

A Potential Therapy Using Antisense Oligonucleotides to Treat Autosomal Recessive Polycystic Kidney Disease
 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Clinical and Laboratory Data
2.2. Genetic Analysis
2.3. In Silico Analysis
2.4. Construction of the Plasmid
2.5. In Vitro Minigene Assay
2.6. Design of Antisense Oligonucleotides
2.7. ASOs Rescue Assays
3. Results
3.1. Clinical Analysis
3.2. Analysis of PKHD1 Variants
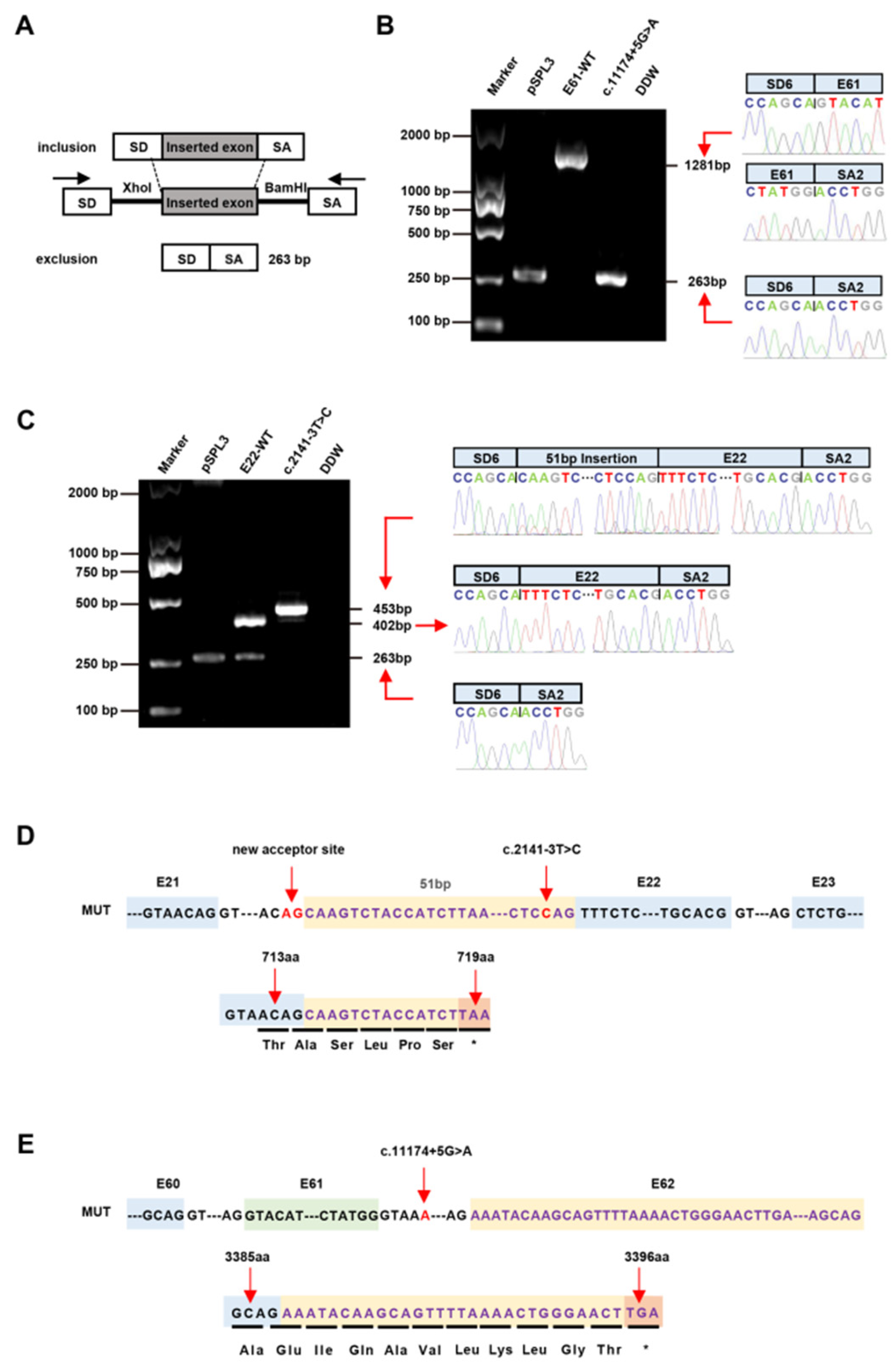
3.3. Splicing Analysis of the PKHD1 Minigene
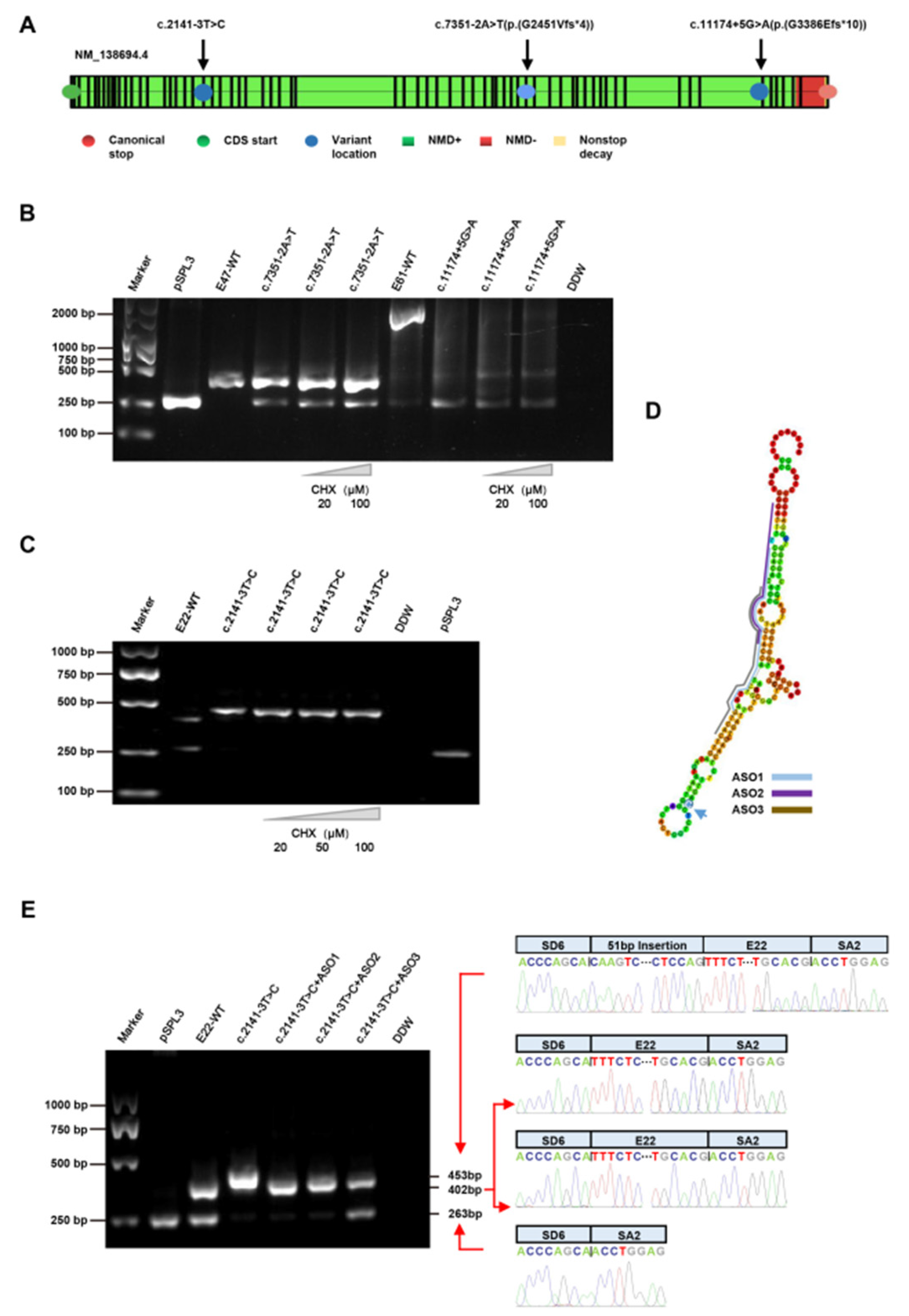
3.4. Treatment with CHX
3.5. Treatment with ASOs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Goggolidou, P.; Richards, T. The genetics of Autosomal Recessive Polycystic Kidney Disease (ARPKD). Biochim. Biophys. Acta Mol. Basis Dis. 2022, 1868, 166348. [Google Scholar] [CrossRef] [PubMed]
- Besse, W.; Roosendaal, C.; Tuccillo, L.; Roy, S.G.; Gallagher, A.R.; Somlo, S. Adult Inactivation of the Recessive Polycystic Kidney Disease Gene Causes Polycystic Liver Disease. Kidney360 2020, 1, 1068–1076. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.; Ao, W.; Fang, J.; Mao, G.; Chen, C.; Yu, L.; Cai, H.; Xu, C. Imaging manifestations of Caroli disease with autosomal recessive polycystic kidney disease: A case report and literature review. BMC Pregnancy Childbirth 2021, 21, 294. [Google Scholar] [CrossRef] [PubMed]
- Hoyer, P.F. Clinical manifestations of autosomal recessive polycystic kidney disease. Curr. Opin. Pediatr. 2015, 27, 186–192. [Google Scholar] [CrossRef]
- Ma, M. Cilia and polycystic kidney disease. Semin Cell Dev Biol. 2021, 110, 139–148. [Google Scholar] [CrossRef]
- Leebens-Mack, J.; Milligan, B. Pollination biology in hybridizing Baptisia (Fabaceae) populations. Am. J. Bot. 1998, 85, 500. [Google Scholar] [CrossRef]
- Tang, R.; Xu, Z. Gene therapy: A double-edged sword with great powers. Mol. Cell Biochem. 2020, 474, 73–81. [Google Scholar] [CrossRef]
- Zu, H.; Gao, D. Non-viral Vectors in Gene Therapy: Recent Development, Challenges, and Prospects. AAPS J. 2021, 23, 78. [Google Scholar] [CrossRef]
