
The Role of Tight Junctions in Atopic Dermatitis: A Systematic Review


{kind=link}
{kind=link}
Abstract
:1. Introduction
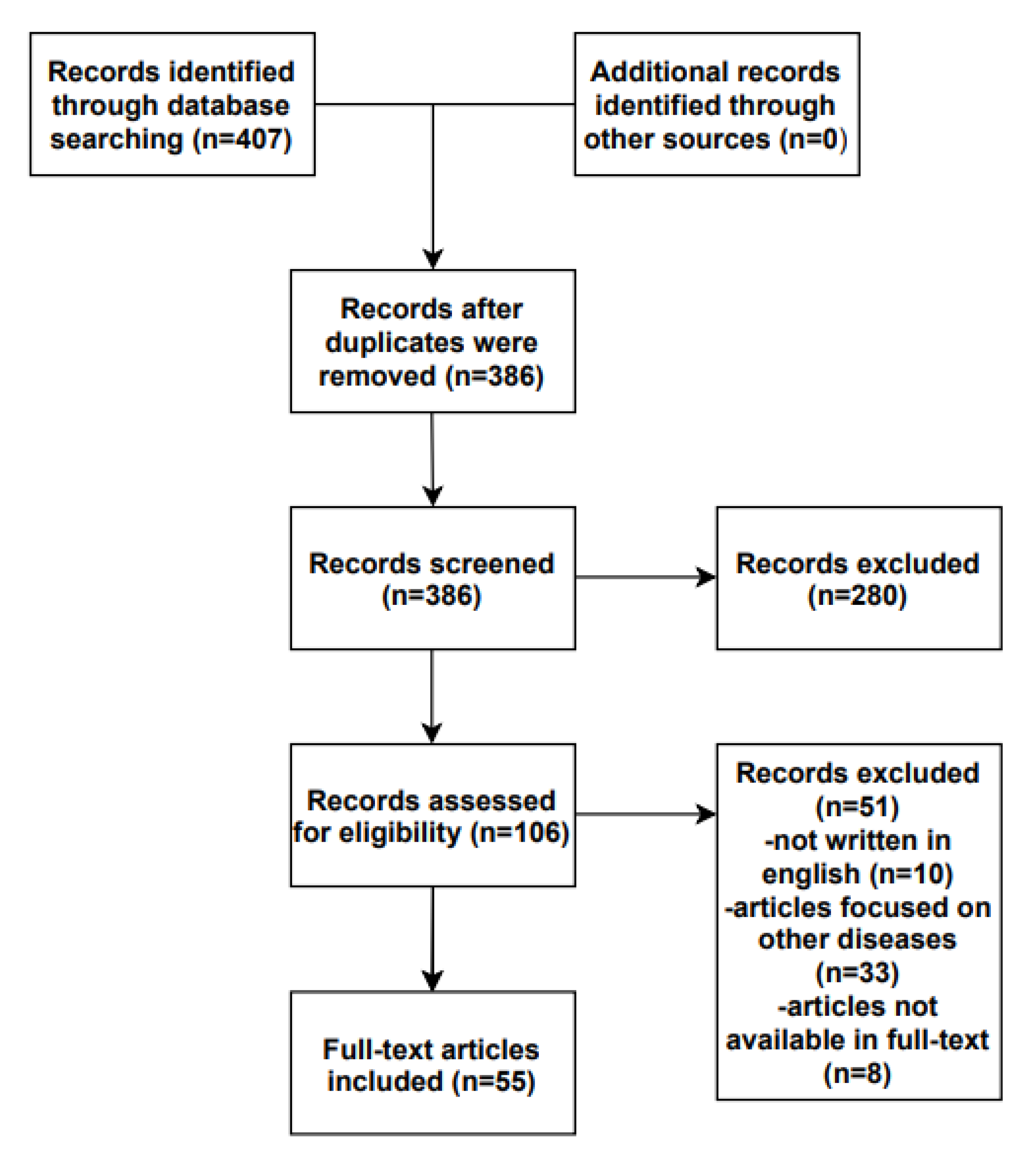
2. Materials and Methods
3. Results
3.1. The “Anatomy” of TJs
Tight Junctions in Skin Appendages
3.2. The Role in Healthy Skin
3.3. Tight Junctions in Atopic Dermatitis
3.3.1. Claudins Regulation in Atopic Dermatitis
3.3.2. The Role of P63
3.3.3. Host Defense Peptides and Tjs
3.3.4. Increased Susceptibility to Infections
3.3.5. Langerhans Cells and TJs
3.3.6. Inflammatory Cytokines and TJs
3.3.7. TJs and Potential Therapeutic Interventions
3.4. Future Perspectives
3.5. Limitations
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AD | atopic dermatitis |
| TJs | tight junctions |
| Cldn | claudin |
| JAM-A | junctional adhesion molecule A |
| TAMP | TJ-associated marvel proteins |
| Ocln | occluding |
| ZO | zonula occludens |
| SC | stratum corneum |
| SB | stratum basale |
| SG | stratum granulosum |
| SSP | stratum spinosum |
| HF | hair follicles |
| SNP | single nucleotide polymorphism |
| DC | Dendritic cell |
| LC | Langerhans cell |
References
- Bylund, S.; Kobyletzki, L.; Svalstedt, M. Prevalence and Incidence of Atopic Dermatitis: A Systematic Review. Acta Derm. Venereol. 2020, 100, adv00160. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhang, Z.; Zhang, H.; Guo, Y.; Yao, Z. Update on the Pathogenesis and Therapy of Atopic Dermatitis. Clin. Rev. Allergy Immunol. 2021, 61, 324–338. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, K.; Kubo, A.; Fujita, H.; Yokouchi, M.; Ishii, K.; Kawasaki, H.; Nomura, T.; Shimizu, H.; Kouyama, K.; Ebihara, T.; et al. Distinct Behavior of Human Langerhans Cells and Inflammatory Dendritic Epidermal Cells at Tight Junctions in Patients with Atopic Dermatitis. J. Allergy Clin. Immunol. 2014, 134, 856–864. [Google Scholar] [CrossRef] [PubMed]
- Bäsler, K.; Bergmann, S.; Heisig, M.; Naegel, A.; Zorn-Kruppa, M.; Brandner, J.M. The Role of Tight Junctions in Skin Barrier Function and Dermal Absorption. J. Control. Release 2016, 242, 105–118. [Google Scholar] [CrossRef] [PubMed]
- Kirschner, N.; Rosenthal, R.; Günzel, D.; Moll, I.; Brandner, J.M. Tight Junctions and Differentiation—A Chicken or the Egg Question? Exp. Dermatol. 2012, 21, 171–175. [Google Scholar] [CrossRef] [PubMed]
- Sasson, E.; Anzi, S.; Bell, B.; Yakovian, O.; Zorsky, M.; Deutsch, U.; Engelhardt, B.; Sherman, E.; Vatine, G.; Dzikowski, R.; et al. Nano-scale architecture of blood-brain barrier tight-junctions. eLife 2021, 10, e63253. [Google Scholar] [CrossRef]
