver inflammation and fibrosis. J. Clin. Invest. 2017, 127, 55–64. [Google Scholar] [CrossRef]
- Pradere, J.P.; Kluwe, J.; De Minicis, S.; Jiao, J.J.; Gwak, G.Y.; Dapito, D.H.; Jang, M.K.; Guenther, N.D.; Mederacke, I.; Friedman, R.; et al. Hepatic macrophages but not dendritic cells contribute to liver fibrosis by promoting the survival of activated hepatic stellate cells in mice. Hepatology 2013, 58, 1461–1473. [Google Scholar] [CrossRef] [Green Version]
- Oakley, F.; Meso, M.; Iredale, J.P.; Green, K.; Marek, C.J.; Zhou, X.; May, M.J.; Millward-Sadler, H.; Wright, M.C.; Mann, D.A. Inhibition of inhibitor of kappaB kinases stimulates hepatic stellate cell apoptosis and accelerated recovery from rat liver fibrosis. Gastroenterology 2005, 128, 108–120. [Google Scholar] [CrossRef]
- Matsuda, M.; Tsurusaki, S.; Miyata, N.; Saijou, E.; Okochi, H.; Miyajima, A.; Tanaka, M. Oncostatin M causes liver fibrosis by regulating cooperation between hepatic stellate cells and macrophages in mice. Hepatology 2018, 67, 296–312. [Google Scholar] [CrossRef] [Green Version]
- Guillot, A.; Tacke, F. Liver macrophages: Old dogmas and new insights. Hepatol. Commun. 2019, 3, 730–743. [Google Scholar] [CrossRef]
- Ahmad, R.; Ahmed, S.; Khan, N.U.; Hasnain, A. Operculina turpethum attenuates N-nitrosodimethylamine induced toxic liver injury and clastogenicity in rats. Chem. Biol. Interact. 2009, 18, 145–153. [Google Scholar] [CrossRef]
- Husain, H.; Latief, U.; Ahmad, R. Pomegranate action in curbing the incidence of liver injury triggered by Diethylnitrosamine by declining oxidative stress via Nrf2 and NFκB regulation. Sci. Rep. 2018, 8, 8606. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Łotowska, J.M.; Sobaniec-Łotowska, M.E.; Bobrus-Chociej, A.; Sobaniec, P. The Ultrastructure of Hepatic Stellate Cell–Macrophage Intercellular Crosstalk as a New Morphological Insight into Phenomenon of Fibrogenesis in Pediatric Autoimmune Hepatitis. J. Clin. Med. 2023, 12, 1024. https://doi.org/10.3390/jcm12031024
Łotowska JM, Sobaniec-Łotowska ME, Bobrus-Chociej A, Sobaniec P. The Ultrastructure of Hepatic Stellate Cell–Macrophage Intercellular Crosstalk as a New Morphological Insight into Phenomenon of Fibrogenesis in Pediatric Autoimmune Hepatitis. Journal of Clinical Medicine. 2023; 12(3):1024. https://doi.org/10.3390/jcm12031024
Chicago/Turabian StyleŁotowska, Joanna Maria, Maria Elżbieta Sobaniec-Łotowska, Anna Bobrus-Chociej, and Piotr Sobaniec. 2023. "The Ultrastructure of Hepatic Stellate Cell–Macrophage Intercellular Crosstalk as a New Morphological Insight into Phenomenon of Fibrogenesis in Pediatric Autoimmune Hepatitis" Journal of Clinical Medicine 12, no. 3: 1024. https://doi.org/10.3390/jcm12031024