

Chondrosarcoma: A Clinical Review

Abstract
:1. Introduction
Epidemiology
2. Clinical Presentation
2.1. Location
2.2. Signs and Symptoms
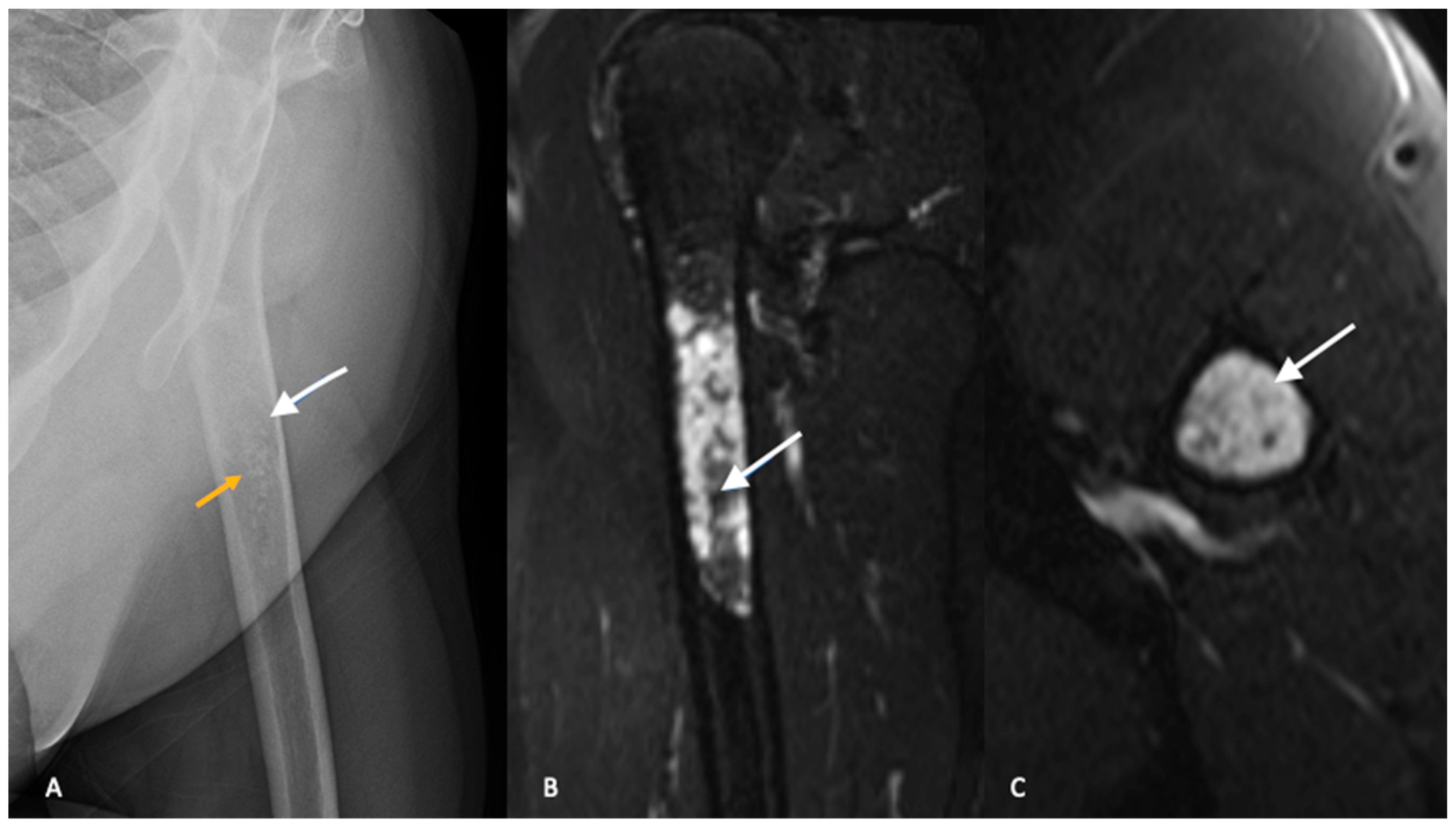
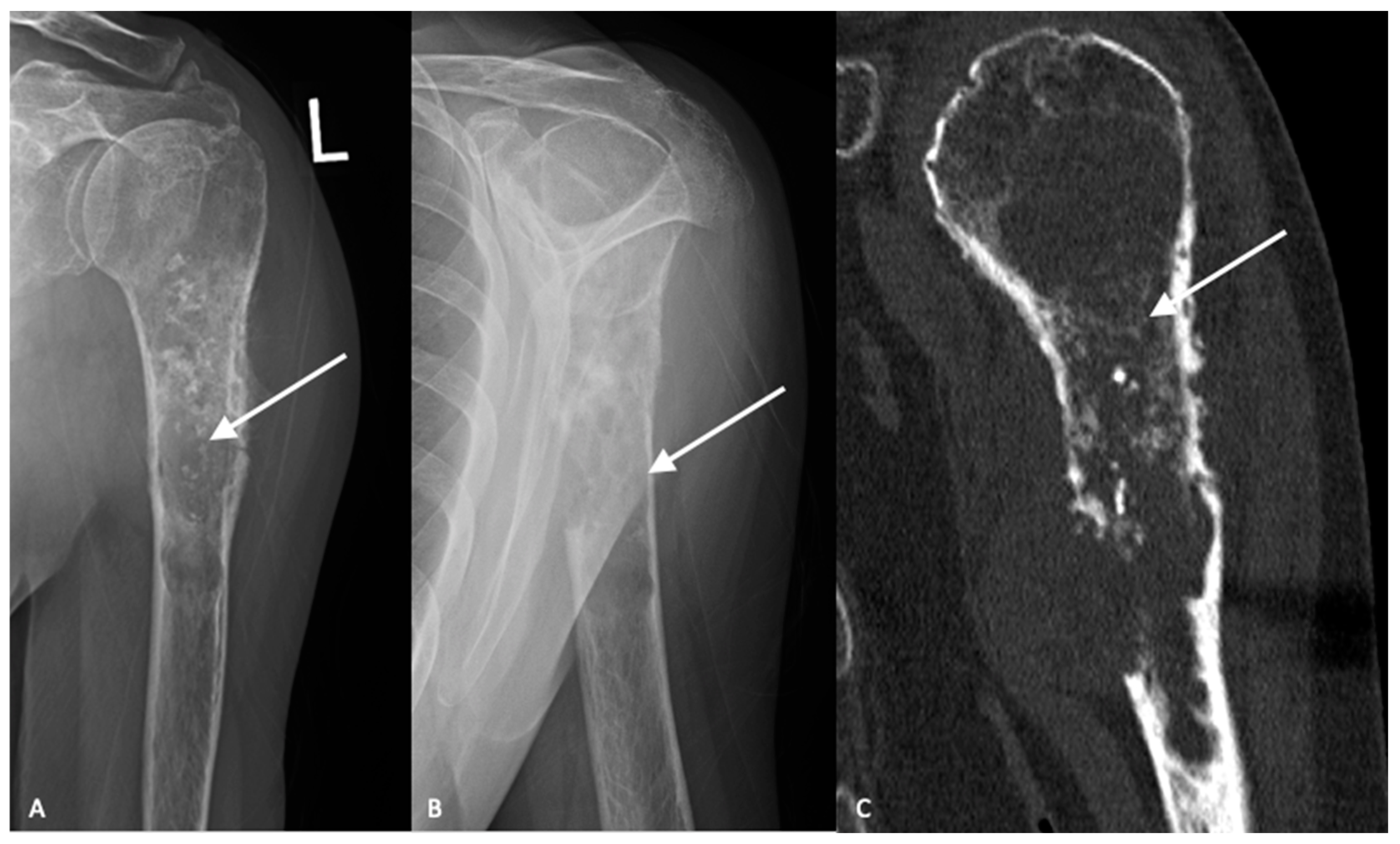
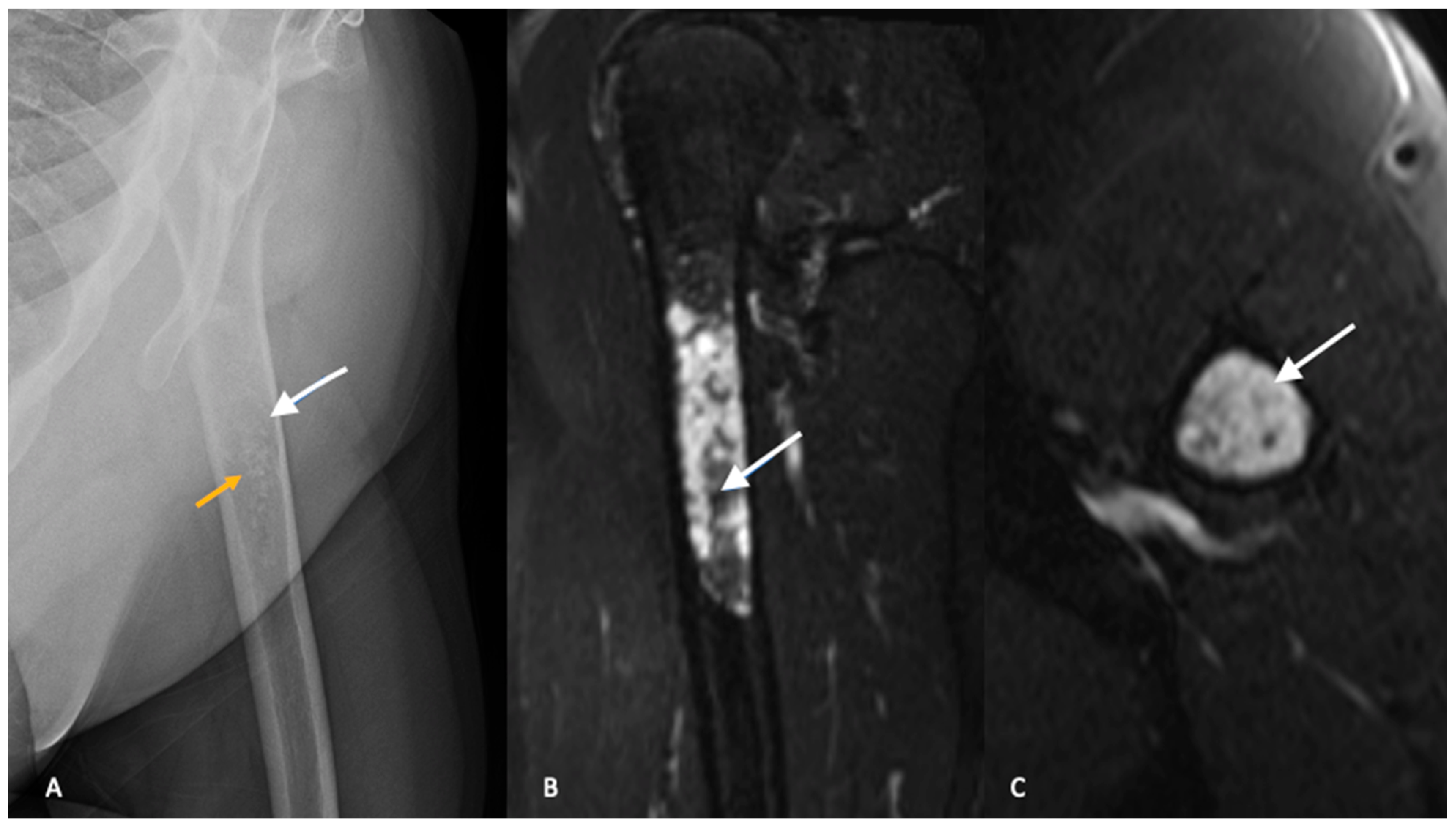
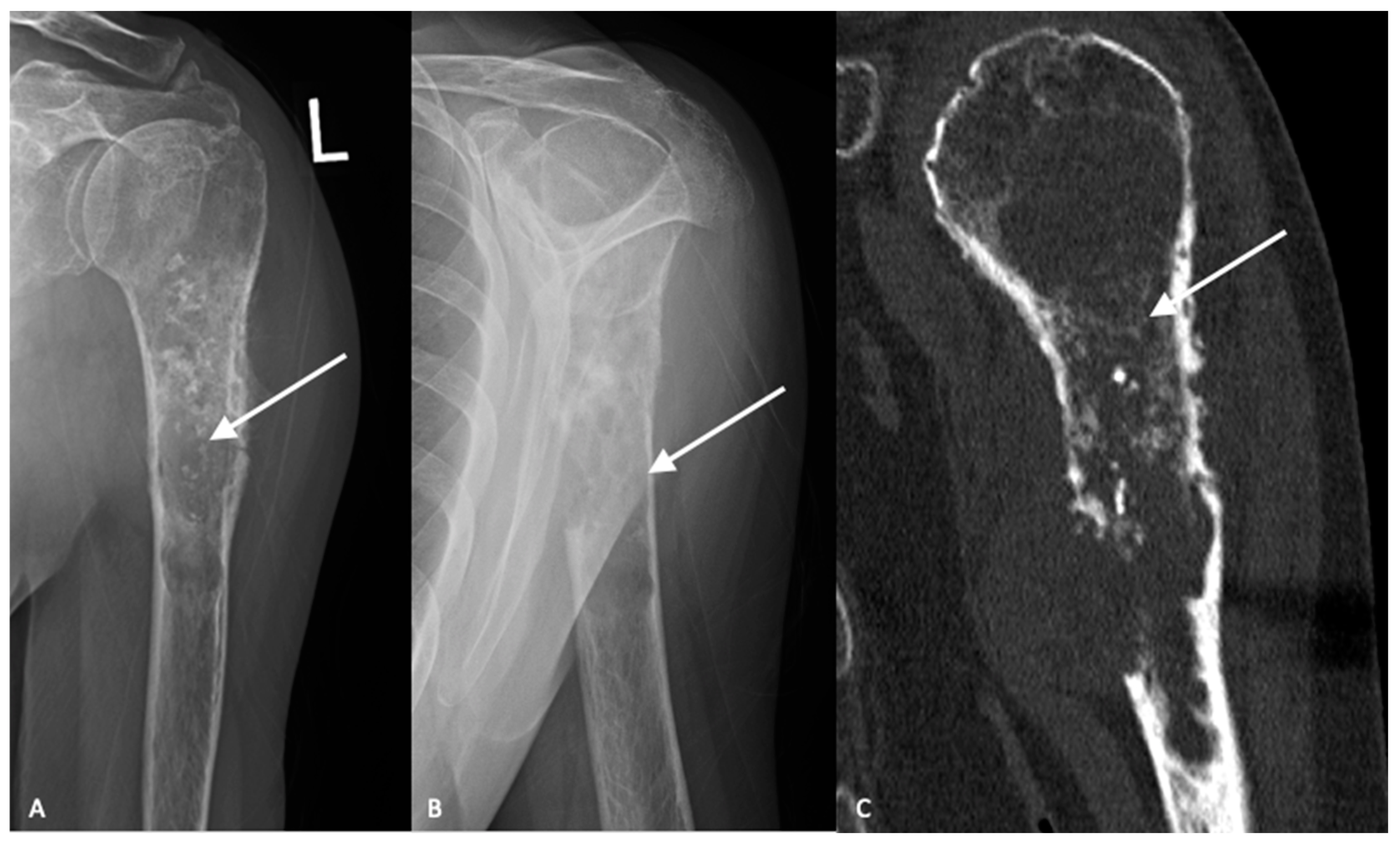
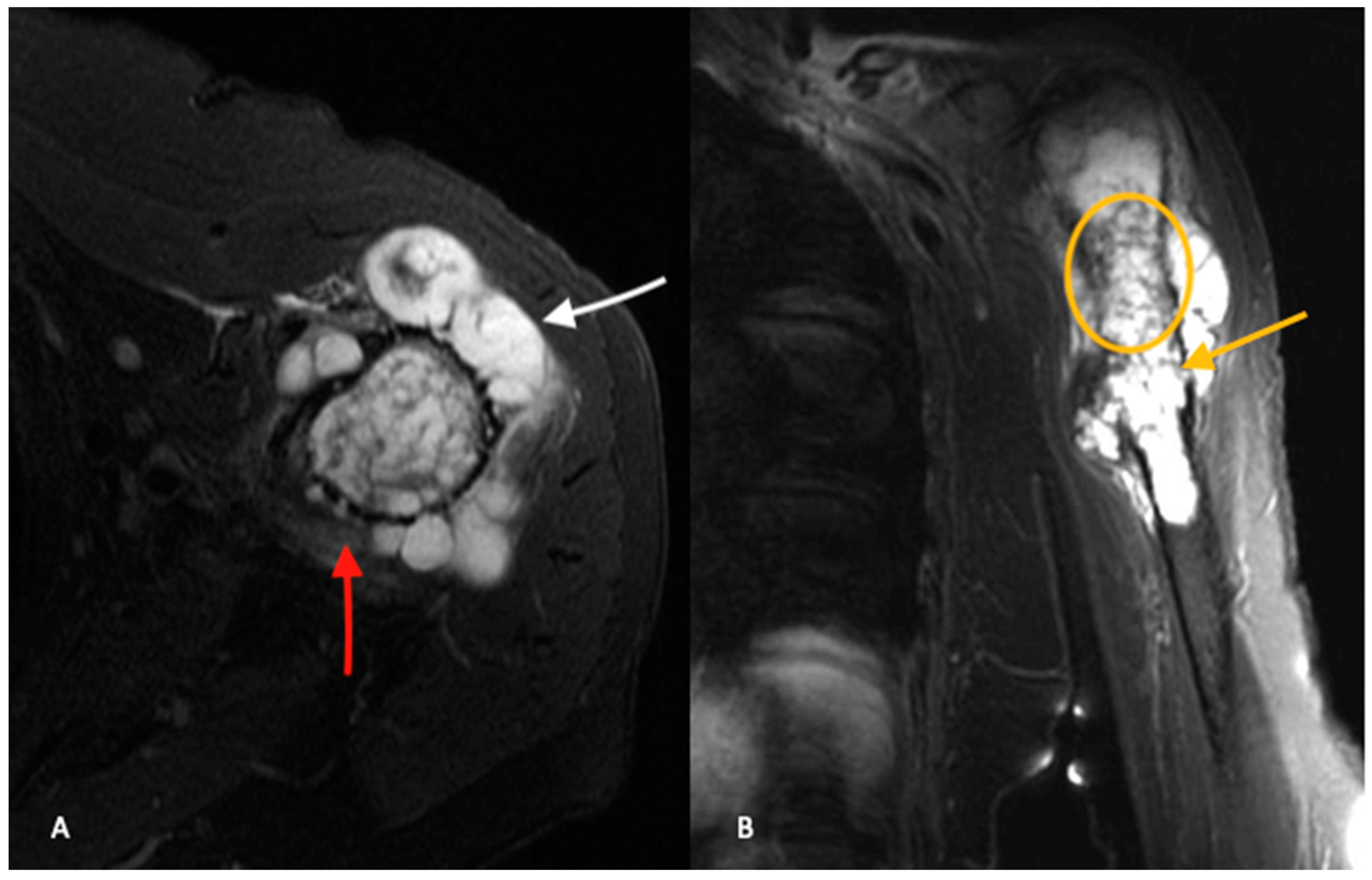
3. Imaging
3.1. Plain Radiographs
3.2. Cross-Sectional Imaging
4. Diagnosis and Staging
4.1. Biopsy
4.2. Staging
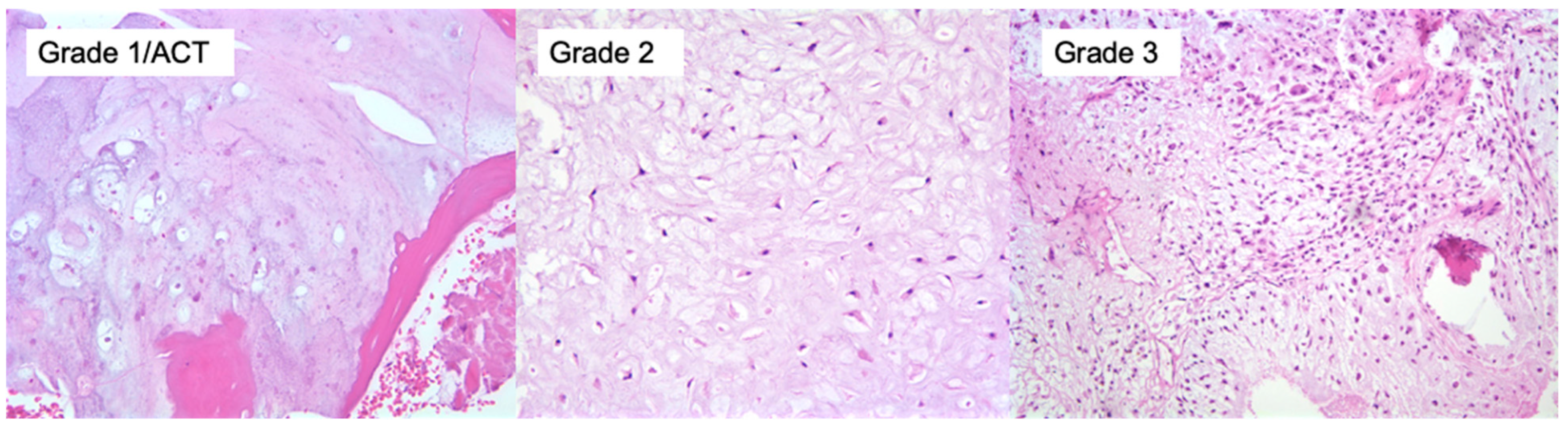
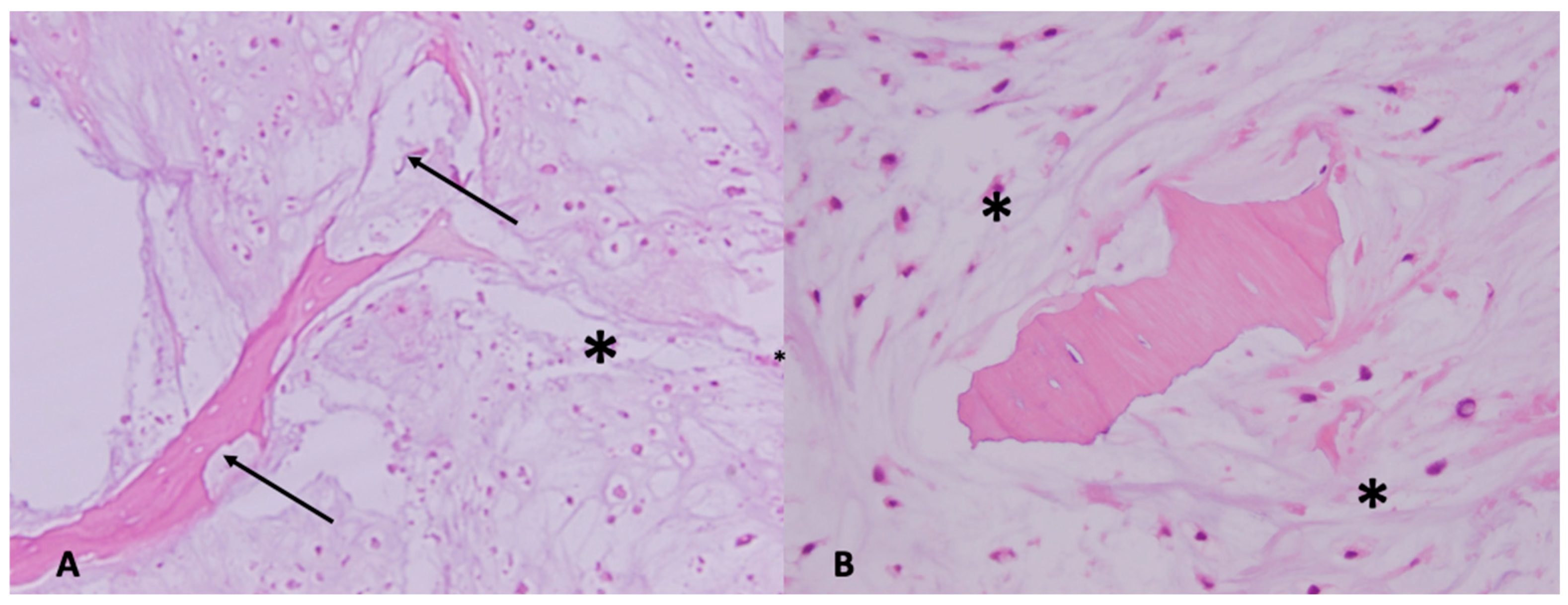
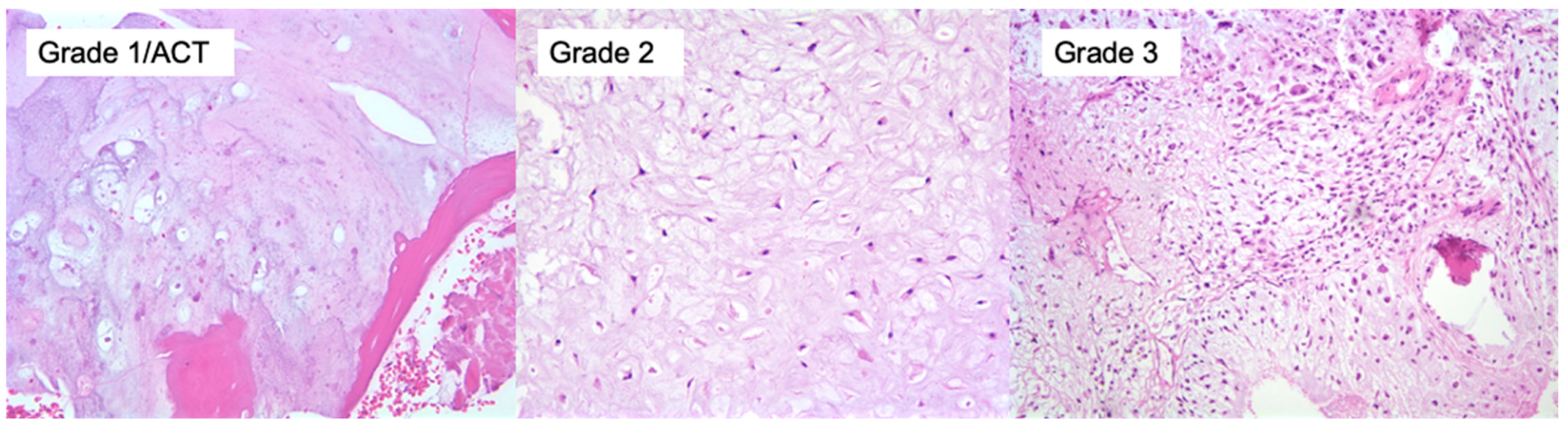
5. Subtypes, Diagnostic and Molecular Pathology
5.1. Conventional Central Chondrosarcoma
5.2. Secondary Chondrosarcomas
5.3. Rare Subtypes
6. Molecular Characteristics
7. Management
7.1. Intrapelvic Tumors
7.2. Atypical Cartilaginous Tumors
8. Metastatic Disease
Systemic Therapy and Advanced Disease
9. Prognosis
Subtypes
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Anfinsen, K.P.; Devesa, S.S.; Bray, F.; Troisi, R.; Jonasdottir, T.J.; Bruland, O.S.; Grotmol, T. Age-Period-Cohort Analysis of Primary Bone Cancer Incidence Rates in the United States (1976–2005). Cancer Epidemiol. Prev. Biomark. 2011, 20, 1770–1777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giuffrida, A.Y.; Burgueno, J.E.; Koniaris, L.G.; Gutierrez, J.C.; Duncan, R.; Scully, S.P. Chondrosarcoma in the United States (1973 to 2003): An Analysis of 2890 Cases from the SEER Database. JBJS 2009, 91, 1063–1072. [Google Scholar] [CrossRef] [PubMed]
- Thorkildsen, J.; Taksdal, I.; Bjerkehagen, B.; Haugland, H.K.; Børge Johannesen, T.; Viset, T.; Norum, O.-J.; Bruland, Ø.; Zaikova, O. Chondrosarcoma in Norway 1990–2013; an Epidemiological and Prognostic Observational Study of a Complete National Cohort. Acta Oncol. 2019, 58, 273–282. [Google Scholar] [CrossRef] [Green Version]
- Van Praag, V.M.; Rueten-Budde, A.J.; Ho, V.; Dijkstra, P.D.S.; van der Geest, I.C.; Bramer, J.A.; Schaap, G.R.; Jutte, P.C.; Schreuder, H.B.; Ploegmakers, J.J.W. Incidence, Outcomes and Prognostic Factors during 25 Years of Treatment of Chondrosarcomas. Surg. Oncol. 2018, 27, 402–408. [Google Scholar] [CrossRef]