- Paradis, T.; Bègue, H.; Basmaciyan, L.; Dalle, F.; Bon, F. Tight Junctions as a Key for Pathogens Invasion in Intestinal Epithelial Cells. Int. J. Mol. Sci. 2021, 22, 2506. [Google Scholar] [CrossRef]
- Tsukita, S.; Furuse, M.; Itoh, M. Multifunctional Strands in Tight Junctions. Nat. Rev. Mol. Cell Biol. 2001, 2, 285–293. [Google Scholar] [CrossRef]
- De Benedetto, A.; Slifka, M.K.; Rafaels, N.M.; Kuo, I.-H.; Georas, S.N.; Boguniewicz, M.; Hata, T.; Schneider, L.C.; Hanifin, J.M.; Gallo, R.L.; et al. Reductions in Claudin-1 May Enhance Susceptibility to Herpes Simplex Virus 1 Infections in Atopic Dermatitis. J. Allergy Clin. Immunol. 2011, 128, 242–246.e5. [Google Scholar] [CrossRef] [Green Version]
- Zihni, C.; Mills, C.; Matter, K.; Balda, M.S. Tight Junctions: From Simple Barriers to Multifunctional Molecular Gates. Nat. Rev. Mol. Cell Biol. 2016, 17, 564–580. [Google Scholar] [CrossRef]
- Tamura, A.; Tsukita, S. Paracellular Barrier and Channel Functions of TJ Claudins in Organizing Biological Systems: Advances in the Field of Barriology Revealed in Knockout Mice. Semin. Cell Dev. Biol. 2014, 36, 177–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandner, J.; Zorn-Kruppa, M.; Yoshida, T.; Moll, I.; Beck, L.; De Benedetto, A. Epidermal Tight Junctions in Health and Disease. Tissue Barriers 2014, 3, e974451. [Google Scholar] [CrossRef] [Green Version]
- Morita, K.; Miyachi, Y. Tight Junctions in the Skin. J. Dermatol. Sci. 2003, 31, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Kirschner, N.; Brandner, J.M. Barriers and More: Functions of Tight Junction Proteins in the Skin. Ann. New York Acad. Sci. 2012, 1257, 158–166. [Google Scholar] [CrossRef] [PubMed]
- Tokumasu, R.; Yamaga, K.; Yamazaki, Y.; Murota, H.; Suzuki, K.; Tamura, A.; Bando, K.; Furuta, Y.; Katayama, I.; Tsukita, S. Dose-Dependent Role of Claudin-1 in Vivo in Orchestrating Features of Atopic Dermatitis. Proc. Natl. Acad. Sci. USA 2016, 113, E4061–E4068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kubo, T.; Sugimoto, K.; Kojima, T.; Sawada, N.; Sato, N.; Ichimiya, S. Tight Junction Protein Claudin-4 Is Modulated via ΔNp63 in Human Keratinocytes. Biochem. Biophys. Res. Commun. 2014, 455, 205–211. [Google Scholar] [CrossRef]
- Zorn-Kruppa, M.; Vidal-y-Sy, S.; Houdek, P.; Wladykowski, E.; Grzybowski, S.; Gruber, R.; Gorzelanny, C.; Harcup, J.; Schneider, S.W.; Majumdar, A.; et al. Tight Junction Barriers in Human Hair Follicles—Role of Claudin-1. Sci. Rep. 2018, 8, 12800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashimoto, K. Intercellular spaces of the human epidermis as demonstrated with lanthanum. J. Investig. Dermatol. 1971, 57, 17–31. [Google Scholar] [CrossRef] [Green Version]