- Chen, C.; Yang, Z.; Tang, X. Chemical modifications of nucleic acid drugs and their delivery systems for gene-based therapy. Med. Res. Rev. 2018, 38, 829–869. [Google Scholar] [CrossRef]
- Alshaer, W.; Zureigat, H.; Al Karaki, A.; Al-Kadash, A.; Gharaibeh, L.; Hatmal, M.M.; Aljabali, A.A.A.; Awidi, A. siRNA: Mechanism of action, challenges, and therapeutic approaches. Eur. J. Pharmacol. 2021, 905, 174178. [Google Scholar] [CrossRef]
- Shadid, M.; Badawi, M.; Abulrob, A. Antisense oligonucleotides: Absorption, distribution, metabolism, and excretion. Expert Opin. Drug Metab. Toxicol. 2021, 17, 1281–1292. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Huang, L.; Luo, M.; Xia, X. Bacterial translocation in acute pancreatitis. Crit. Rev. Microbiol. 2019, 45, 539–547. [Google Scholar] [CrossRef] [PubMed]
- Hagedorn, P.H.; Brown, J.M.; Easton, A.; Pierdomenico, M.; Jones, K.; Olson, R.E.; Mercer, S.E.; Li, D.; Loy, J.; Hog, A.M.; et al. Acute Neurotoxicity of Antisense Oligonucleotides After Intracerebroventricular Injection Into Mouse Brain Can Be Predicted from Sequence Features. Nucleic Acid Ther. 2022, 32, 151–162. [Google Scholar] [CrossRef]
- Fish, L.; Khoroshkin, M.; Navickas, A.; Garcia, K.; Culbertson, B.; Hanisch, B.; Zhang, S.; Nguyen, H.C.B.; Soto, L.M.; Dermit, M.; et al. A prometastatic splicing program regulated by SNRPA1 interactions with structured RNA elements. Science 2021, 372, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Bennett, C.F. Therapeutic Antisense Oligonucleotides Are Coming of Age. Annu. Rev. Med. 2019, 70, 307–321. [Google Scholar] [CrossRef]
- Bennett, C.F.; Krainer, A.R.; Cleveland, D.W. Antisense Oligonucleotide Therapies for Neurodegenerative Diseases. Annu. Rev. Neurosci. 2019, 42, 385–406. [Google Scholar] [CrossRef]
- Finkel, R.S.; Mercuri, E.; Darras, B.T.; Connolly, A.M.; Kuntz, N.L.; Kirschner, J.; Chiriboga, C.A.; Saito, K.; Servais, L.; Tizzano, E.; et al. Nusinersen versus Sham Control in Infantile-Onset Spinal Muscular Atrophy. N. Engl. J. Med. 2017, 377, 1723–1732. [Google Scholar] [CrossRef] [PubMed]