- Damron, T.A.; Ward, W.G.; Stewart, A. Osteosarcoma, Chondrosarcoma, and Ewing’s Sarcoma: National Cancer Data Base Report. Clin. Orthop. Relat. Res. 2007, 459, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Collins, M.S.; Koyama, T.; Swee, R.G.; Inwards, C.Y. Clear Cell Chondrosarcoma: Radiographic, Computed Tomographic, and Magnetic Resonance Findings in 34 Patients with Pathologic Correlation. Skelet. Radiol. 2003, 32, 687–694. [Google Scholar] [CrossRef] [PubMed]
- Shakked, R.J.; Geller, D.S.; Gorlick, R.; Dorfman, H.D. Mesenchymal Chondrosarcoma: Clinicopathologic Study of 20 Cases. Arch. Pathol. Amp Lab. Med. 2012, 136, 61. [Google Scholar] [CrossRef] [Green Version]
- Leddy, L.R.; Holmes, R.E. Chondrosarcoma of Bone. In Orthopaedic Oncology; Springer: Berlin/Heidelberg, Germany, 2014; pp. 117–130. [Google Scholar]
- Rougraff, B.T.; Davis, K.; Lawrence, J. Does Length of Symptoms Before Diagnosis of Sarcoma Affect Patient Survival? Clin. Orthop. Relat. Res. 2007, 462, 181–189. [Google Scholar] [CrossRef]
- Albergo, J.I.; Gaston, C.L.; Jeys, L.M.; Khajuria, A.; Carter, S.R.; Tillman, R.M.; Abudu, A.T.; Grimer, R.J. Management and Prognostic Significance of Pathological Fractures through Chondrosarcoma of the Femur. Int. Orthop. SICOT 2015, 39, 943–946. [Google Scholar] [CrossRef]
- Lin, P.P.; Moussallem, C.D.; Deavers, M.T. Secondary Chondrosarcoma. JAAOS J. Am. Acad. Orthop. Surg. 2010, 18, 608–615. [Google Scholar] [CrossRef]
- Gutteridge, A.; Rathbone, V.M.; Gibbons, R.; Bi, M.; Archard, N.; Davies, K.E.J.; Brown, J.; Plagnol, V.; Pillay, N.; Amary, F.; et al. Digital PCR Analysis of Circulating Tumor DNA: A Biomarker for Chondrosarcoma Diagnosis, Prognostication, and Residual Disease Detection. Cancer Med. 2017, 6, 2194–2202. [Google Scholar] [CrossRef]
- Douis, H.; Saifuddin, A. The Imaging of Cartilaginous Bone Tumours. II. Chondrosarcoma. Skelet. Radiol. 2013, 42, 611–626. [Google Scholar] [CrossRef]
- Geirnaerdt, M.J.; Hermans, J.; Bloem, J.L.; Kroon, H.M.; Pope, T.L.; Taminiau, A.H.; Hogendoorn, P.C. Usefulness of Radiography in Differentiating Enchondroma from Central Grade 1 Chondrosarcoma. AJR. Am. J. Roentgenol. 1997, 169, 1097–1104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skeletal Lesions Interobserver Correlation among Expert Diagnosticians (SLICED) Study Group. Reliability of Histopathologic and Radiologic Grading of Cartilaginous Neoplasms in Long Bones. JBJS 2007, 89, 2113–2123. [Google Scholar] [CrossRef]
