
Controls of Central and Peripheral Blood Pressure and Hemorrhagic/Hypovolemic Shock

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Central and Peripheral Blood Pressure
3. Regulation of Central and Peripheral Blood Pressure
3.1. Neural Controls
3.1.1. Sensory Receptors (Baroreceptors and Chemoreceptors)
3.1.2. Peripheral Nervous System (PNS)
3.1.3. Central Nervous System (CNS)
3.1.4. Spinal Cord
3.2. Hormonal (Endocrine) and Enzymatic Controls
3.2.1. Catecholamines
3.2.2. Renin–Angiotensin–Aldosterone–Antidiuretic Hormone System (RAAAS)
3.2.3. Natriuretic Peptides
3.2.4. Erythropoietin
3.3. Vascular Endothelial Cell-Mediated Controls
4. Regulation of Blood Pressure after Hemorrhagic Shock
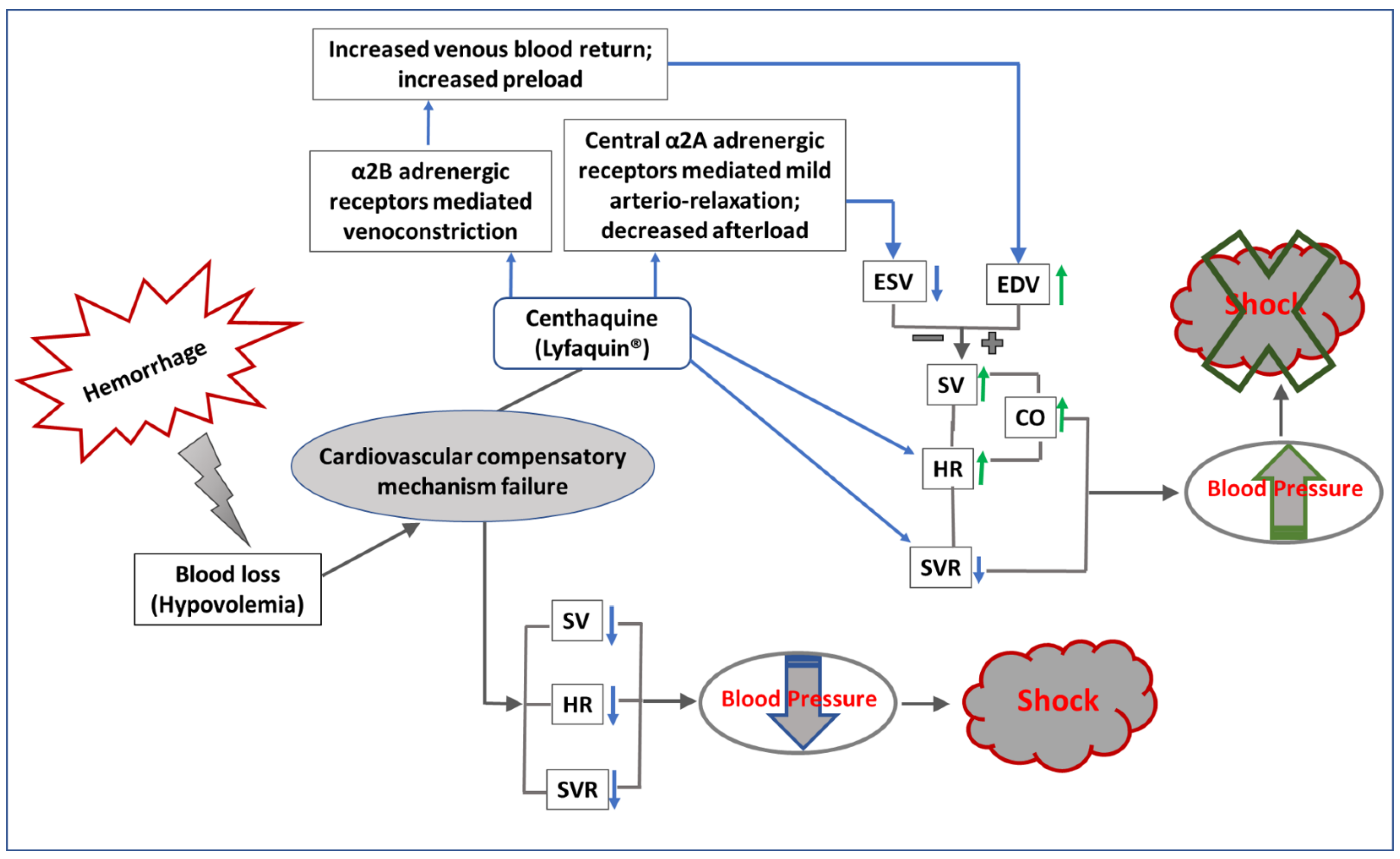
Regulation of Blood Pressure by Centhaquine (Lyfaquin®) after Hypovolemic/Hemorrhagic Shock
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rogers, M.E.; Oliver, M.F.; Matthews, B.E. “Circulatory System”. Encyclopedia Britannica, 20 October 2022. Available online: https://www.britannica.com/science/circulatory-system (accessed on 30 October 2022).