- Charleston, J.S.; Schnell, F.J.; Dworzak, J.; Donoghue, C.; Lewis, S.; Chen, L.; Young, G.D.; Milici, A.J.; Voss, J.; DeAlwis, U.; et al. Eteplirsen treatment for Duchenne muscular dystrophy: Exon skipping and dystrophin production. Neurology 2018, 90, e2146–e2154. [Google Scholar] [CrossRef] [PubMed]
- Aartsma-Rus, A.; Corey, D.R. The 10th Oligonucleotide Therapy Approved: Golodirsen for Duchenne Muscular Dystrophy. Nucleic Acid Ther. 2020, 30, 67–70. [Google Scholar] [CrossRef]
- Frank, D.E.; Schnell, F.J.; Akana, C.; El-Husayni, S.H.; Desjardins, C.A.; Morgan, J.; Charleston, J.S.; Sardone, V.; Domingos, J.; Dickson, G.; et al. Increased dystrophin production with golodirsen in patients with Duchenne muscular dystrophy. Neurology 2020, 94, e2270–e2282. [Google Scholar] [CrossRef] [Green Version]
- Zakeri, S.E.; Pradeep, S.P.; Kasina, V.; Laddha, A.P.; Manautou, J.E.; Bahal, R. Casimersen for the treatment of Duchenne muscular dystrophy. Trends Pharmacol. Sci. 2022, 43, 607–608. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Han, Y.; Zhou, J.; Zheng, B.; Zhou, W.; Bao, H.; Jia, Z.; Zhang, A.; Huang, S.; Ding, G.; et al. Splicing Characterization of CLCNKB Variants in Four Patients With Type III Bartter Syndrome. Front. Genet. 2020, 11, 81. [Google Scholar] [CrossRef] [PubMed]
- Krall, P.; Pineda, C.; Ruiz, P.; Ejarque, L.; Vendrell, T.; Camacho, J.A.; Mendizabal, S.; Oliver, A.; Ballarin, J.; Torra, R.; et al. Cost-effective PKHD1 genetic testing for autosomal recessive polycystic kidney disease. Pediatr. Nephrol. 2014, 29, 223–234. [Google Scholar] [CrossRef]
- Losekoot, M.; Haarloo, C.; Ruivenkamp, C.; White, S.J.; Breuning, M.H.; Peters, D.J. Analysis of missense variants in the PKHD1-gene in patients with autosomal recessive polycystic kidney disease (ARPKD). Hum. Genet. 2005, 118, 185–206. [Google Scholar] [CrossRef] [PubMed]
- Hao, X.; Liu, S.; Dong, Q.; Zhang, H.; Zhao, J.; Su, L. Whole exome sequencing identifies recessive PKHD1 mutations in a Chinese twin family with Caroli disease. PLoS ONE 2014, 9, e92661. [Google Scholar] [CrossRef] [PubMed]
- Melchionda, S.; Palladino, T.; Castellana, S.; Giordano, M.; Benetti, E.; De Bonis, P.; Zelante, L.; Bisceglia, L. Expanding the mutation spectrum in 130 probands with ARPKD: Identification of 62 novel PKHD1 mutations by sanger sequencing and MLPA analysis. J. Hum. Genet. 2016, 61, 811–821. [Google Scholar] [CrossRef]