- Biermann, J.S.; Chow, W.; Reed, D.R.; Lucas, D.; Adkins, D.R.; Agulnik, M.; Benjamin, R.S.; Brigman, B.; Budd, G.T.; Curry, W.T. NCCN Guidelines Insights: Bone Cancer, Version 2.2017. J. Natl. Compr. Cancer Netw. 2017, 15, 155–167. [Google Scholar] [CrossRef] [Green Version]
- Casali, P.G.; Bielack, S.; Abecassis, N.; Aro, H.T.; Bauer, S.; Biagini, R.; Bonvalot, S.; Boukovinas, I.; Bovee, J.; Brennan, B. Bone Sarcomas: ESMO–PaedCan–EURACAN Clinical Practice Guidelines for Diagnosis, Treatment and Follow-Up. Ann. Oncol. 2018, 29, iv79–iv95. [Google Scholar] [CrossRef]
- Deckers, C.; Steyvers, M.J.; Hannink, G.; Schreuder, H.B.; de Rooy, J.W.; Van Der Geest, I.C. Can MRI Differentiate between Atypical Cartilaginous Tumors and High-Grade Chondrosarcoma? A Systematic Review. Acta Orthop. 2020, 91, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Crim, J.; Schmidt, R.; Layfield, L.; Hanrahan, C.; Manaster, B.J. Can Imaging Criteria Distinguish Enchondroma from Grade 1 Chondrosarcoma? Eur. J. Radiol. 2015, 84, 2222–2230. [Google Scholar] [CrossRef]
- Yoshimura, Y.; Isobe, K.; Arai, H.; Aoki, K.; Kito, M.; Kato, H. Preoperative Radiographic and Histopathologic Evaluation of Central Chondrosarcoma. Arch. Orthop. Trauma Surg. 2013, 133, 1225–1231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsuda, Y.; Gregory, J.J.; Fujiwara, T.; Abudu, S. Secondary Chondrosarcoma Arising from Osteochondroma: Outcomes and Prognostic Factors. Bone Jt. J. 2019, 101, 1313–1320. [Google Scholar] [CrossRef]
- Eefting, D.; Schrage, Y.M.; Geirnaerdt, M.J.; Le Cessie, S.; Taminiau, A.H.; Bovée, J.V.; Hogendoorn, P.C. Assessment of Interobserver Variability and Histologic Parameters to Improve Reliability in Classification and Grading of Central Cartilaginous Tumors. Am. J. Surg. Pathol. 2009, 33, 50–57. [Google Scholar] [CrossRef]
- Hodel, S.; Laux, C.; Farei-Campagna, J.; Götschi, T.; Bode-Lesniewska, B.; Müller, D.A. The Impact of Biopsy Sampling Errors and the Quality of Surgical Margins on Local Recurrence and Survival in Chondrosarcoma. Cancer Manag. Res. 2018, 10, 3765–3771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saifuddin, A.; Oliveira, I.; Singla, N.; Chavda, A.; Khoo, M.; O’Donnell, P. The Importance of MRI Review Following the Diagnosis of Atypical Cartilaginous Tumour Using Image-Guided Needle Biopsy. Skelet. Radiol. 2021, 50, 407–415. [Google Scholar] [CrossRef]
