
Prognostic Prediction Model for Glioblastoma: A Ferroptosis-Related Gene Prediction Model and Independent External Validation
 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Design and Data Acquisition
2.2. Identification of DEGs and Ferroptosis-Related DEGs
2.3. Selection of Ferroptosis-Related Prognostic DEGs
2.4. Establishment of the Predictive Model of Ferroptosis-Related Prognostic DEGs
2.5. Validation of the Predictive Prognostic Model
2.6. Functional and Immune Enrichment Analysis
2.7. Validation of Key Genes by the Quantitative Real-Time Polymerase Chain Reaction
2.8. Statistical Analysis
3. Results
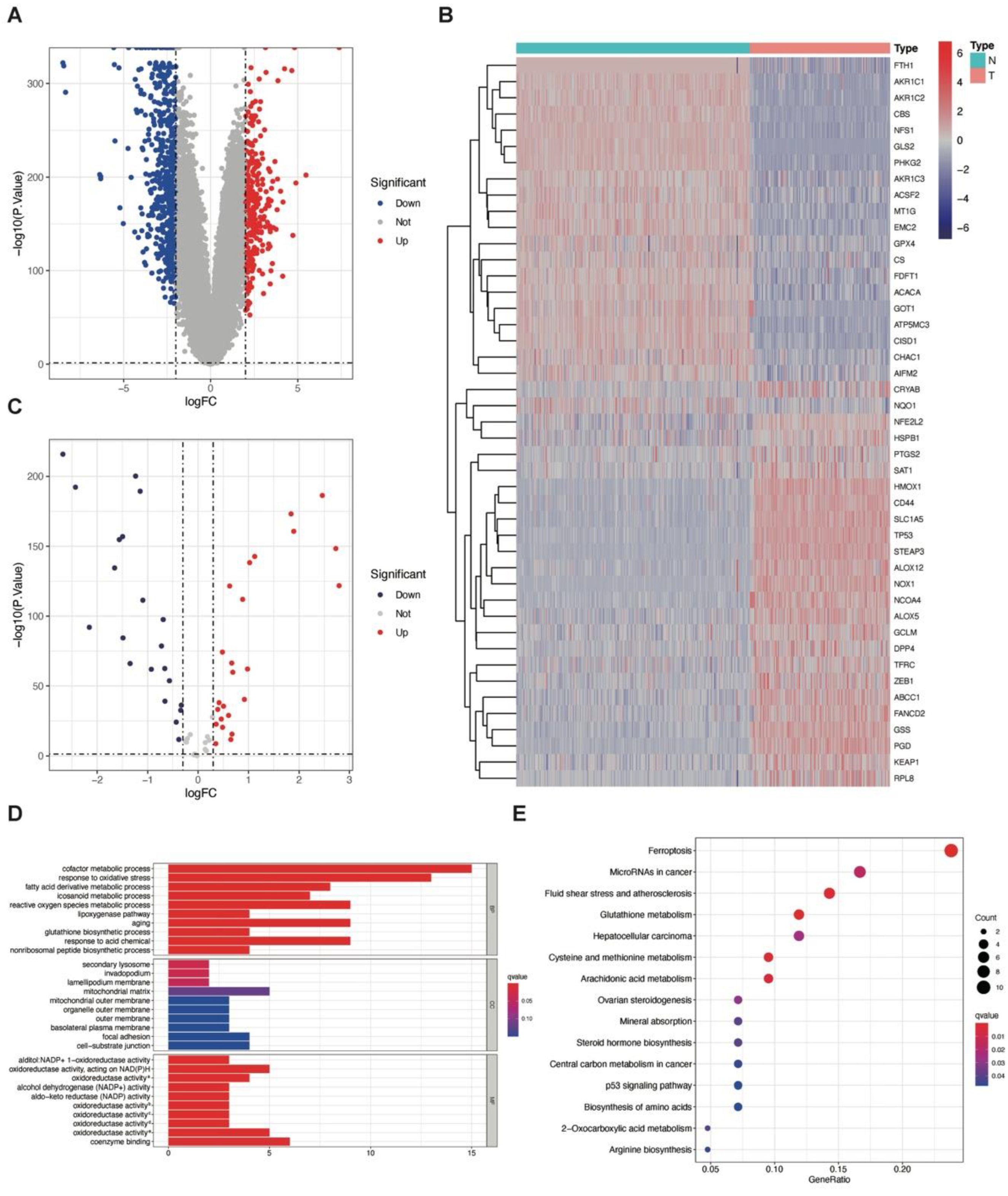
3.1. Identification of DEGs between GBM and Normal Brain Tissue
3.2. Identification and Gene Set Enrichment Analysis of Ferroptosis-Related DEGs between GBM and Normal Brain Tissue
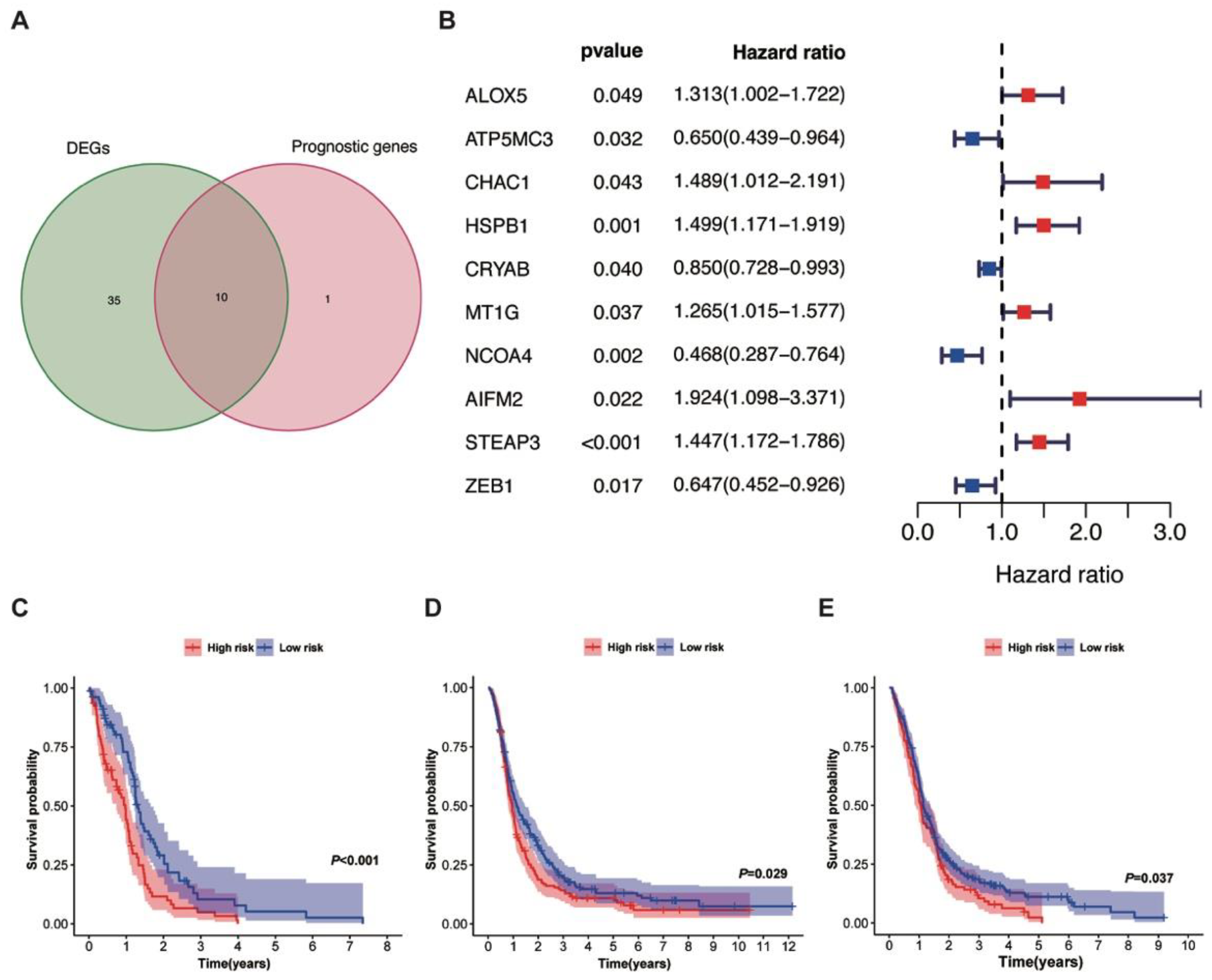
3.3. Lasso Regression Model and Kaplan–Meier Analysis of the Training and Validation Cohorts
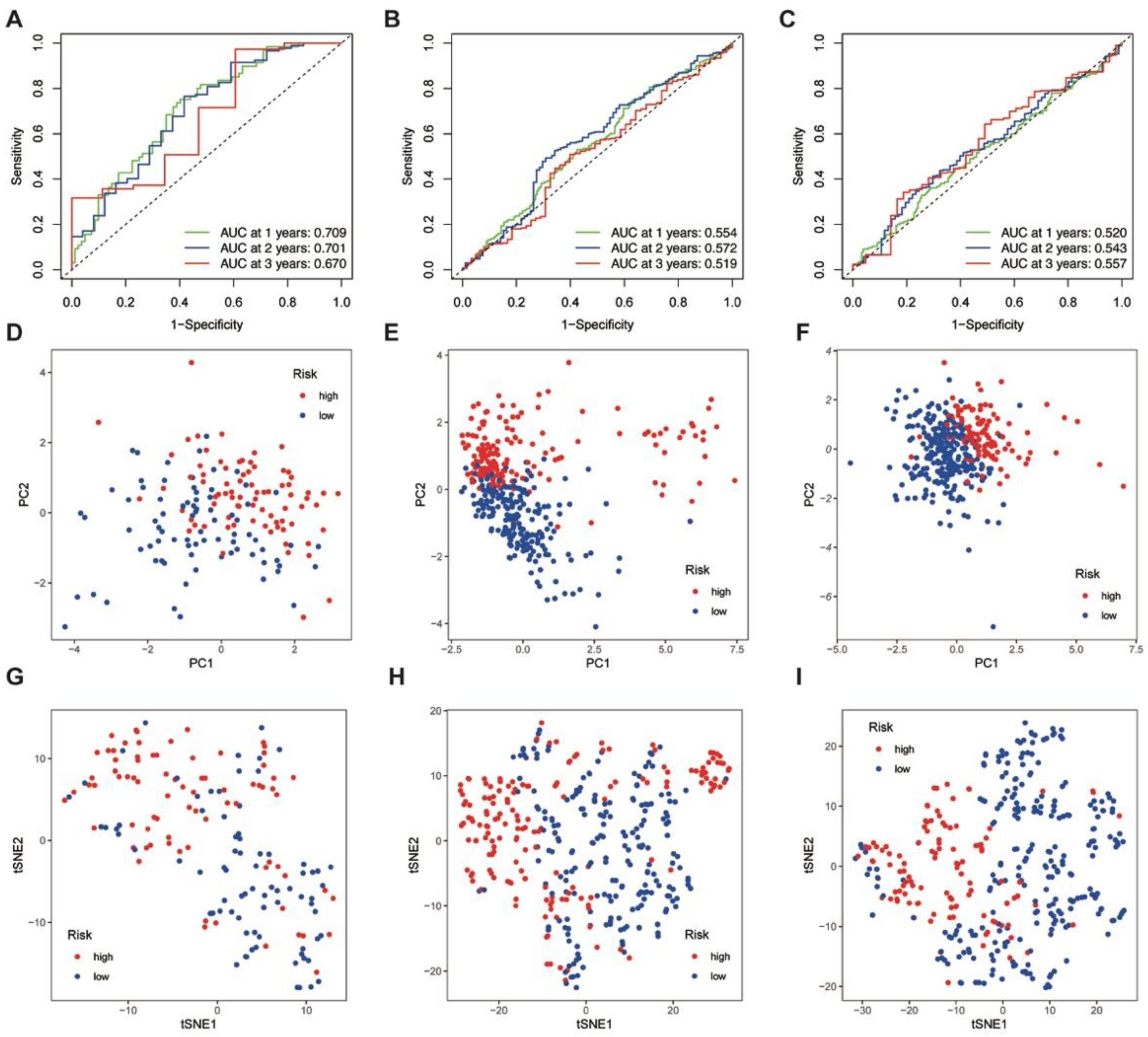
3.4. ROC Analysis, PCA, and t-SNE Analyses of the Training and Validation Cohorts
3.5. Multivariate Cox Regression Analysis and Gene Set Enrichment Analysis of DEGs between Risk Groups