- Yamaga, K.; Murota, H.; Tamura, A.; Miyata, H.; Ohmi, M.; Kikuta, J.; Ishii, M.; Tsukita, S.; Katayama, I. Claudin-3 Loss Causes Leakage of Sweat from the Sweat Gland to Contribute to the Pathogenesis of Atopic Dermatitis. J. Investig. Dermatol. 2018, 138, 1279–1287. [Google Scholar] [CrossRef] [Green Version]
- Tsukita, S.; Furuse, M. Claudin-Based Barrier in Simple and Stratified Cellular Sheets. Curr. Opin. Cell Biol. 2002, 14, 531–536. [Google Scholar] [CrossRef]
- Akiyama, T.; Niyonsaba, F.; Kiatsurayanon, C.; Nguyen, T.T.; Ushio, H.; Fujimura, T.; Ueno, T.; Okumura, K.; Ogawa, H.; Ikeda, S. The Human Cathelicidin LL-37 Host Defense Peptide Upregulates Tight Junction-Related Proteins and Increases Human Epidermal Keratinocyte Barrier Function. J. Innate Immun. 2014, 6, 739–753. [Google Scholar] [CrossRef] [PubMed]
- Sugita, K.; Kabashima, K. Tight Junctions in the Development of Asthma, Chronic Rhinosinusitis, Atopic Dermatitis, Eosinophilic Esophagitis, and Inflammatory Bowel Diseases. J. Leukoc. Biol. 2020, 107, 749–762. [Google Scholar] [CrossRef] [PubMed]
- Kuo, I.-H.; Carpenter-Mendini, A.; Yoshida, T.; McGirt, L.Y.; Ivanov, A.I.; Barnes, K.C.; Gallo, R.L.; Borkowski, A.W.; Yamasaki, K.; Leung, D.Y.; et al. Activation of Epidermal Toll-like Receptor 2 Enhances Tight Junction Function: Implications for Atopic Dermatitis and Skin Barrier Repair. J. Investig. Dermatol. 2013, 133, 988–998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuki, T.; Komiya, A.; Kusaka, A.; Kuze, T.; Sugiyama, Y.; Inoue, S. Impaired Tight Junctions Obstruct Stratum Corneum Formation by Altering Polar Lipid and Profilaggrin Processing. J. Dermatol. Sci. 2013, 69, 148–158. [Google Scholar] [CrossRef]
- Yokouchi, M.; Kubo, A.; Kawasaki, H.; Yoshida, K.; Ishii, K.; Furuse, M.; Amagai, M. Epidermal Tight Junction Barrier Function Is Altered by Skin Inflammation, but Not by Filaggrin-Deficient Stratum Corneum. J. Dermatol. Sci. 2015, 77, 28–36. [Google Scholar] [CrossRef]
- De Benedetto, A.; Rafaels, N.M.; Leung, D.Y.M.; Ivanov, A.I.; Hand, T.; Gao, L.; Yang, M.; Boguniewicz, M.; Hata, T.R.; Schneider, L. Variants in the Tight Junction Gene, Claudin-1 are Associated with Atopic Dermatitis in Two American Populations and May Contribute to Skin Barrier Dysfunction. J. Allergy Clin. Immunol. 2009, 123, S150. [Google Scholar] [CrossRef]