- Bulatovic, I.; Mansson-Broberg, A.; Sylven, C.; Grinnemo, K.H. Human fetal cardiac progenitors: The role of stem cells and progenitors in the fetal and adult heart. Best Pract. Res. Clin. Obs. Gynaecol. 2016, 31, 58–68. [Google Scholar] [CrossRef]
- Buckingham, M.; Meilhac, S.; Zaffran, S. Building the mammalian heart from two sources of myocardial cells. Nat. Rev. Genet. 2005, 6, 826–835. [Google Scholar] [CrossRef]
- Tan, C.M.J.; Lewandowski, A.J. The Transitional Heart: From Early Embryonic and Fetal Development to Neonatal Life. Fetal Diagn. Ther. 2020, 47, 373–386. [Google Scholar] [CrossRef]
- Rushmer, R.F. Regulation of the heart’s functions. Circulation 1960, 21, 744–748. [Google Scholar] [CrossRef] [Green Version]
- Scalco, A.; Moro, N.; Mongillo, M.; Zaglia, T. Neurohumoral Cardiac Regulation: Optogenetics Gets into the Groove. Front. Physiol. 2021, 12, 726895. [Google Scholar] [CrossRef]
- Prothero, J.W.; Burton, A.C. The physics of blood flood in capillaries. II. The capillary resistance to flow. Biophys. J. 1962, 2, 199–212. [Google Scholar] [CrossRef] [Green Version]
- Prothero, J.; Burton, A.C. The physics of blood flow in capillaries. I. The nature of the motion. Biophys. J. 1961, 1, 565–579. [Google Scholar] [CrossRef] [Green Version]
- Kumar, B.V.; Naidu, K.B. Finite element analysis of nonlinear pulsatile suspension flow dynamics in blood vessels with aneurysm. Comput. Biol. Med. 1995, 25, 1–20. [Google Scholar] [CrossRef]
- Peterson, L.H. The dynamics of pulsatile blood flow. Circ. Res. 1954, 2, 127–139. [Google Scholar] [CrossRef] [Green Version]
- Thomas, G.D. Neural control of the circulation. Adv. Physiol. Educ. 2011, 35, 28–32. [Google Scholar] [CrossRef]
- Estanol, B.; Porras-Betancourt, M.; Sanchez-Torres, G.; Martinez-Memije, R.; Infante, O.; Senties-Madrid, H. [Neural control of the peripheral circulation and blood pressure]. Arch. Cardiol. Mex. 2009, 79 (Suppl. S2), 109–116. [Google Scholar]
- Garcia Ramos, J. [Neural control of the coronary circulation]. Bol. Sanid. Mil. 1951, 4, 83–95. [Google Scholar] [PubMed]
- Haig, A. Capillary Circulation and Blood-Pressure and the Conditions that Control them. Med. Chir. Trans. 1906, 89, 205–237. [Google Scholar] [PubMed]
- Mensah, G.A.; Croft, J.B.; Giles, W.H. The heart, kidney, and brain as target organs in hypertension. Cardiol. Clin. 2002, 20, 225–247. [Google Scholar] [CrossRef]
- Metzler, M.; Duerr, S.; Granata, R.; Krismer, F.; Robertson, D.; Wenning, G.K. Neurogenic orthostatic hypotension: Pathophysiology, evaluation, and management. J. Neurol. 2013, 260, 2212–2219. [Google Scholar] [CrossRef] [Green Version]
- Marazzato, J.; Blasi, F.; Golino, M.; Verdecchia, P.; Angeli, F.; De Ponti, R. Hypertension and Arrhythmias: A Clinical Overview of the Pathophysiology-Driven Management of Cardiac Arrhythmias in Hypertensive Patients. J. Cardiovasc. Dev. Dis. 2022, 9, 110. [Google Scholar] [CrossRef]
- Cheng, Y.B.; Li, Y.; Cheng, H.M.; Siddique, S.; Huynh, M.V.; Sukonthasarn, A.; Chen, C.H.; Wang, J.G. Central hypertension is a non-negligible cardiovascular risk factor. J. Clin. Hypertens. 2022, 24, 1174–1179. [Google Scholar] [CrossRef] [PubMed]
- Graham, C.A.; Parke, T.R.J. Critical care in the emergency department: Shock and circulatory support. Emerg. Med. J. 2005, 22, 17–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roman, M.J.; Devereux, R.B. Association of central and peripheral blood pressures with intermediate cardiovascular phenotypes. Hypertension 2014, 63, 1148–1153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.Y.; Wang, X.H.; Lu, L.C.; Li, H. Discrepancy of blood pressure between the brachial artery and radial artery. World J. Emerg. Med. 2013, 4, 294–297. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Kim, I.C.; Hwang, J.; Lee, C.H.; Cho, Y.K.; Park, H.S.; Chung, J.W.; Nam, C.W.; Han, S.; Hur, S.H. Features and implications of higher systolic central than peripheral blood pressure in patients at very high risk of atherosclerotic cardiovascular disease. J. Hum. Hypertens. 2021, 35, 994–1002. [Google Scholar] [CrossRef]
- Terentes-Printzios, D.; Gardikioti, V.; Vlachopoulos, C. Central Over Peripheral Blood Pressure: An Emerging Issue in Hypertension Research. Heart. Lung. Circ. 2021, 30, 1667–1674. [Google Scholar] [CrossRef] [PubMed]
- Flores Geronimo, J.; Corvera Poire, E.; Chowienczyk, P.; Alastruey, J. Estimating Central Pulse Pressure from Blood Flow by Identifying the Main Physical Determinants of Pulse Pressure Amplification. Front. Physiol. 2021, 12, 608098. [Google Scholar] [CrossRef] [PubMed]
- Kelly, R.P.; Gibbs, H.H.; O’Rourke, M.F.; Daley, J.E.; Mang, K.; Morgan, J.J.; Avolio, A.P. Nitroglycerin has more favourable effects on left ventricular afterload than apparent from measurement of pressure in a peripheral artery. Eur. Heart J. 1990, 11, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Avolio, A.P.; Van Bortel, L.M.; Boutouyrie, P.; Cockcroft, J.R.; McEniery, C.M.; Protogerou, A.D.; Roman, M.J.; Safar, M.E.; Segers, P.; Smulyan, H. Role of pulse pressure amplification in arterial hypertension: Experts’ opinion and review of the data. Hypertension 2009, 54, 375–383. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Yuan, J.; Li, B. SBP Is Superior to MAP to Reflect Tissue Perfusion and Hemodynamic Abnormality Perioperatively. Front. Physiol. 2021, 12, 705558. [Google Scholar] [CrossRef]