- Sharp, A.M.; Messiaen, L.M.; Page, G.; Antignac, C.; Gubler, M.C.; Onuchic, L.F.; Somlo, S.; Germino, G.G.; Guay-Woodford, L.M. Comprehensive genomic analysis of PKHD1 mutations in ARPKD cohorts. J. Med. Genet. 2005, 42, 336–349. [Google Scholar] [CrossRef]
- Qiu, L.R.; Xu, R.R.; Tang, J.H.; Zhou, J.H. Possible PKHD1 Hot-spot Mutations Related to Early Kidney Function Failure or Hepatofibrosis in Chinese Children with ARPKD: A Retrospective Single Center Cohort Study and Literature Review. Curr. Med. Sci. 2020, 40, 835–844. [Google Scholar] [CrossRef]
- Yon, D.; Frith, C.D. Precision and the Bayesian brain. Curr. Biol. 2021, 31, R1026–R1032. [Google Scholar] [CrossRef]
- Song, M.H.; Cho, H.J.; Lee, H.K.; Kwon, T.J.; Lee, W.S.; Oh, S.; Bok, J.; Choi, J.Y.; Kim, U.K. CHD7 mutational analysis and clinical considerations for auditory rehabilitation in deaf patients with CHARGE syndrome. PLoS ONE 2011, 6, e24511. [Google Scholar] [CrossRef] [Green Version]
- Green, I.D.; Pinello, N.; Song, R.; Lee, Q.; Halstead, J.M.; Kwok, C.T.; Wong, A.C.H.; Nair, S.S.; Clark, S.J.; Roediger, B.; et al. Macrophage development and activation involve coordinated intron retention in key inflammatory regulators. Nucleic Acids Res. 2020, 48, 6513–6529. [Google Scholar] [CrossRef] [PubMed]
- Sergeeva, O.V.; Shcherbinina, E.Y.; Shomron, N.; Zatsepin, T.S. Modulation of RNA Splicing by Oligonucleotides: Mechanisms of Action and Therapeutic Implications. Nucleic Acid Ther. 2022, 32, 123–138. [Google Scholar] [CrossRef] [PubMed]
- Yabe, S.G.; Nishida, J.; Fukuda, S.; Takeda, F.; Nasiro, K.; Yasuda, K.; Iwasaki, N.; Okochi, H. Expression of mutant mRNA and protein in pancreatic cells derived from MODY3- iPS cells. PLoS ONE 2019, 14, e0217110. [Google Scholar] [CrossRef] [PubMed]
- Leier, A.; Moore, M.; Liu, H.; Daniel, M.; Hyde, A.M.; Messiaen, L.; Korf, B.R.; Selvakumaran, J.; Ciszewski, L.; Lambert, L.; et al. Targeted exon skipping of NF1 exon 17 as a therapeutic for neurofibromatosis type I. Mol. Ther. Nucleic Acids 2022, 28, 261–278. [Google Scholar] [CrossRef] [PubMed]