- Asad, S.; Winge, M.C.G.; Wahlgren, C.-F.; Bilcha, K.D.; Nordenskjöld, M.; Taylan, F.; Bradley, M. The Tight Junction Gene Claudin-1 Is Associated with Atopic Dermatitis among Ethiopians. J. Eur. Acad. Dermatol. Venereol. JEADV 2016, 30, 1939–1941. [Google Scholar] [CrossRef]
- Yu, H.-S.; Kang, M.-J.; Kwon, J.-W.; Lee, S.-Y.; Lee, E.; Yang, S.-I.; Jung, Y.-H.; Hong, K.; Kim, Y.-J.; Lee, S.-H.; et al. Claudin-1 Polymorphism Modifies the Effect of Mold Exposure on the Development of Atopic Dermatitis and Production of IgE. J. Allergy Clin. Immunol. 2015, 135, 827–830.e5. [Google Scholar] [CrossRef]
- Sugawara, T.; Iwamoto, N.; Akashi, M.; Kojima, T.; Hisatsune, J.; Sugai, M.; Furuse, M. Tight Junction Dysfunction in the Stratum Granulosum Leads to Aberrant Stratum Corneum Barrier Function in Claudin-1-Deficient Mice. J. Dermatol. Sci. 2013, 70, 12–18. [Google Scholar] [CrossRef]
- Brandner, J. Organization and Formation of the Tight Junction System in Human Epidermis and Cultured Keratinocytes. Eur. J. Cell Biol. 2002, 81, 253–263. [Google Scholar] [CrossRef]
- Bäsler, K.; Brandner, J.M. Tight Junctions in Skin Inflammation. Pflügers Arch. Eur. J. Physiol. 2016, 469, 3–14. [Google Scholar] [CrossRef] [PubMed]
- De Benedetto, A.; Rafaels, N.M.; McGirt, L.Y.; Ivanov, A.I.; Georas, S.N.; Cheadle, C.; Berger, A.E.; Zhang, K.; Vidyasagar, S.; Yoshida, T.; et al. Tight Junction Defects in Patients with Atopic Dermatitis. J. Allergy Clin. Immunol. 2011, 127, 773–786.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DE Benedetto, A.; Latchney, L.; McGirt, L.; Vidyasagar, S.; Cheadle, C.; Barnes, K.; Beck, L. The Tight Junction Protein, Claudin-1 Is Dysregulated in Atopic Dermatitis. J. Allergy Clin. Immunol. 2008, 121, S32. [Google Scholar] [CrossRef]
- Bergmann, S.; von Buenau, B.; Vidal-y-Sy, S.; Haftek, M.; Wladykowski, E.; Houdek, P.; Lezius, S.; Duplan, H.; Bäsler, K.; Dähnhardt-Pfeiffer, S.; et al. Claudin-1 Decrease Impacts Epidermal Barrier Function in Atopic Dermatitis Lesions Dose-Dependently. Sci. Rep. 2020, 10, 2024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gruber, R.; Börnchen, C.; Rose, K.; Daubmann, A.; Volksdorf, T.; Wladykowski, E.; Vidal-y-Sy, S.; Peters, E.M.; Danso, M.; Bouwstra, J.A.; et al. Diverse Regulation of Claudin-1 and Claudin-4 in Atopic Dermatitis. Am. J. Pathol. 2015, 185, 2777–2789. [Google Scholar] [CrossRef] [PubMed]
- Esaki, H.; Ewald, D.A.; Ungar, B.; Rozenblit, M.; Zheng, X.; Xu, H.; Estrada, Y.D.; Peng, X.; Mitsui, H.; Litman, T.; et al. Identification of Novel Immune and Barrier Genes in Atopic Dermatitis by Means of Laser Capture Microdissection. J. Allergy Clin. Immunol. 2015, 135, 153–163. [Google Scholar] [CrossRef] [Green Version]