- Pohlig, F.; Kirchhoff, C.; Lenze, U.; Schauwecker, J.; Burgkart, R.; Rechl, H.; von Eisenhart-Rothe, R. Percutaneous Core Needle Biopsy versus Open Biopsy in Diagnostics of Bone and Soft Tissue Sarcoma: A Retrospective Study. Eur. J. Med. Res. 2012, 17, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Saghieh, S.; Masrouha, K.Z.; Musallam, K.M.; Mahfouz, R.; Abboud, M.; Khoury, N.J.; Haidar, R. The Risk of Local Recurrence along the Core-Needle Biopsy Tract in Patients with Bone Sarcomas. Iowa Orthop. J. 2010, 30, 80. [Google Scholar]
- Kiatisevi, P.; Thanakit, V.; Sukunthanak, B.; Boonthatip, M.; Bumrungchart, S.; Witoonchart, K. Computed Tomography-Guided Core Needle Biopsy versus Incisional Biopsy in Diagnosing Musculoskeletal Lesions. J. Orthop. Surg. Hong Kong 2013, 21, 204–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roitman, P.D.; Farfalli, G.L.; Ayerza, M.A.; Múscolo, D.L.; Milano, F.E.; Aponte-Tinao, L.A. Is Needle Biopsy Clinically Useful in Preoperative Grading of Central Chondrosarcoma of the Pelvis and Long Bones? Clin. Orthop. Relat. Res. 2017, 475, 808–814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jennings, R.; Riley, N.; Rose, B.; Rossi, R.; Skinner, J.A.; Cannon, S.R.; Briggs, T.W.; Pollock, R.; Saifuddin, A. An Evaluation of the Diagnostic Accuracy of the Grade of Preoperative Biopsy Compared to Surgical Excision in Chondrosarcoma of the Long Bones. Int. J. Surg. Oncol. 2010, 2010, 270195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Enneking, W.F.; Spanier, S.S.; Goodman, M.A. A System for the Surgical Staging of Musculoskeletal Sarcoma. Clin. Orthop. Relat. Res. 1980, 153, 106–120. [Google Scholar] [CrossRef]
- Amin, M.B.; Edge, S.B. AJCC Cancer Staging Manual; Springer: Berlin/Heidelberg, Germany, 2017. [Google Scholar]
- Czerniak, B. Dorfman and Czerniak’s Bone Tumors E-Book; Elsevier Health Sciences: Amsterdam, The Netherlands, 2015. [Google Scholar]
- Ottesen, T.D.; Shultz, B.N.; Munger, A.M.; Amick, M.; Toombs, C.S.; Friedaender, G.E.; Grauer, J.N. Chondrosarcoma Patient Characteristics, Management, and Outcomes Based on over 5,000 Cases from the National Cancer Database (NCDB). PLoS ONE 2022, 17, e0268215. [Google Scholar] [CrossRef]
- WHO. Soft Tissue and Bone Tumours, 5th ed.; WHO Classification of Tumours Editorial; IARC Press: Lyon, France, 2020; ISBN 978-92-832-4502-5. [Google Scholar]