- Curtis, L.J.; Rabkin, D.C.; Cabreriza, S.E.; Spotnitz, H.M. Relation between mean arterial pressure (MAP) and cardiac output (CO) in open chest pigs. ASAIO J. 2003, 49, 173. [Google Scholar] [CrossRef]
- Grand, J.; Wiberg, S.; Kjaergaard, J.; Wanscher, M.; Hassager, C. Increasing mean arterial pressure or cardiac output in comatose out-of-hospital cardiac arrest patients undergoing targeted temperature management: Effects on cerebral tissue oxygenation and systemic hemodynamics. Resuscitation 2021, 168, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Berner, M.; Oberhansli, I.; Rouge, J.C.; Jaccard, C.; Friedli, B. Chronotropic and inotropic supports are both required to increase cardiac output early after corrective operations for tetralogy of Fallot. J. Thorac. Cardiovasc. Surg. 1989, 97, 297–302. [Google Scholar] [CrossRef] [PubMed]
- Tanigawa, T.; Yano, M.; Kohno, M.; Yamamoto, T.; Hisaoka, T.; Ono, K.; Ueyama, T.; Kobayashi, S.; Hisamatsu, Y.; Ohkusa, T.; et al. Mechanism of preserved positive lusitropy by cAMP-dependent drugs in heart failure. Am. J. Physiol. Heart Circ. Physiol. 2000, 278, H313–H320. [Google Scholar] [CrossRef]
- Raff, G.L.; Glantz, S.A. Volume loading slows left ventricular isovolumic relaxation rate. Evidence of load-dependent relaxation in the intact dog heart. Circ. Res. 1981, 48, 813–824. [Google Scholar] [CrossRef]
- Sloop, G.D.; Weidman, J.J.; St Cyr, J.A. The systemic vascular resistance response: A cardiovascular response modulating blood viscosity with implications for primary hypertension and certain anemias. Ther. Adv. Cardiovasc. Dis. 2015, 9, 403–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thrasher, T.N. Baroreceptors, baroreceptor unloading, and the long-term control of blood pressure. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005, 288, R819–R827. [Google Scholar] [CrossRef] [PubMed]
- Thrasher, T.N. Baroreceptors and the long-term control of blood pressure. Exp. Physiol. 2004, 89, 331–335. [Google Scholar] [CrossRef] [PubMed]
- Low, P.A.; Tomalia, V.A. Orthostatic Hypotension: Mechanisms, Causes, Management. J. Clin. Neurol. 2015, 11, 220–226. [Google Scholar] [CrossRef] [Green Version]
- Minisi, A.J. Vagal cardiopulmonary reflexes after total cardiac deafferentation. Circulation 1998, 98, 2615–2620. [Google Scholar] [CrossRef] [PubMed]
- Sata, Y.; Head, G.A.; Denton, K.; May, C.N.; Schlaich, M.P. Role of the Sympathetic Nervous System and Its Modulation in Renal Hypertension. Front. Med. 2018, 5, 82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahdi, A.; Sturdy, J.; Ottesen, J.T.; Olufsen, M.S. Modeling the afferent dynamics of the baroreflex control system. PLoS Comput. Biol. 2013, 9, e1003384. [Google Scholar] [CrossRef] [Green Version]
- Dampney, R.A.L. Resetting of the Baroreflex Control of Sympathetic Vasomotor Activity during Natural Behaviors: Description and Conceptual Model of Central Mechanisms. Front. Neurosci. 2017, 11, 461. [Google Scholar] [CrossRef] [Green Version]
- Guyenet, P.G. Regulation of breathing and autonomic outflows by chemoreceptors. Compr. Physiol. 2014, 4, 1511–1562. [Google Scholar] [CrossRef] [Green Version]
- Prabhakar, N.R.; Peng, Y.J. Peripheral chemoreceptors in health and disease. J. Appl. Physiol. (1985) 2004, 96, 359–366. [Google Scholar] [CrossRef]
- Purves, M.J. Chemoreceptors and their reflexes with special reference to the fetus and newborn. J. Dev. Physiol. 1981, 3, 21–57. [Google Scholar] [PubMed]
- Panneton, W.M.; Gan, Q. The Mammalian Diving Response: Inroads to Its Neural Control. Front. Neurosci. 2020, 14, 524. [Google Scholar] [CrossRef]
- Sekizawa, S.I.; Tsubone, H. Nasal receptors responding to noxious chemical irritants. Respir. Physiol. 1994, 96, 37–48. [Google Scholar] [CrossRef]
- Panneton, W.M.; Gan, Q.; Le, J.; Livergood, R.S.; Clerc, P.; Juric, R. Activation of brainstem neurons by underwater diving in the rat. Front. Physiol. 2012, 3, 111. [Google Scholar] [CrossRef] [Green Version]
- Fahim, M. Cardiovascular sensory receptors and their regulatory mechanisms. Indian J. Physiol. Pharm. 2003, 47, 124–146. [Google Scholar]
- Jin, G.S.; Li, X.L.; Jin, Y.Z.; Kim, M.S.; Park, B.R. Role of peripheral vestibular receptors in the control of blood pressure following hypotension. Korean J. Physiol. Pharm. 2018, 22, 363–368. [Google Scholar] [CrossRef]
- Murphy, M.N.; Mizuno, M.; Mitchell, J.H.; Smith, S.A. Cardiovascular regulation by skeletal muscle reflexes in health and disease. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H1191–H1204. [Google Scholar] [CrossRef]
- Sacco, M.; Meschi, M.; Regolisti, G.; Detrenis, S.; Bianchi, L.; Bertorelli, M.; Pioli, S.; Magnano, A.; Spagnoli, F.; Giuri, P.G.; et al. The relationship between blood pressure and pain. J. Clin. Hypertens. 2013, 15, 600–605. [Google Scholar] [CrossRef]
- Fahmy, L.M.; Chen, Y.; Xuan, S.; Haacke, E.M.; Hu, J.; Jiang, Q. All Central Nervous System Neuro- and Vascular-Communication Channels Are Surrounded with Cerebrospinal Fluid. Front. Neurol. 2021, 12, 614636. [Google Scholar] [CrossRef]
- McCorry, L.K. Physiology of the autonomic nervous system. Am. J. Pharm. Educ. 2007, 71, 78. [Google Scholar] [CrossRef] [Green Version]
- Bankenahally, R.; Krovvidi, H. Autonomic nervous system: Anatomy, physiology, and relevance in anaesthesia and critical care medicine. BJA Educ. 2016, 16, 381–387. [Google Scholar] [CrossRef]