- Sait, H.; Srivastava, S.; Saxena, D. Integrated Management Strategies for Epidermolysis Bullosa: Current Insights. Int. J. Gen. Med. 2022, 15, 5133–5144. [Google Scholar] [CrossRef]
- Han, Z.; Chen, C.; Christiansen, A.; Ji, S.; Lin, Q.; Anumonwo, C.; Liu, C.; Leiser, S.C.; Meena; Aznarez, I.; et al. Antisense oligonucleotides increase Scn1a expression and reduce seizures and SUDEP incidence in a mouse model of Dravet syndrome. Sci. Transl. Med. 2020, 12, 1–14. [Google Scholar] [CrossRef]
- Radke, M.H.; Badillo-Lisakowski, V.; Britto-Borges, T.; Kubli, D.A.; Juttner, R.; Parakkat, P.; Carballo, J.L.; Huttemeister, J.; Liss, M.; Hansen, A.; et al. Therapeutic inhibition of RBM20 improves diastolic function in a murine heart failure model and human engineered heart tissue. Sci. Transl. Med. 2021, 13, eabe8952. [Google Scholar] [CrossRef]
- Adeva, M.; El-Youssef, M.; Rossetti, S.; Kamath, P.S.; Kubly, V.; Consugar, M.B.; Milliner, D.M.; King, B.F.; Torres, V.E.; Harris, P.C. Clinical and molecular characterization defines a broadened spectrum of autosomal recessive polycystic kidney disease (ARPKD). Medicine 2006, 85, 1–21. [Google Scholar] [CrossRef]
- Burgmaier, K.; Brinker, L.; Erger, F.; Beck, B.B.; Benz, M.R.; Bergmann, C.; Boyer, O.; Collard, L.; Dafinger, C.; Fila, M.; et al. Refining genotype-phenotype correlations in 304 patients with autosomal recessive polycystic kidney disease and PKHD1 gene variants. Kidney Int. 2021, 100, 650–659. [Google Scholar] [CrossRef]
- Outeda, P.; Menezes, L.; Hartung, E.A.; Bridges, S.; Zhou, F.; Zhu, X.; Xu, H.; Huang, Q.; Yao, Q.; Qian, F.; et al. A novel model of autosomal recessive polycystic kidney questions the role of the fibrocystin C-terminus in disease mechanism. Kidney Int. 2017, 92, 1130–1144. [Google Scholar] [CrossRef]
- Tsunoda, T.; Kakinuma, S.; Miyoshi, M.; Kamiya, A.; Kaneko, S.; Sato, A.; Tsuchiya, J.; Nitta, S.; Kawai-Kitahata, F.; Murakawa, M.; et al. Loss of fibrocystin promotes interleukin-8-dependent proliferation and CTGF production of biliary epithelium. J. Hepatol. 2019, 71, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Society for Maternal-Fetal, M.; Swanson, K. Autosomal recessive polycystic kidney disease. Am. J. Obstet. Gynecol. 2021, 225, B7–B8. [Google Scholar] [CrossRef]