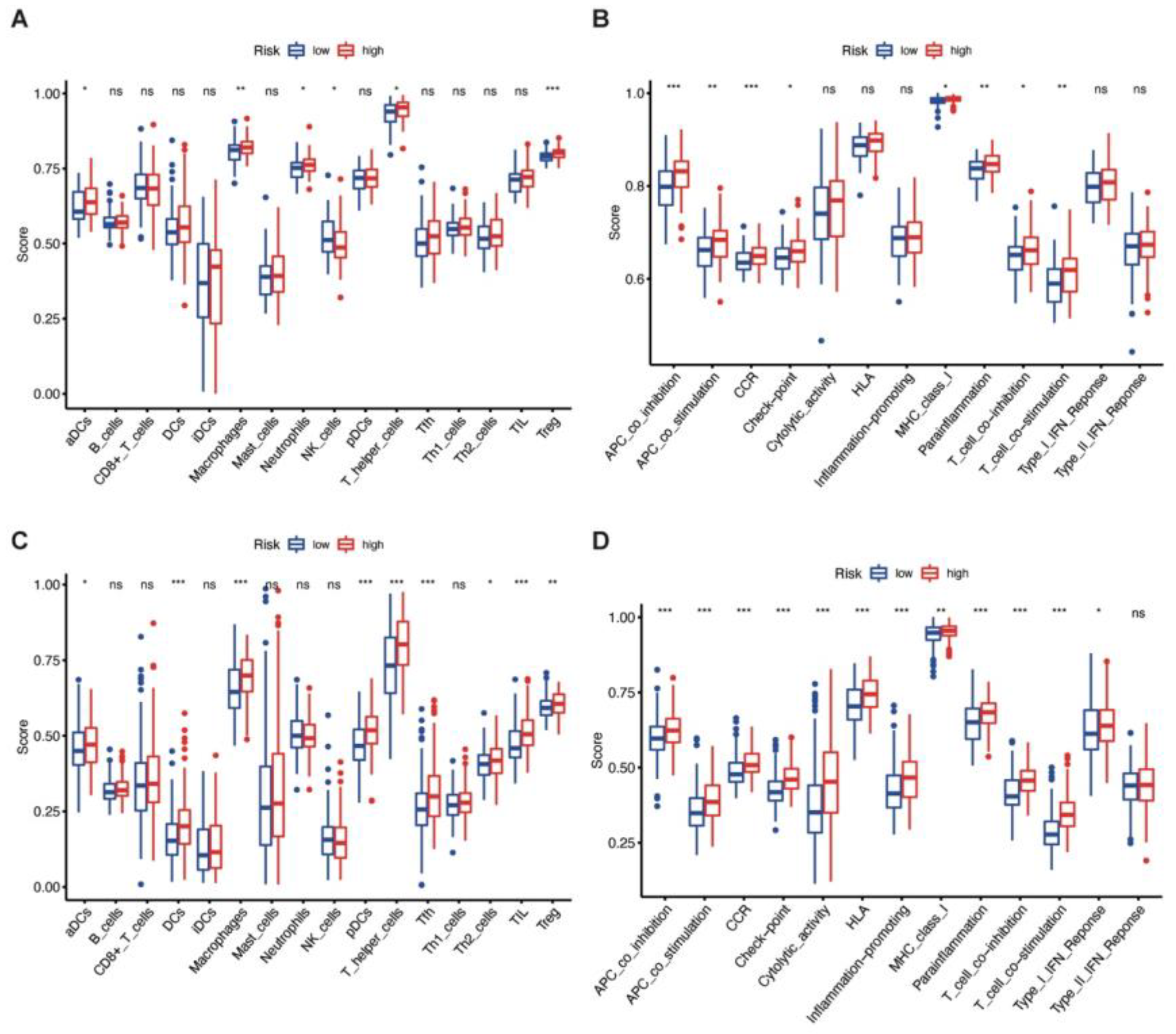
3.6. Immune Enrichment Score of High-Risk and Low-Risk Groups
3.7. Validation of the Key Genes and Related Proteins in Human Tissues
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ostrom, Q.T.; Patil, N.; Cioffi, G.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2013–2017. Neuro Oncol. 2020, 22, iv1–v96. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Reifenberger, G.; Von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A Summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [PubMed]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro Oncol. 2021, 23, 1231–1251. [Google Scholar] [CrossRef]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [PubMed]
- White, K.; Connor, K.; Clerkin, J.; Murphy, B.M.; Salvucci, M.; O’Farrell, A.C.; Rehm, M.; O’Brien, D.; Prehn, J.H.M.; Niclou, S.P.; et al. New hints towards a precision medicine strategy for IDH wild-type glioblastoma. Ann. Oncol. 2020, 31, 1679–1692. [Google Scholar] [CrossRef]
- NCCN. Central Nervous System Cancers (Version 2.2022). Available online: https://www.nccn.org/professionals/physician_gls/pdf/cns.pdf (accessed on 27 December 2022).