- Yuki, T.; Tobiishi, M.; Kusaka-Kikushima, A.; Ota, Y.; Tokura, Y. Impaired Tight Junctions in Atopic Dermatitis Skin and in a Skin-Equivalent Model Treated with Interleukin-17. PLoS ONE 2016, 11, e0161759. [Google Scholar] [CrossRef] [Green Version]
- Hu, X.Q.; Tang, Y.; Ju, Y.; Zhang, X.Y.; Yan, J.J.; Wang, C.M.; Yang, Y.; Zhu, C.; Tang, Z.X.; Zhou, Y.; et al. Scratching Damages Tight Junctions through the Akt–Claudin 1 Axis in Atopic Dermatitis. Clin. Exp. Dermatol. 2020, 46, 74–81. [Google Scholar] [CrossRef]
- Rizzo, J.M.; Oyelakin, A.; Min, S.; Smalley, K.; Bard, J.; Luo, W.; Nyquist, J.; Guttman-Yassky, E.; Yoshida, T.; De Benedetto, A.; et al. ΔΝp63 regulates IL-33 and IL-31 signaling in atopic dermatitis. Cell Death Differ. 2016, 23, 1073–1085. [Google Scholar] [CrossRef]
- Kojima, T.; Kohno, T.; Kubo, T.; Kaneko, Y.; Kakuki, T.; Kakiuchi, A.; Kurose, M.; Takano, K.; Ogasawara, N.; Obata, K.; et al. Regulation of Claudin-4 via P63 in Human Epithelial Cells. Ann. New York Acad. Sci. 2017, 1405, 25–31. [Google Scholar] [CrossRef]
- De Benedetto, A.; Rafaels, N.M.; Dubois, M.; Bolognino, M.; Leung, D.Y.; Barnes, K.C.; Slifka, M.K.; Beck, L.A. Reductions in an Epidermal Tight Junction Protein Enhance Atopic Dermatitis Subjects’ Susceptibility to HSV Infections. J. Allergy Clin. Immunol. 2011, 127, AB137. [Google Scholar] [CrossRef]
- Geoghegan, J.A.; Irvine, A.D.; Foster, T.J. Staphylococcus aureus and atopic dermatitis: A complex and evolving relationship. Trends Microbiol. 2018, 26, 484–497, ISSN 0966-842X. [Google Scholar] [CrossRef] [PubMed]
- Bäsler, K.; Galliano, M.-F.; Bergmann, S.; Rohde, H.; Wladykowski, E.; Vidal-y-Sy, S.; Guiraud, B.; Houdek, P.; Schüring, G.; Volksdorf, T.; et al. Biphasic Influence Of Staphylococcus Aureuson Human Epidermal Tight Junctions. Ann. N. Y. Acad. Sci. 2017, 1405, 53–70. [Google Scholar] [CrossRef]
- Van Dalen, R.; De La Cruz Diaz, J.S.; Rumpret, M.; Fuchsberger, F.F.; van Teijlingen, N.H.; Hanske, J.; van Sorge, N.M. Langerhans Cells Sense Staphylococcus aureus Wall Teichoic Acid through Langerin To Induce Inflammatory Responses. mBio 2019, 10, e00330-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizutani, Y.; Takagi, N.; Nagata, H.; Inoue, S. Interferon-γ Downregulates Tight Junction Function, Which Is Rescued by Interleukin-17A. Exp. Dermatol. 2021, 30, 1754–1763. [Google Scholar] [CrossRef] [PubMed]