- Aravind, L.; Koonin, E.V. Gleaning non-trivial structural, functional and evolutionary information about proteins by iterative database searches. J. Mol. Biol. 1999, 287, 1023–1040. [Google Scholar] [CrossRef] [PubMed]
- Bork, P.; Doerks, T.; Springer, T.A.; Snel, B. Domains in plexins: Links to integrins and transcription factors. Trends Biochem. Sci. 1999, 24, 261–263. [Google Scholar] [CrossRef]
- He, Q.Y.; Liu, X.H.; Li, Q.; Studholme, D.J.; Li, X.W.; Liang, S.P. G8: A novel domain associated with polycystic kidney disease and non-syndromic hearing loss. Bioinformatics 2006, 22, 2189–2191. [Google Scholar] [CrossRef]
- Kim, I.; Fu, Y.; Hui, K.; Moeckel, G.; Mai, W.; Li, C.; Liang, D.; Zhao, P.; Ma, J.; Chen, X.Z.; et al. Fibrocystin/polyductin modulates renal tubular formation by regulating polycystin-2 expression and function. J. Am. Soc. Nephrol. 2008, 19, 455–468. [Google Scholar] [CrossRef]
- Nagano, J.; Kitamura, K.; Hujer, K.M.; Ward, C.J.; Bram, R.J.; Hopfer, U.; Tomita, K.; Huang, C.; Miller, R.T. Fibrocystin interacts with CAML, a protein involved in Ca2+ signaling. Biochem. Biophys. Res. Commun. 2005, 338, 880–889. [Google Scholar] [CrossRef]
- Sweeney, W.E.; Frost, P.; Avner, E.D. Tesevatinib ameliorates progression of polycystic kidney disease in rodent models of autosomal recessive polycystic kidney disease. World J. Nephrol. 2017, 6, 188–200. [Google Scholar] [CrossRef]
- Parisi, M.A. The molecular genetics of Joubert syndrome and related ciliopathies: The challenges of genetic and phenotypic heterogeneity. Transl. Sci. Rare Dis. 2019, 4, 25–49. [Google Scholar] [CrossRef]
- Imai, E.; Isaka, Y. Gene electrotransfer: Potential for gene therapy of renal diseases. Kidney Int. 2002, 61, S37–S41. [Google Scholar] [CrossRef] [Green Version]