- Weller, M.; van den Bent, M.; Preusser, M.; Le Rhun, E.; Tonn, J.C.; Minniti, G.; Bendszus, M.; Balana, C.; Chinot, O.; Dirven, L.; et al. EANO guidelines on the diagnosis and treatment of diffuse gliomas of adulthood. Nat. Rev. Clin. Oncol. 2020, 18, 170–186. [Google Scholar] [CrossRef]
- Jiang, T.; Mao, Y.; Ma, W.; Mao, Q.; You, Y.; Yang, X.; Jiang, C.; Kang, C.; Li, X.; Chen, L.; et al. CGCG clinical practice guidelines for the management of adult diffuse gliomas. Cancer Lett. 2016, 375, 263–273. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Friedmann Angeli, J.P.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Tyurin, V.A.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Eggenhofer, E.; et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 2014, 16, 1180–1191. [Google Scholar] [CrossRef]
- Jiang, L.; Kon, N.; Li, T.; Wang, S.J.; Su, T.; Hibshoosh, H.; Baer, R.; Gu, W. Ferroptosis as a p53-mediated activity during tumour suppression. Nature 2015, 520, 57–62. [Google Scholar] [CrossRef] [Green Version]
- Doll, S.; Freitas, F.P.; Shah, R.; Aldrovandi, M.; da Silva, M.C.; Ingold, I.; Goya Grocin, A.; Xavier da Silva, T.N.; Panzilius, E.; Scheel, C.H.; et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature 2019, 575, 693–698. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Kang, R.; Kroemer, G.; Tang, D. Broadening horizons: The role of ferroptosis in cancer. Nat. Rev. Clin. Oncol. 2021, 18, 280–296. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Green, M.; Choi, J.E.; Gijón, M.; Kennedy, P.D.; Johnson, J.K.; Liao, P.; Lang, X.; Kryczek, I.; Sell, A.; et al. CD8(+) T cells regulate tumour ferroptosis during cancer immunotherapy. Nature 2019, 569, 270–274. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, M.; Freigang, S.; Schneider, C.; Conrad, M.; Bornkamm, G.W.; Kopf, M. T cell lipid peroxidation induces ferroptosis and prevents immunity to infection. J. Exp. Med. 2015, 212, 555–568. [Google Scholar] [CrossRef] [PubMed]
- Fan, Z.; Wirth, A.K.; Chen, D.; Wruck, C.J.; Rauh, M.; Buchfelder, M.; Savaskan, N. Nrf2-Keap1 pathway promotes cell proliferation and diminishes ferroptosis. Oncogenesis 2017, 6, e371. [Google Scholar] [CrossRef]
- Li, S.; He, Y.; Chen, K.; Sun, J.; Zhang, L.; He, Y.; Yu, H.; Li, Q. RSL3 Drives Ferroptosis through NF-κB Pathway Activation and GPX4 Depletion in Glioblastoma. Oxidative Med. Cell. Longev. 2021, 2021, 2915019. [Google Scholar] [CrossRef]