- Lumb, R.; Tata, M.; Xu, X.; Joyce, A.; Marchant, C.; Harvey, N.; Ruhrberg, C.; Schwarz, Q. Neuropilins guide preganglionic sympathetic axons and chromaffin cell precursors to establish the adrenal medulla. Development 2018, 145, dev.162552. [Google Scholar] [CrossRef] [Green Version]
- Ernsberger, U.; Deller, T.; Rohrer, H. The sympathies of the body: Functional organization and neuronal differentiation in the peripheral sympathetic nervous system. Cell Tissue Res. 2021, 386, 455–475. [Google Scholar] [CrossRef]
- Gilbey, M.P.; Spyer, K.M. Essential organization of the sympathetic nervous system. Baillieres Clin. Endocrinol. Metab. 1993, 7, 259–278. [Google Scholar] [CrossRef]
- Szulczyk, P. Functional organization of the sympathetic nervous system. Acta Physiol. Pol. 1981, 32, 155–157. [Google Scholar]
- Breit, S.; Kupferberg, A.; Rogler, G.; Hasler, G. Vagus Nerve as Modulator of the Brain-Gut Axis in Psychiatric and Inflammatory Disorders. Front. Psychiatry 2018, 9, 44. [Google Scholar] [CrossRef] [Green Version]
- Dampney, R.A. Central neural control of the cardiovascular system: Current perspectives. Adv. Physiol. Educ. 2016, 40, 283–296. [Google Scholar] [CrossRef] [Green Version]
- Spyer, K.M.; Lambert, J.H.; Thomas, T. Central nervous system control of cardiovascular function: Neural mechanisms and novel modulators. Clin. Exp. Pharm. Physiol. 1997, 24, 743–747. [Google Scholar] [CrossRef]
- Tjen, A.L.S.C.; Guo, Z.L.; Li, M.; Longhurst, J.C. Medullary GABAergic mechanisms contribute to electroacupuncture modulation of cardiovascular depressor responses during gastric distention in rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2013, 304, R321–R332. [Google Scholar] [CrossRef] [Green Version]
- Zanutto, B.S.; Valentinuzzi, M.E.; Segura, E.T. Neural set point for the control of arterial pressure: Role of the nucleus tractus solitarius. Biomed. Eng. Online 2010, 9, 4. [Google Scholar] [CrossRef] [Green Version]
- Boone, M.; Deen, P.M.T. Physiology and pathophysiology of the vasopressin-regulated renal water reabsorption. Pflügers Arch. -Eur. J. Physiol. 2008, 456, 1005–1024. [Google Scholar] [CrossRef] [Green Version]
- Harlan, S.M.; Rahmouni, K. Neuroanatomical determinants of the sympathetic nerve responses evoked by leptin. Clin. Auton. Res. 2013, 23, 1–7. [Google Scholar] [CrossRef]
- Wei, S.G.; Zhang, Z.H.; Beltz, T.G.; Yu, Y.; Johnson, A.K.; Felder, R.B. Subfornical organ mediates sympathetic and hemodynamic responses to blood-borne proinflammatory cytokines. Hypertension 2013, 62, 118–125. [Google Scholar] [CrossRef] [Green Version]
- Di Spiezio, A.; Sandin, E.S.; Dore, R.; Muller-Fielitz, H.; Storck, S.E.; Bernau, M.; Mier, W.; Oster, H.; Johren, O.; Pietrzik, C.U.; et al. The LepR-mediated leptin transport across brain barriers controls food reward. Mol. Metab. 2018, 8, 13–22. [Google Scholar] [CrossRef]
- Minic, Z.; O’Leary, D.S.; Reynolds, C.A. Spinal Reflex Control of Arterial Blood Pressure: The Role of TRP Channels and Their Endogenous Eicosanoid Modulators. Front. Physiol. 2022, 13, 838175. [Google Scholar] [CrossRef]
- Spencer, N.J.; Kyloh, M.; Duffield, M. Identification of different types of spinal afferent nerve endings that encode noxious and innocuous stimuli in the large intestine using a novel anterograde tracing technique. PLoS ONE 2014, 9, e112466. [Google Scholar] [CrossRef] [Green Version]
- Ozdemir, R.A.; Perez, M.A. Afferent input and sensory function after human spinal cord injury. J. Neurophysiol. 2018, 119, 134–144. [Google Scholar] [CrossRef] [Green Version]
- Moynes, D.M.; Lucas, G.H.; Beyak, M.J.; Lomax, A.E. Effects of inflammation on the innervation of the colon. Toxicol. Pathol. 2014, 42, 111–117. [Google Scholar] [CrossRef] [Green Version]
- Czubkowski, P.; Osiecki, M.; Szymańska, E.; Kierkuś, J. The risk of cardiovascular complications in inflammatory bowel disease. Clin. Exp. Med. 2020, 20, 481–491. [Google Scholar] [CrossRef] [PubMed]
- Jensen, A.B.; Ajslev, T.A.; Brunak, S.; Sorensen, T.I. Long-term risk of cardiovascular and cerebrovascular disease after removal of the colonic microbiota by colectomy: A cohort study based on the Danish National Patient Register from 1996 to 2014. BMJ Open 2015, 5, e008702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chopra, S.; Baby, C.; Jacob, J.J. Neuro-endocrine regulation of blood pressure. Indian J. Endocrinol. Metab. 2011, 15 (Suppl. S4), S281–S288. [Google Scholar] [CrossRef] [PubMed]
- Emamian, M.; Hasanian, S.M.; Tayefi, M.; Bijari, M.; Movahedian Far, F.; Shafiee, M.; Avan, A.; Heidari-Bakavoli, A.; Moohebati, M.; Ebrahimi, M.; et al. Association of hematocrit with blood pressure and hypertension. J. Clin. Lab. Anal. 2017, 31. [Google Scholar] [CrossRef] [PubMed]
- Wurtman, R.J. Catecholamines. N. Engl. J. Med. 1965, 273, 637–646. [Google Scholar] [CrossRef]
- Slotkin, T.A. Development of the Sympathoadrenal Axis. In Developmental Neurobiology of the Autonomic Nervous System; Gootman, P.M., Ed.; Humana Press: Totowa, NJ, USA, 1986; pp. 69–96. [Google Scholar]
- Graham, R.M.; Perez, D.M.; Hwa, J.; Piascik, M.T. alpha 1-adrenergic receptor subtypes. Molecular structure, function, and signaling. Circ. Res. 1996, 78, 737–749. [Google Scholar] [CrossRef]
- Paravati, S.R.A.; Warrington, S.J. Physiology, Catecholamines; StatPearls Publishing: Treasure Island, FL, USA, 2021. Available online: https://www.ncbi.nlm.nih.gov/books/NBK507716/ (accessed on 20 May 2022).