- Schmale, G.A.; Conrad, E.U., 3rd; Raskind, W.H. The Natural History of Hereditary Multiple Exostoses. JBJS 1994, 76, 986–992. [Google Scholar] [CrossRef]
- Legeai-Mallet, L.; Munnich, A.; Maroteaux, P.; Merrer, M.L. Incomplete Penetrance and Expressivity Skewing in Hereditary Multiple Exostoses. Clin. Genet. 1997, 52, 12–16. [Google Scholar] [CrossRef]
- Amer, K.M.; Munn, M.; Congiusta, D.; Abraham, J.A.; Mallick, A.B. Survival and Prognosis of Chondrosarcoma Subtypes: SEER Database Analysis. J. Orthop. Res. 2020, 38, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Cleven, A.H.; Zwartkruis, E.; Hogendoorn, P.C.; Kroon, H.M.; Briaire-de Bruijn, I.; Bovée, J.V. Periosteal Chondrosarcoma: A Histopathological and Molecular Analysis of a Rare Chondrosarcoma Subtype. Histopathology 2015, 67, 483–490. [Google Scholar] [CrossRef] [PubMed]
- Zając, A.E.; Kopeć, S.; Szostakowski, B.; Spałek, M.J.; Fiedorowicz, M.; Bylina, E.; Filipowicz, P.; Szumera-Ciećkiewicz, A.; Tysarowski, A.; Czarnecka, A.M.; et al. Chondrosarcoma-from Molecular Pathology to Novel Therapies. Cancers 2021, 13, 2390. [Google Scholar] [CrossRef]
- Tiet, T.D.; Hopyan, S.; Nadesan, P.; Gokgoz, N.; Poon, R.; Lin, A.C.; Yan, T.; Andrulis, I.L.; Alman, B.A.; Wunder, J.S. Constitutive Hedgehog Signaling in Chondrosarcoma Up-Regulates Tumor Cell Proliferation. Am. J. Pathol. 2006, 168, 321–330. [Google Scholar] [CrossRef] [Green Version]
- Campbell, V.T.; Nadesan, P.; Ali, S.A.; Wang, C.Y.Y.; Whetstone, H.; Poon, R.; Wei, Q.; Keilty, J.; Proctor, J.; Wang, L.W. Hedgehog Pathway Inhibition in Chondrosarcoma Using the Smoothened Inhibitor IPI-926 Directly Inhibits Sarcoma Cell Growth. Mol. Cancer Ther. 2014, 13, 1259–1269. [Google Scholar] [CrossRef] [Green Version]
- Wagner, A.J.; Hohenberger, P.; Okuno, S.; Eriksson, M.; Patel, S.; Ferrari, S.; Gasali, P.G.; Chawla, S.P.; Woehr, M.; Ross, R. Results from a Phase 2 Randomized, Placebo-Controlled, Double Blind Study of the Hedgehog Pathway Antagonist IPI-926 in Patients with Advanced Chondrosarcoma. In Proceedings of the Connective Tissue Oncology Society Annual Meeting, New York, NY, USA, 30 October–2 November 2013. [Google Scholar]
- Italiano, A.; Le Cesne, A.; Bellera, C.; Piperno-Neumann, S.; Duffaud, F.; Penel, N.; Cassier, P.; Domont, J.; Takebe, N.; Kind, M. GDC-0449 in Patients with Advanced Chondrosarcomas: A French Sarcoma Group/US and French National Cancer Institute Single-Arm Phase II Collaborative Study. Ann. Oncol. 2013, 24, 2922–2926. [Google Scholar] [CrossRef] [PubMed]