- Zheng, X.J.; Chen, W.L.; Yi, J.; Li, W.; Liu, J.Y.; Fu, W.Q.; Ren, L.W.; Li, S.; Ge, B.B.; Yang, Y.H.; et al. Apolipoprotein C1 promotes glioblastoma tumorigenesis by reducing KEAP1/NRF2 and CBS-regulated ferroptosis. Acta Pharmacol. Sin. 2022, 43, 2977–2992. [Google Scholar] [CrossRef]
- Yee, P.P.; Wei, Y.; Kim, S.Y.; Lu, T.; Chih, S.Y.; Lawson, C.; Tang, M.; Liu, Z.; Anderson, B.; Thamburaj, K.; et al. Neutrophil-induced ferroptosis promotes tumor necrosis in glioblastoma progression. Nat. Commun. 2020, 11, 5424. [Google Scholar] [CrossRef]
- Feng, L.; Qian, H.; Yu, X.; Liu, K.; Xiao, T.; Zhang, C.; Kuang, M.; Cheng, S.; Li, X.; Wan, J.; et al. Heterogeneity of tumor-infiltrating lymphocytes ascribed to local immune status rather than neoantigens by multi-omics analysis of glioblastoma multiforme. Sci. Rep. 2017, 7, 6968. [Google Scholar] [CrossRef]
- Yu, X.; Feng, L.; Liu, D.; Zhang, L.; Wu, B.; Jiang, W.; Han, Z.; Cheng, S. Quantitative proteomics reveals the novel co-expression signatures in early brain development for prognosis of glioblastoma multiforme. Oncotarget 2016, 7, 14161–14171. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.; Scheck, A.C.; Cloughesy, T.F.; Lai, A.; Dong, J.; Farooqi, H.K.; Liau, L.M.; Horvath, S.; Mischel, P.S.; Nelson, S.F. Gene expression analysis of glioblastomas identifies the major molecular basis for the prognostic benefit of younger age. BMC Med. Genom. 2008, 1, 52. [Google Scholar] [CrossRef]
- Tibshirani, R. The lasso method for variable selection in the Cox model. Stat. Med. 1997, 16, 385–395. [Google Scholar] [CrossRef]
- Yu, H.; Guo, P.; Xie, X.; Wang, Y.; Chen, G. Ferroptosis, a new form of cell death, and its relationships with tumourous diseases. J. Cell. Mol. Med. 2017, 21, 648–657. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.J.; Hu, H.M.; Li, G.Z.; Zhang, Y.; Wu, F.; Liu, X.; Wang, K.Y.; Zhang, C.B.; Jiang, T. Ferroptosis-Related Gene Signature Predicts Glioma Cell Death and Glioma Patient Progression. Front. Cell Dev. Biol. 2020, 8, 538. [Google Scholar] [CrossRef] [PubMed]
- Zhuo, S.; Chen, Z.; Yang, Y.; Zhang, J.; Tang, J.; Yang, K. Clinical and Biological Significances of a Ferroptosis-Related Gene Signature in Glioma. Front. Oncol. 2020, 10, 590861. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Xu, Z.; Jin, T.; Xu, K.; Liu, M.; Xu, H. Ferroptosis in Low-Grade Glioma: A New Marker for Diagnosis and Prognosis. Med. Sci. Monit. 2020, 26, e921947. [Google Scholar] [CrossRef]