- Sprague, J.E.; Arbelaez, A.M. Glucose counterregulatory responses to hypoglycemia. Pediatr. Endocrinol. Rev. 2011, 9, 463–473. [Google Scholar]
- Roh, E.; Song, D.K.; Kim, M.-S. Emerging role of the brain in the homeostatic regulation of energy and glucose metabolism. Exp. Mol. Med. 2016, 48, e216. [Google Scholar] [CrossRef] [Green Version]
- Frith, K.; Smith, J.; Joshi, P.; Ford, L.S.; Vale, S. Updated anaphylaxis guidelines: Management in infants and children. Aust. Prescr. 2021, 44, 91–95. [Google Scholar] [CrossRef]
- Hsueh, W.A.; Baxter, J.D. Human prorenin. Hypertension 1991, 17, 469–477. [Google Scholar] [CrossRef] [Green Version]
- Nabi, A.H.M.N.; Suzuki, F. Biochemical properties of renin and prorenin binding to the (pro)renin receptor. Hypertens. Res. 2010, 33, 91–97. [Google Scholar] [CrossRef] [Green Version]
- Aldehni, F.; Tang, T.; Madsen, K.; Plattner, M.; Schreiber, A.; Friis, U.G.; Hammond, H.K.; Han, P.L.; Schweda, F. Stimulation of renin secretion by catecholamines is dependent on adenylyl cyclases 5 and 6. Hypertension 2011, 57, 460–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chappell, M.C.; Marshall, A.C.; Alzayadneh, E.M.; Shaltout, H.A.; Diz, D.I. Update on the Angiotensin converting enzyme 2-Angiotensin (1-7)-MAS receptor axis: Fetal programing, sex differences, and intracellular pathways. Front. Endocrinol. 2014, 4, 201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forrester, S.J.; Booz, G.W.; Sigmund, C.D.; Coffman, T.M.; Kawai, T.; Rizzo, V.; Scalia, R.; Eguchi, S. Angiotensin II Signal Transduction: An Update on Mechanisms of Physiology and Pathophysiology. Physiol. Rev. 2018, 98, 1627–1738. [Google Scholar] [CrossRef] [PubMed]
- Tsilosani, A.; Gao, C.; Zhang, W. Aldosterone-Regulated Sodium Transport and Blood Pressure. Front. Physiol. 2022, 13, 770375. [Google Scholar] [CrossRef]
- Yatabe, J.; Yoneda, M.; Yatabe, M.S.; Watanabe, T.; Felder, R.A.; Jose, P.A.; Sanada, H. Angiotensin III stimulates aldosterone secretion from adrenal gland partially via angiotensin II type 2 receptor but not angiotensin II type 1 receptor. Endocrinology 2011, 152, 1582–1588. [Google Scholar] [CrossRef] [Green Version]
- Goswami, N.; Blaber, A.P.; Hinghofer-Szalkay, H.; Convertino, V.A. Lower Body Negative Pressure: Physiological Effects, Applications, and Implementation. Physiol. Rev. 2019, 99, 807–851. [Google Scholar] [CrossRef]
- Boldyreff, B.; Wehling, M. Aldosterone: Refreshing a slow hormone by swift action. News Physiol. Sci. 2004, 19, 97–100. [Google Scholar] [CrossRef] [Green Version]
- Castrop, H.; Schiessl, I.M. Physiology and pathophysiology of the renal Na-K-2Cl cotransporter (NKCC2). Am. J. Physiol. Ren. Physiol. 2014, 307, F991–F1002. [Google Scholar] [CrossRef] [Green Version]
- Salyer, S.A.; Parks, J.; Barati, M.T.; Lederer, E.D.; Clark, B.J.; Klein, J.D.; Khundmiri, S.J. Aldosterone regulates Na(+), K(+) ATPase activity in human renal proximal tubule cells through mineralocorticoid receptor. Biochim. Biophys. Acta 2013, 1833, 2143–2152. [Google Scholar] [CrossRef] [Green Version]
- Zieg, J. Pathophysiology of Hyponatremia in Children. Front. Pediatr. 2017, 5, 213. [Google Scholar] [CrossRef] [Green Version]
- Ares, G.R.; Caceres, P.S.; Ortiz, P.A. Molecular regulation of NKCC2 in the thick ascending limb. Am. J. Physiol. Ren. Physiol. 2011, 301, F1143–F1159. [Google Scholar] [CrossRef] [Green Version]
- Lifton, R.P.; Gharavi, A.G.; Geller, D.S. Molecular mechanisms of human hypertension. Cell 2001, 104, 545–556. [Google Scholar] [CrossRef] [Green Version]
- Martins, L.C.; Figueiredo, V.N.; Quinaglia, T.; Boer-Martins, L.; Yugar-Toledo, J.C.; Martin, J.F.; Demacq, C.; Pimenta, E.; Calhoun, D.A.; Moreno, H., Jr. Characteristics of resistant hypertension: Ageing, body mass index, hyperaldosteronism, cardiac hypertrophy and vascular stiffness. J. Hum. Hypertens. 2011, 25, 532–538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calhoun, D.A.; Sharma, K. The role of aldosteronism in causing obesity-related cardiovascular risk. Cardiol. Clin. 2010, 28, 517–527. [Google Scholar] [CrossRef]
- Conn, J.W. Aldosterone in clinical medicine; past, present, and future. AMA Arch. Intern. Med. 1956, 97, 135–144. [Google Scholar] [CrossRef]