- Ricker, J.L.; Mata, J.E.; Iversen, P.L.; Gattone, V.H. c-myc antisense oligonucleotide treatment ameliorates murine ARPKD. Kidney Int. 2002, 61, S125–S131. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Liu, S.; Liu, G.; Ozturk, A.; Hicks, G.G. ALS-associated FUS mutations result in compromised FUS alternative splicing and autoregulation. PLoS Genet. 2013, 9, e1003895. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.C.; Valencia, T.; Allerson, C.; Schairer, A.; Flaten, A.; Yheskel, M.; Kersjes, K.; Li, J.; Gatto, S.; Takhar, M.; et al. Discovery and preclinical evaluation of anti-miR-17 oligonucleotide RGLS4326 for the treatment of polycystic kidney disease. Nat. Commun. 2019, 10, 4148. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
| Patient | Age (Year) | Sex | Ethnicity | Family History | Renal Cysts | Kidney Enlargement | Caroli Disease | Hepatic Cysts | Splenomegaly | Renal Dysfunction | Liver Dysfunction |
|---|---|---|---|---|---|---|---|---|---|---|---|
| P1 | 7 | Female | Han | No | Yes | Yes | No | No | No | No | No |
| P2 | 1.6 | Female | Han | No | Yes | No | No | No | No | No | No |
| P3 | 9 | Male | Han | Yes | Yes | Yes | Yes | Yes | Yes | No | Elevated liver enzymes |
| P4 | 14.3 | Female | Han | No | Yes | No | No | No | No | No | No |
| P5 | 0.7 | Male | Han | No | Yes | No | No | No | No | No | No |
| P6 | 1.6 | Male | Han | No | Yes | No | No | No | No | No, with urinary occult blood | No |
| P7 | 1 | Male | Han | No | Yes | Yes | No | No | No | No | No |
| P8 | 0.8 | Female | Han | No | Yes | No | No | No | No | No | No |
| P9 | 12 | Female | Han | Yes | Yes | No | No | No | No | No | No |
| P10 | 1.1 | Female | Han | No | Yes | Yes | No | Yes | No | Elevated blood urea and uric acid | Elevated liver enzymes |
| P11 | 5 | Male | Han | Yes | Yes | No | No | No | No | No | No |
| Patient | Nucleotide Change | Amino Acid Change | Hom/Het | Location | Domain | Mutation Type | ACMG | Reported |
|---|---|---|---|---|---|---|---|---|
| P1 | c.2141-3T>C | - | Het | I21 | - | Splicing | VUS (PM2 + PP3 + PM5) | No |
| P2, P9 | c.7942G>A | p.Gly2648Ser | Het | E50 | Extracellular | Missense | VUS (PM2) | [23] |
| P2 | c.7351-2A>T | - | Het | I46 | - | Splicing | P (PVS1 + PM2 + PM5) | [24] |
| P3 | c.3500T>C | p.Leu1167Pro | Het | E30 | IPT/TIG 6; atypical | Missense | VUS (PM2 + PP3) | No |
| P3 | c.11174+5G>A | - | Het | I61 | - | Splicing | VUS (PM2 + PP3) | No |