- Amary, M.F.; Bacsi, K.; Maggiani, F.; Damato, S.; Halai, D.; Berisha, F.; Pollock, R.; O’Donnell, P.; Grigoriadis, A.; Diss, T. IDH1 and IDH2 Mutations Are Frequent Events in Central Chondrosarcoma and Central and Periosteal Chondromas but Not in Other Mesenchymal Tumours. J. Pathol. 2011, 224, 334–343. [Google Scholar] [CrossRef]
- Mellinghoff, I.K.; Touat, M.; Maher, E. AG-120, a First-in-Class Mutant IDH1 Inhibitor in Patients with Recurrent or Progressive IDH1 Mutant Glioma: Updated Results from the Phase I Nonenhancing Glioma Population. Neuro-Oncol 2017, 19, vi10–vi11. [Google Scholar] [CrossRef]
- Tap, W.; Villalobos, V.M.; Cote, G.M.; Burris, H.; Janku, F.; Mir, O.; Beeram, M.; Wagner, A.; Auer, J.; Liu, H. A Phase 1 Study of AG-120, an IDH1 Mutant Enzyme Inhibitor: Results from the Chondrosarcoma Dose Escalation and Expansion Cohorts. In Proceedings of the Connective Tissue Oncology Society 21st Annual Meeting, Lisbon, Portugal, 9–12 November 2016. [Google Scholar]
- Cojocaru, E.; Wilding, C.; Engelman, B.; Huang, P.; Jones, R.L. Is the IDH Mutation a Good Target for Chondrosarcoma Treatment? Curr. Mol. Biol. Rep. 2020, 6, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.-X.; van Oosterwijk, J.G.; Sicinska, E.; Moss, S.; Remillard, S.P.; van Wezel, T.; Bühnemann, C.; Hassan, A.B.; Demetri, G.D.; Bovée, J.V.M.G.; et al. Functional Profiling of Receptor Tyrosine Kinases and Downstream Signaling in Human Chondrosarcomas Identifies Pathways for Rational Targeted Therapy. Clin. Cancer Res. 2013, 19, 3796–3807. [Google Scholar] [CrossRef] [Green Version]
- Bernstein-Molho, R.; Kollender, Y.; Issakov, J.; Bickels, J.; Dadia, S.; Flusser, G.; Meller, I.; Sagi-Eisenberg, R.; Merimsky, O. Clinical Activity of MTOR Inhibition in Combination with Cyclophosphamide in the Treatment of Recurrent Unresectable Chondrosarcomas. Cancer Chemother. Pharm. 2012, 70, 855–860. [Google Scholar] [CrossRef]
- Van Oosterwijk, J.G.; van Ruler, M.A.J.H.; Briaire-de Bruijn, I.H.; Herpers, B.; Gelderblom, H.; van de Water, B.; Bovée, J.V.M.G. Src Kinases in Chondrosarcoma Chemoresistance and Migration: Dasatinib Sensitises to Doxorubicin in TP53 Mutant Cells. Br. J. Cancer 2013, 109, 1214–1222. [Google Scholar] [CrossRef] [Green Version]