- Pitt, B.; Zannad, F.; Remme, W.J.; Cody, R.; Castaigne, A.; Perez, A.; Palensky, J.; Wittes, J. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized Aldactone Evaluation Study Investigators. N. Engl. J. Med. 1999, 341, 709–717. [Google Scholar] [CrossRef] [Green Version]
- Pitt, B.; Remme, W.; Zannad, F.; Neaton, J.; Martinez, F.; Roniker, B.; Bittman, R.; Hurley, S.; Kleiman, J.; Gatlin, M.; et al. Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction. N. Engl. J. Med. 2003, 348, 1309–1321. [Google Scholar] [CrossRef] [PubMed]
- Rossignol, P.; Menard, J.; Fay, R.; Gustafsson, F.; Pitt, B.; Zannad, F. Eplerenone survival benefits in heart failure patients post-myocardial infarction are independent from its diuretic and potassium-sparing effects. Insights from an EPHESUS (Eplerenone Post-Acute Myocardial Infarction Heart Failure Efficacy and Survival Study) substudy. J. Am. Coll. Cardiol. 2011, 58, 1958–1966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmieder, R.E.; Martus, P.; Klingbeil, A. Reversal of left ventricular hypertrophy in essential hypertension. A meta-analysis of randomized double-blind studies. JAMA 1996, 275, 1507–1513. [Google Scholar] [CrossRef]
- Viberti, G.; Wheeldon, N.M.; MicroAlbuminuria Reduction With, V.S.I. Microalbuminuria reduction with valsartan in patients with type 2 diabetes mellitus: A blood pressure-independent effect. Circulation 2002, 106, 672–678. [Google Scholar] [CrossRef] [Green Version]
- Johnston, C.I.; Hodsman, P.G.; Kohzuki, M.; Casley, D.J.; Fabris, B.; Phillips, P.A. Interaction between atrial natriuretic peptide and the renin angiotensin aldosterone system. Endogenous antagonists. Am. J. Med. 1989, 87, 24S–28S. [Google Scholar] [CrossRef]
- Lee, N.S.; Daniels, L.B. Current Understanding of the Compensatory Actions of Cardiac Natriuretic Peptides in Cardiac Failure: A Clinical Perspective. Card. Fail. Rev. 2016, 2, 14–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atsma, F.; Veldhuizen, I.; de Kort, W.; van Kraaij, M.; Pasker-de Jong, P.; Deinum, J. Hemoglobin level is positively associated with blood pressure in a large cohort of healthy individuals. Hypertension 2012, 60, 936–941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoener, B.; Borger, J. Erythropoietin Stimulating Agents; StatPearls: Treasure Island, FL, USA, 2022. [Google Scholar]
- Suresh, S.; Rajvanshi, P.K.; Noguchi, C.T. The Many Facets of Erythropoietin Physiologic and Metabolic Response. Front. Physiol. 2019, 10, 1534. [Google Scholar] [CrossRef] [PubMed]
- Sandoo, A.; van Zanten, J.J.; Metsios, G.S.; Carroll, D.; Kitas, G.D. The endothelium and its role in regulating vascular tone. Open Cardiovasc. Med. J. 2010, 4, 302–312. [Google Scholar] [CrossRef] [Green Version]
- Loscalzo, J. The identification of nitric oxide as endothelium-derived relaxing factor. Circ. Res. 2013, 113, 100–103. [Google Scholar] [CrossRef] [Green Version]
- Raij, L. Nitric oxide in the pathogenesis of cardiac disease. J. Clin. Hypertens. 2006, 8, 30–39. [Google Scholar] [CrossRef]
- Katusic, Z.S.; Santhanam, A.V.; He, T. Vascular effects of prostacyclin: Does activation of PPARdelta play a role? Trends Pharm. Sci. 2012, 33, 559–564. [Google Scholar] [CrossRef] [Green Version]
- Yanagisawa, M.; Masaki, T. Molecular biology and biochemistry of the endothelins. Trends Pharm. Sci. 1989, 10, 374–378. [Google Scholar] [CrossRef]
- Davenport, A.P.; Maguire, J.J. Endothelin. Handb. Exp. Pharm. 2006, 176/I, 295–329. [Google Scholar] [CrossRef]
- Ribeiro-Oliveira, A., Jr.; Nogueira, A.I.; Pereira, R.M.; Boas, W.W.; Dos Santos, R.A.; Simoes e Silva, A.C. The renin-angiotensin system and diabetes: An update. Vasc. Health. Risk. Manag. 2008, 4, 787–803. [Google Scholar] [PubMed]
- Iwasaki, H.; Eguchi, S.; Ueno, H.; Marumo, F.; Hirata, Y. Endothelin-mediated vascular growth requires p42/p44 mitogen-activated protein kinase and p70 S6 kinase cascades via transactivation of epidermal growth factor receptor. Endocrinology 1999, 140, 4659–4668. [Google Scholar] [CrossRef] [PubMed]
- Robin, P.; Boulven, I.; Desmyter, C.; Harbon, S.; Leiber, D. ET-1 stimulates ERK signaling pathway through sequential activation of PKC and Src in rat myometrial cells. Am. J. Physiol. Cell. Physiol. 2002, 283, C251–C260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutierrez, G.; Reines, H.; Wulf-Gutierrez, M.E. Clinical review: Hemorrhagic shock. Crit. Care 2004, 8, 373. [Google Scholar] [CrossRef]