| P4 | c.325G>A | p.Ala109Thr | Het | E5 | IPT/TIG 1; atypical | Missense | LP (PS2 + PM2) | No |
| P5 | c.6001G>T | p.Glu2001 * | Het | E37 | G8 1 | Nonsense | P (PVS1 + PM2 + PM5) | No |
| P1, P5, P6 | c.2507T>C | p.Val836Ala | Het | E24 | Extracellular | Missense | VUS (PM2 + PP3 + PM3) | [25] |
| P6 | c.5869G>A | p.Asp1957Asn | Het | E36 | G8 1 | Missense | LP (PS2 + PM2 + PP3) | No |
| P7 | c.6245C>T | p.Thr2082Ile | Het | E38 | Extracellular | Missense | VUS (PM2 + PP3) | No |
| P8 | c.6910C>T | p.Gln2304 * | Het | E43 | PbH1 3′ | Nonsense | P (PVS1 + PM2 + PM3) | [26] |
| P8 | c.4199C>T | p.Ser1400Leu | Het | E32 | IPT/TIG 9 | Missense | VUS (PM2 + PP3) | [26] |
| P9 | c.11525G>A | p.Arg3842Gln | Het | E65 | Extracellular | Missense | VUS (PM2 + PP3) | No |
| P10 | c.9780G>A | p.Trp3260 * | Het | E58 | Extracellular | Nonsense | P (PVS1 + PM2 + PM3) | No |
| P10 | c.8518C>T | p.Arg2840Cys | Het | E54 | G8 2 | Missense | LP (PS1 + PM2 + PM3 + PP3) | [27] |
| P11 | c.10756_10759delAACT | p.Asn3586Serfs *22 | Het | E61 | Extracellular | Frameshift | P (PVS1 + PM2 + PP3) | [28] |
| P11 | c.10072G>A | p.Asp3358Asn | Het | E60 | Extracellular | Missense | LP (PS2 + PM2 + PM2 + PP3) | No |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, H.; Wang, C.; Che, R.; Zheng, B.; Zhou, W.; Huang, S.; Jia, Z.; Zhang, A.; Zhao, F.; Ding, G. A Potential Therapy Using Antisense Oligonucleotides to Treat Autosomal Recessive Polycystic Kidney Disease. J. Clin. Med. 2023, 12, 1428. https://doi.org/10.3390/jcm12041428
Li H, Wang C, Che R, Zheng B, Zhou W, Huang S, Jia Z, Zhang A, Zhao F, Ding G. A Potential Therapy Using Antisense Oligonucleotides to Treat Autosomal Recessive Polycystic Kidney Disease. Journal of Clinical Medicine. 2023; 12(4):1428. https://doi.org/10.3390/jcm12041428
Chicago/Turabian StyleLi, Huixia, Chunli Wang, Ruochen Che, Bixia Zheng, Wei Zhou, Songming Huang, Zhanjun Jia, Aihua Zhang, Fei Zhao, and Guixia Ding. 2023. "A Potential Therapy Using Antisense Oligonucleotides to Treat Autosomal Recessive Polycystic Kidney Disease" Journal of Clinical Medicine 12, no. 4: 1428. https://doi.org/10.3390/jcm12041428