- Polychronidou, G.; Karavasilis, V.; Pollack, S.M.; Huang, P.H.; Lee, A.; Jones, R.L. Novel Therapeutic Approaches in Chondrosarcoma. Future Oncol. 2017, 13, 637–648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, D.R.; Lazarides, A.L.; Visgauss, J.D.; Somarelli, J.A.; Blazer, D.G.; Brigman, B.E.; Eward, W.C. Limb Salvage versus Amputation in Patients with Osteosarcoma of the Extremities: An Update in the Modern Era Using the National Cancer Database. BMC Cancer 2020, 20, 995. [Google Scholar] [CrossRef]
- Malek, F.; Somerson, J.S.; Mitchel, S.; Williams, R.P. Does Limb-Salvage Surgery Offer Patients Better Quality of Life and Functional Capacity than Amputation? Clin. Orthop. Relat. Res. 2012, 470, 2000–2006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erstad, D.J.; Ready, J.; Abraham, J.; Ferrone, M.L.; Bertagnolli, M.M.; Baldini, E.H.; Raut, C.P. Amputation for Extremity Sarcoma: Contemporary Indications and Outcomes. Ann. Surg. Oncol. 2018, 25, 394–403. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, J.D.; Laitinen, M.K.; Parry, M.C.; Sumathi, V.; Grimer, R.J.; Jeys, L.M. The Role of Surgical Margins in Chondrosarcoma. Eur. J. Surg. Oncol. 2018, 44, 1412–1418. [Google Scholar] [CrossRef] [Green Version]
- Fiorenza, F.; Abudu, A.; Grimer, R.J.; Carter, S.R.; Tillman, R.M.; Ayoub, K.; Mangham, D.C.; Davies, A.M. Risk Factors for Survival and Local Control in Chondrosarcoma of Bone. J. Bone Jt. Surg. Br. Vol. 2002, 84-B, 93–99. [Google Scholar] [CrossRef]
- Tsuda, Y.; Evans, S.; Stevenson, J.D.; Parry, M.; Fujiwara, T.; Laitinen, M.; Outani, H.; Jeys, L. Is the Width of a Surgical Margin Associated with the Outcome of Disease in Patients with Peripheral Chondrosarcoma of the Pelvis? A Multicenter Study. Clin. Orthop. Relat. Res. 2019, 477, 2432–2440. [Google Scholar] [CrossRef] [PubMed]
- Ghert, M.A.; Abudu, A.; Driver, N.; Davis, A.M.; Griffin, A.M.; Pearce, D.; White, L.; O’Sullivan, B.; Catton, C.N.; Bell, R.S.; et al. The Indications for and the Prognostic Significance of Amputation as the Primary Surgical Procedure for Localized Soft Tissue Sarcoma of the Extremity. Ann. Surg. Oncol. 2005, 12, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Calderón, S.A.L.; Kuechle, J.; Raskin, K.A.; Hornicek, F.J. Lower Extremity Megaprostheses in Orthopaedic Oncology. JAAOS-J. Am. Acad. Orthop. Surg. 2018, 26, e249–e257. [Google Scholar] [CrossRef] [Green Version]