- Kreimeier, U. Pathophysiology of fluid imbalance. Crit. Care 2000, 4, S3. [Google Scholar] [CrossRef] [Green Version]
- Patel, K.; Rossler, A.; Lackner, H.K.; Trozic, I.; Laing, C.; Lorr, D.; Green, D.A.; Hinghofer-Szalkay, H.; Goswami, N. Effect of postural changes on cardiovascular parameters across gender. Medicine 2016, 95, e4149. [Google Scholar] [CrossRef]
- de Lucia, C.; Femminella, G.D.; Gambino, G.; Pagano, G.; Allocca, E.; Rengo, C.; Silvestri, C.; Leosco, D.; Ferrara, N.; Rengo, G. Adrenal adrenoceptors in heart failure. Front. Physiol. 2014, 5, 246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coble, J.P.; Grobe, J.L.; Johnson, A.K.; Sigmund, C.D. Mechanisms of brain renin angiotensin system-induced drinking and blood pressure: Importance of the subfornical organ. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2015, 308, R238–R249. [Google Scholar] [CrossRef] [Green Version]
- Harfstrand, A. Brain neuropeptide Y mechanisms. Basic aspects and involvement in cardiovascular and neuroendocrine regulation. Acta Physiol. Scand. Suppl. 1987, 565, 1–83. [Google Scholar]
- Torda, T.; Cruciani, R.A.; Saavedra, J.M. Localization of neuropeptide Y binding sites in the zona glomerulosa of the bovine adrenal gland. Neuroendocrinology 1988, 48, 207–210. [Google Scholar] [CrossRef]
- Xiang, L.; Hinojosa-Laborde, C.; Ryan, K.L.; Rickards, C.A.; Convertino, V.A. Time course of compensatory physiological responses to central hypovolemia in high- and low-tolerant human subjects. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2018, 315, R408–R416. [Google Scholar] [CrossRef] [PubMed]
- Bakker, J.; Nijsten, M.W.N.; Jansen, T.C. Clinical use of lactate monitoring in critically ill patients. Ann. Intensive Care. 2013, 3, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Standl, T.; Annecke, T.; Cascorbi, I.; Heller, A.R.; Sabashnikov, A.; Teske, W. The Nomenclature, Definition and Distinction of Types of Shock. Dtsch. Arztebl. Int. 2018, 115, 757–768. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Liu, F.; Yan, K.; Pan, W.; Xu, L.; Liu, H.; Yan, C.; Chen, C.; Zhu, H. Effects of resuscitation with polymerized porcine hemoglobin (pPolyHb) on hemodynamic stability and oxygen delivery in a rat model of hemorrhagic shock. Artif. Cells Nanomed. Biotechnol. 2017, 45, 51–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strandenes, G.; Cap, A.P.; Cacic, D.; Lunde, T.H.; Eliassen, H.S.; Hervig, T.; Spinella, P.C. Blood Far Forward—A whole blood research and training program for austere environments. Transfusion. 2013, 53 (Suppl. S1), 124S–130S. [Google Scholar] [CrossRef] [PubMed]
- Smart, L.; Hughes, D. The Effects of Resuscitative Fluid Therapy on the Endothelial Surface Layer. Front. Vet. Sci. 2021, 8, 661660. [Google Scholar] [CrossRef]
- Khan, F.; Singh, K.; Friedman, M.T. Artificial Blood: The History and Current Perspectives of Blood Substitutes. Discoveries 2020, 8, e104. [Google Scholar] [CrossRef]
- Mozzarelli, A.; Ronda, L.; Faggiano, S.; Bettati, S.; Bruno, S. Haemoglobin-based oxygen carriers: Research and reality towards an alternative to blood transfusions. Blood. Transfus. 2010, 8 (Suppl. S3), s59–s68. [Google Scholar] [CrossRef]
- Cao, M.; Wang, G.; He, H.; Yue, R.; Zhao, Y.; Pan, L.; Huang, W.; Guo, Y.; Yin, T.; Ma, L.; et al. Hemoglobin-Based Oxygen Carriers: Potential Applications in Solid Organ Preservation. Front. Pharm. 2021, 12, 760215. [Google Scholar] [CrossRef]
- Cao, M.; Zhao, Y.; He, H.; Yue, R.; Pan, L.; Hu, H.; Ren, Y.; Qin, Q.; Yi, X.; Yin, T.; et al. New Applications of HBOC-201: A 25-Year Review of the Literature. Front. Med. 2021, 8, 794561. [Google Scholar] [CrossRef]
- Edgworth, E.; Ernst, L.; Czigany, Z.; Saritas, T.; Zarnitz, L.S.; Wiartalla, M.; Boor, P.; Buhl, E.M.; Rossaint, R.; Tolba, R.H.; et al. HBOC-301 in Porcine Kidney Normothermic Machine Perfusion and the Effect of Vitamin C on Methemoglobin Formation. Antioxidants 2022, 11, 1329. [Google Scholar] [CrossRef] [PubMed]