- Brewer, M.G.; Yoshida, T.; Kuo, F.I.; Fridy, S.; Beck, L.A.; De Benedetto, A. Antagonistic Effects of IL-4 on IL-17A-Mediated Enhancement of Epidermal Tight Junction Function. Int. J. Mol. Sci. 2019, 20, 4070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryu, W.-I.; Lee, H.; Bae, H.C.; Jeon, J.; Ryu, H.J.; Kim, J.; Kim, J.H.; Son, J.W.; Kim, J.; Imai, Y.; et al. IL-33 Down-Regulates CLDN1 Expression through the ERK/STAT3 Pathway in Keratinocytes. J. Dermatol. Sci. 2018, 90, 313–322. [Google Scholar] [CrossRef] [Green Version]
- Beck, L.A.; Cork, M.J.; Amagai, M.; De Benedetto, A.; Kabashima, K.; Hamilton, J.D.; Rossi, A.B. Type 2 Inflammation Contributes To Skin Barrier Dysfunction In Atopic Dermatitis. JID Innov. 2022, 2, 100131. [Google Scholar] [CrossRef]
- Peltonen, S.; Riehokainen, J.; Pummi, K.; Peltonen, J. Tight junction components occludin, ZO-1, and Claudin-1, -4 and -5 in active and healing psoriasis. Br. J. Dermatol. 2007, 156, 466–472. [Google Scholar] [CrossRef]
- Donetti, E.; Cornaghi, L.; Gualerzi, A.; Baruffaldi Preis, F.W.; Prignano, F. An innovative three-dimensional model of normal human skin to study the proinflammatory psoriatic effect of tumor necrosis factor-alpha and interleukin-17. Cytocine 2014, 68, 1–8. [Google Scholar] [CrossRef]
- Gutowska-Owsiak, D.; Schaupp, A.L.; Salimi, M.; Selvakumar, T.A.; McPherson, T.; Taylor, S.; Ogg, G.S. IL-17 downregulates fillagrin and affects keratinocyte expression of genes associated with cellular adhesion. Exp. Dermatol. 2012, 21, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-S.; Ryu, H.W.; Yang, W.-K.; Park, M.H.; Park, Y.-C.; Kim, D.-Y.; Kwon, H.J.; Kim, S.-Y.; Oh, S.-R.; Kim, S.-H. A Combination of Olea Europaea Leaf Extract and Spirodela Polyrhiza Extract Alleviates Atopic Dermatitis by Modulating Immune Balance and Skin Barrier Function in a 1-Chloro-2,4-Dinitrobenzene-Induced Murine Model. Phytomedicine 2021, 82, 153407. [Google Scholar] [CrossRef] [PubMed]
- Galliano, M.F.; Bäsler, K.; Caruana, A.; Mias, C.; Bessou-Touya, S.; Brandner, J.M.; Duplan, H. Protective Effect of Aquaphilus Dolomiae Extract-G1, ADE-G1, on Tight Junction Barrier Function in a Staphylococcus Aureus -Infected Atopic Dermatitis Model. J. Eur. Acad. Dermatol. Venereol. 2020, 34, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.; Matsushita, K.; Wang, J.; Kanekura, T. Topical Glucose Induces Claudin-1 and Filaggrin Expression in a Mouse Model of Atopic Dermatitis and in Keratinocyte Culture, Exerting Anti-Inflammatory Effects by Repairing Skin Barrier Function. Acta Derm. Venereol. 2018, 98, 19–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Jiang, X.; Yu, X.; Liu, H.; Tao, Y.; Jiang, G.; Hong, M. Cimifugin Suppresses Allergic Inflammation by Reducing Epithelial Derived Initiative Key FactorsViaRegulating Tight Junctions. J. Cell. Mol. Med. 2017, 21, 2926–2936. [Google Scholar] [CrossRef]