- Zhang, Y.; Kong, Y.; Ma, Y.; Ni, S.; Wikerholmen, T.; Xi, K.; Zhao, F.; Zhao, Z.; Wang, J.; Huang, B.; et al. Loss of COPZ1 induces NCOA4 mediated autophagy and ferroptosis in glioblastoma cell lines. Oncogene 2021, 40, 1425–1439. [Google Scholar] [CrossRef]
- Wang, X.; Chen, Y.; Zhang, S.; Zhang, L.; Liu, X.; Zhang, L.; Li, X.; Chen, D. Co-expression of COX-2 and 5-LO in primary glioblastoma is associated with poor prognosis. J. Neuro-Oncol. 2015, 125, 277–285. [Google Scholar] [CrossRef]
- Nathoo, N.; Prayson, R.A.; Bondar, J.; Vargo, L.; Arrigain, S.; Mascha, E.J.; Suh, J.H.; Barnett, G.H.; Golubic, M. Increased expression of 5-lipoxygenase in high-grade astrocytomas. Neurosurgery 2006, 58, 347–354; discussion 347–354. [Google Scholar] [CrossRef]
- Han, M.; Xu, R.; Wang, S.; Yang, N.; Ni, S.; Zhang, Q.; Xu, Y.; Zhang, X.; Zhang, C.; Wei, Y.; et al. Six-Transmembrane Epithelial Antigen of Prostate 3 Predicts Poor Prognosis and Promotes Glioblastoma Growth and Invasion. Neoplasia 2018, 20, 543–554. [Google Scholar] [CrossRef]
- Kore, R.A.; Abraham, E.C. Inflammatory cytokines, interleukin-1 beta and tumor necrosis factor-alpha, upregulated in glioblastoma multiforme, raise the levels of CRYAB in exosomes secreted by U373 glioma cells. Biochem. Biophys. Res. Commun. 2014, 453, 326–331. [Google Scholar] [CrossRef] [PubMed]
- Kore, R.A.; Abraham, E.C. Phosphorylation negatively regulates exosome mediated secretion of cryAB in glioma cells. Biochim. Biophys. Acta 2016, 1863, 368–377. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.H.; Shen, W.L.; Shih, C.M.; Ho, K.H.; Cheng, C.H.; Lin, C.W.; Lee, C.C.; Liu, A.J.; Chen, K.C. The CHAC1-inhibited Notch3 pathway is involved in temozolomide-induced glioma cytotoxicity. Neuropharmacology 2017, 116, 300–314. [Google Scholar] [CrossRef] [PubMed]
- Lim, E.J.; Kim, S.; Oh, Y.; Suh, Y.; Kaushik, N.; Lee, J.H.; Lee, H.J.; Kim, M.J.; Park, M.J.; Kim, R.K.; et al. Crosstalk between GBM cells and mesenchymal stemlike cells promotes the invasiveness of GBM through the C5a/p38/ZEB1 axis. Neuro-Oncol. 2020, 22, 1452–1462. [Google Scholar] [CrossRef]
- Edwards, L.A.; Kim, S.; Madany, M.; Nuno, M.; Thomas, T.; Li, A.; Berel, D.; Lee, B.S.; Liu, M.; Black, K.L.; et al. ZEB1 Is a Transcription Factor That Is Prognostic and Predictive in Diffuse Gliomas. Front. Neurol. 2018, 9, 1199. [Google Scholar] [CrossRef]
- Yi, J.; Zhu, J.; Wu, J.; Thompson, C.B.; Jiang, X. Oncogenic activation of PI3K-AKT-mTOR signaling suppresses ferroptosis via SREBP-mediated lipogenesis. Proc. Natl. Acad. Sci. USA 2020, 117, 31189–31197. [Google Scholar] [CrossRef]