- Pati, S.; Matijevic, N.; Doursout, M.F.; Ko, T.; Cao, Y.; Deng, X.; Kozar, R.A.; Hartwell, E.; Conyers, J.; Holcomb, J.B. Protective effects of fresh frozen plasma on vascular endothelial permeability, coagulation, and resuscitation after hemorrhagic shock are time dependent and diminish between days 0 and 5 after thaw. J. Trauma 2010, 69 (Suppl. S1), S55–S63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stahl, K.; Schmidt, J.J.; Seeliger, B.; Schmidt, B.M.W.; Welte, T.; Haller, H.; Hoeper, M.M.; Budde, U.; Bode, C.; David, S. Effect of therapeutic plasma exchange on endothelial activation and coagulation-related parameters in septic shock. Crit. Care 2020, 24, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouglé, A.; Harrois, A.; Duranteau, J. Resuscitative strategies in traumatic hemorrhagic shock. Ann. Intensive Care 2013, 3, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pusateri, A.E.; Moore, E.E.; Moore, H.B.; Le, T.D.; Guyette, F.X.; Chapman, M.P.; Sauaia, A.; Ghasabyan, A.; Chandler, J.; McVaney, K.; et al. Association of Prehospital Plasma Transfusion with Survival in Trauma Patients With Hemorrhagic Shock When Transport Times Are Longer Than 20 Minutes: A Post Hoc Analysis of the PAMPer and COMBAT Clinical Trials. JAMA Surg. 2020, 155, e195085. [Google Scholar] [CrossRef]
- Bhatnagar, M.; Pande, M.; Dubey, M.P.; Dhawan, B.N. Effect of centhaquine on spontaneous and evoked norepinephrine release from isolated perfused rabbit heart. Arzneimittelforschung 1985, 35, 693–697. [Google Scholar]
- Gulati, A.; Lavhale, M.; Giri, R.; Andurkar, S.V.; Xanthos, T. Centhaquine citrate. Drug Future 2020, 45, 153–163. [Google Scholar] [CrossRef]
- Gulati, A.; Jain, D.; Agrawal, N.; Rahate, P.; Das, S.; Chowdhuri, R.; Prabhu, M.; Haveri, S.; Dhibar, D.; Agarwal, R. Evaluation of Centhaquine, a Novel Resuscitative Agent, in Hemorrhagic Shock Patients. Circulation 2019, 140, A16250. [Google Scholar]
- Gulati, A.; Jain, D.; Agrawal, N.; Rahate, P.; Das, S.; Chowdhuri, R.; Prabhu, M.; Haveri, S.; Dhibar, D.; Lavhale, M. A phase II multicentric randomized controlled study of centhaquine in hemorrhagic shock patients. Crit. Care Med. 2020, 48, 840. [Google Scholar] [CrossRef]
- Gulati, A.; Choudhuri, R.; Gupta, A.; Singh, S.; Ali, S.K.N.; Sidhu, G.K.; Haque, P.D.; Rahate, P.; Bothra, A.R.; Singh, G.P.; et al. A Multicentric, Randomized, Controlled Phase III Study of Centhaquine (Lyfaquin((R))) as a Resuscitative Agent in Hypovolemic Shock Patients. Drugs 2021, 81, 1079–1100. [Google Scholar] [CrossRef]
- Gulati, A.; Jain, D.; Agrawal, N.R.; Rahate, P.; Choudhuri, R.; Das, S.; Dhibar, D.P.; Prabhu, M.; Haveri, S.; Agarwal, R.; et al. Resuscitative Effect of Centhaquine (Lyfaquin((R))) in Hypovolemic Shock Patients: A Randomized, Multicentric, Controlled Trial. Adv. Ther. 2021, 38, 3223–3265. [Google Scholar] [CrossRef]
- Harrois, A.; Soyer, B.; Gauss, T.; Hamada, S.; Raux, M.; Duranteau, J.; Langeron, O.; Paugam-Burtz, C.; Pirracchio, R.; Riou, B.; et al. Prevalence and risk factors for acute kidney injury among trauma patients: A multicenter cohort study. Crit. Care 2018, 22, 344. [Google Scholar] [CrossRef] [Green Version]
- Legrand, M.; Mik, E.G.; Balestra, G.M.; Lutter, R.; Pirracchio, R.; Payen, D.; Ince, C. Fluid resuscitation does not improve renal oxygenation during hemorrhagic shock in rats. Anesthesiology 2010, 112, 119–127. [Google Scholar] [CrossRef] [Green Version]
- Basile, D.P.; Anderson, M.D.; Sutton, T.A. Pathophysiology of acute kidney injury. Compr. Physiol. 2012, 2, 1303–1353. [Google Scholar] [CrossRef] [Green Version]
- Ranjan, A.K.; Zhang, Z.; Briyal, S.; Gulati, A. Centhaquine Restores Renal Blood Flow and Protects Tissue Damage After Hemorrhagic Shock and Renal Ischemia. Front. Pharm. 2021, 12, 616253. [Google Scholar] [CrossRef]
- Ranjan, A.; Briyal, S.; Zhang, Z.; Marwah, M.; Posen, M.; Cherian, V.; Kotsko, M.; Gulati, A. Centhaquine Upregulates Hif1-a and Protects Hemorrhage-Induced Acute Kidney Injury. Crit. Care Med. 2020, 48, 690. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ranjan, A.K.; Gulati, A. Controls of Central and Peripheral Blood Pressure and Hemorrhagic/Hypovolemic Shock. J. Clin. Med. 2023, 12, 1108. https://doi.org/10.3390/jcm12031108
Ranjan AK, Gulati A. Controls of Central and Peripheral Blood Pressure and Hemorrhagic/Hypovolemic Shock. Journal of Clinical Medicine. 2023; 12(3):1108. https://doi.org/10.3390/jcm12031108
Chicago/Turabian StyleRanjan, Amaresh K., and Anil Gulati. 2023. "Controls of Central and Peripheral Blood Pressure and Hemorrhagic/Hypovolemic Shock" Journal of Clinical Medicine 12, no. 3: 1108. https://doi.org/10.3390/jcm12031108