- Na, K.; Lkhagva-Yondon, E.; Kim, M.; Lim, Y.; Shin, E.; Lee, C.; Jeon, M. Oral Treatment with Aloe Polysaccharide Ameliorates Ovalbumin-Induced Atopic Dermatitis by Restoring Tight Junctions in Skin. Scand. J. Immunol. 2019, 91, e12856. [Google Scholar] [CrossRef]
- Anagawa-Nakamura, A.; Ryoke, K.; Yasui, Y.; Shoda, T.; Sugai, S. Effects of Delgocitinib Ointment 0.5% on the Normal Mouse Skin and Epidermal Tight Junction Proteins in Comparison with Topical Corticosteroids. Toxicol. Pathol. 2020, 48, 1008–1016. [Google Scholar] [CrossRef]
- Kim, Y.-E.; Cho, N.; Cheon, S.; Kim, K.K. Bortezomib, a Proteasome Inhibitor, Alleviates Atopic Dermatitis by Increasing Claudin 1 Protein Expression. Biochem. Biophys. Res. Commun. 2017, 493, 744–750. [Google Scholar] [CrossRef]
- Lee, S.E.; Choi, Y.; Kim, S.-E.; Noh, E.B.; Kim, S.-C. Differential Effects of Topical Corticosteroid and Calcineurin Inhibitor on the Epidermal Tight Junction. Exp. Dermatol. 2012, 22, 59–61. [Google Scholar] [CrossRef]
- Rozenblit, M.; Suarez-Farinas, M.; Shemer, A.; Khattri, S.; Gilleaudeau, P.; Sullivan-Whalen, M.; Zheng, X.; Xu, H.; Cardinale, I.; Krueger, J.G.; et al. Residual genomic profile cyclosporine treatment may offer insights into atopic dermatitis reoccurrence. J. Allergy Clin. Immunol. 2014, 134, 955–957. [Google Scholar] [CrossRef]
- Guttman-Yassky, E.; Bissonnette, R.; Ungar, B.; Suarez-Farinas, M.; Ardeleanu, M.; Esaki, H.; Suprun, M.; Estrada, Y.; Xu, H.; Peng, X.; et al. Dupilumab progressively improves systemic and cutaneous abnormalities in patients with atopic dermatitis. J. Allergy Clin. Immunol. 2019, 143, 155–172. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Katsarou, S.; Makris, M.; Vakirlis, E.; Gregoriou, S. The Role of Tight Junctions in Atopic Dermatitis: A Systematic Review. J. Clin. Med. 2023, 12, 1538. https://doi.org/10.3390/jcm12041538
Katsarou S, Makris M, Vakirlis E, Gregoriou S. The Role of Tight Junctions in Atopic Dermatitis: A Systematic Review. Journal of Clinical Medicine. 2023; 12(4):1538. https://doi.org/10.3390/jcm12041538
Chicago/Turabian StyleKatsarou, Spyridoula, Michael Makris, Efstratios Vakirlis, and Stamatios Gregoriou. 2023. "The Role of Tight Junctions in Atopic Dermatitis: A Systematic Review" Journal of Clinical Medicine 12, no. 4: 1538. https://doi.org/10.3390/jcm12041538