- Luo, M.; Wu, L.; Zhang, K.; Wang, H.; Zhang, T.; Gutierrez, L.; O’Connell, D.; Zhang, P.; Li, Y.; Gao, T.; et al. miR-137 regulates ferroptosis by targeting glutamine transporter SLC1A5 in melanoma. Cell Death Differ. 2018, 25, 1457–1472. [Google Scholar] [CrossRef]
- Lin, R.; Zhang, Z.; Chen, L.; Zhou, Y.; Zou, P.; Feng, C.; Wang, L.; Liang, G. Dihydroartemisinin (DHA) induces ferroptosis and causes cell cycle arrest in head and neck carcinoma cells. Cancer Lett. 2016, 381, 165–175. [Google Scholar] [CrossRef]
- Dai, E.; Han, L.; Liu, J.; Xie, Y.; Kroemer, G.; Klionsky, D.J.; Zeh, H.J.; Kang, R.; Wang, J.; Tang, D. Autophagy-dependent ferroptosis drives tumor-associated macrophage polarization via release and uptake of oncogenic KRAS protein. Autophagy 2020, 16, 2069–2083. [Google Scholar] [CrossRef]

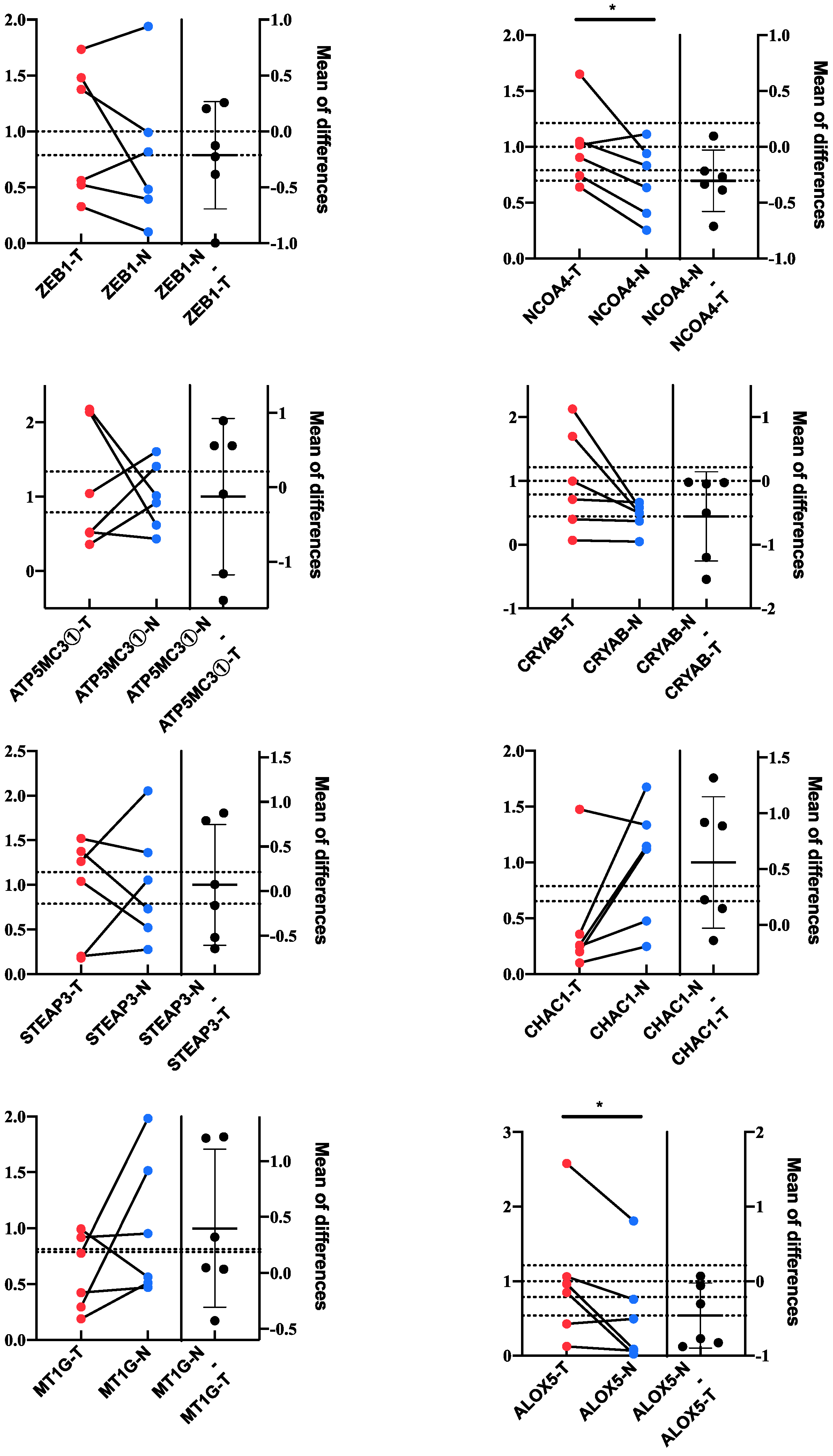

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | Coefficient † | Training Cohort | Validation Cohort | Chromosome Localization | ||
|---|---|---|---|---|---|---|
| Log2FC ‡ | FDR ¶ | Log2FC ‡ | FDR ¶ | |||
| ALOX5 | 0.13724394 | 1.167 | <0.001 * | 0.076 | NS | 10q11.21 |
| CHAC1 | 0.13378105 | 0.250 | NS | 0.085 | NS | 15q15.1 |
| STEAP3 | 0.11443311 | 1.005 | <0.001 * | 0.158 | NS | 2q14.2 |
| MT1G | 0.08304952 | 0.217 | NS | −0.007 | NS | 16q13 |
| CRYAB | −0.0297374 | 2.106 | <0.001 * | −0.059 | NS | 11q23.1 |
| ZEB1 | −0.2148601 | −0.143 | NS | −0.119 | NS | 10p11.22 |
| ATP5MC3 | −0.4763973 | −0.136 | NS | −0.075 | NS | 2q31.1 |
| NCOA4 | −0.5506693 | −0.097 | NS | −0.107 | NS | 10q11.22 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, W.; Lei, C.; Wang, Y.; Guo, D.; Zhang, S.; Wang, X.; Zhang, Z.; Wang, Y.; Ma, W. Prognostic Prediction Model for Glioblastoma: A Ferroptosis-Related Gene Prediction Model and Independent External Validation. J. Clin. Med. 2023, 12, 1341. https://doi.org/10.3390/jcm12041341
Chen W, Lei C, Wang Y, Guo D, Zhang S, Wang X, Zhang Z, Wang Y, Ma W. Prognostic Prediction Model for Glioblastoma: A Ferroptosis-Related Gene Prediction Model and Independent External Validation. Journal of Clinical Medicine. 2023; 12(4):1341. https://doi.org/10.3390/jcm12041341
Chicago/Turabian StyleChen, Wenlin, Chuxiang Lei, Yuekun Wang, Dan Guo, Sumei Zhang, Xiaoxi Wang, Zixin Zhang, Yu Wang, and Wenbin Ma. 2023. "Prognostic Prediction Model for Glioblastoma: A Ferroptosis-Related Gene Prediction Model and Independent External Validation" Journal of Clinical Medicine 12, no. 4: 1341. https://doi.org/10.3390/jcm12041341