- Hennessy, D.W.; Raskin, K.A.; Schwab, J.H.; Lozano-Calderón, S.A. Endoprosthetic Reconstruction of the Upper Extremity in Oncologic Surgery. JAAOS-J. Am. Acad. Orthop. Surg. 2020, 28, e319–e327. [Google Scholar] [CrossRef]
- Mavrogenis, A.F.; Angelini, A.; Drago, G.; Merlino, B.; Ruggieri, P. Survival Analysis of Patients with Chondrosarcomas of the Pelvis. J. Surg. Oncol. 2013, 108, 19–27. [Google Scholar] [CrossRef]
- Bus, M.P.A.; Campanacci, D.A.; Albergo, J.I.; Leithner, A.; van de Sande, M.A.J.; Gaston, C.L.; Caff, G.; Mettelsiefen, J.; Capanna, R.; Tunn, P.-U.; et al. Conventional Primary Central Chondrosarcoma of the Pelvis: Prognostic Factors and Outcome of Surgical Treatment in 162 Patients. JBJS 2018, 100, 316–325. [Google Scholar] [CrossRef]
- Schwartz, A.J.; Kiatisevi, P.; Eilber, F.C.; Eilber, F.R.; Eckardt, J.J. The Friedman-Eilber Resection Arthroplasty of the Pelvis. Clin. Orthop. Relat. Res. 2009, 467, 2825–2830. [Google Scholar] [CrossRef] [Green Version]
- Fisher, N.E.; Patton, J.T.; Grimer, R.J.; Porter, D.; Jeys, L.; Tillman, R.M.; Abudu, A.; Carter, S.R. Ice-Cream Cone Reconstruction of the Pelvis: A New Type of Pelvic Replacement. J. Bone Jt. Surg. Br. Vol. 2011, 93-B, 684–688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erol, B.; Sofulu, O.; Sirin, E.; Saglam, F.; Buyuktopcu, O. Reconstruction after Periacetabular Tumor Resection with Lumic® Endoprosthesis: What Are the Midterm Results? J. Surg. Oncol. 2021, 123, 532–543. [Google Scholar] [CrossRef]
- Fujiwara, T.; Kaneuchi, Y.; Stevenson, J.; Parry, M.; Kurisunkal, V.; Clark, R.; Tsuda, Y.; Laitinen, M.; Grimer, R.; Jeys, L. Navigation-Assisted Pelvic Resections and Reconstructions for Periacetabular Chondrosarcomas. Eur. J. Surg. Oncol. 2021, 47, 416–423. [Google Scholar] [CrossRef]
- Bosma, S.E.; Cleven, A.H.G.; Dijkstra, P.D.S. Can Navigation Improve the Ability to Achieve Tumor-Free Margins in Pelvic and Sacral Primary Bone Sarcoma Resections? A Historically Controlled Study. Clin. Orthop. Relat. Res. 2019, 477, 1548–1559. [Google Scholar] [CrossRef]
- Grimer, R.J.; Chandrasekar, C.R.; Carter, S.R.; Abudu, A.; Tillman, R.M.; Jeys, L. Hindquarter Amputation: Is It Still Needed and What Are the Outcomes? Bone Jt. J. 2013, 95, 127–131. [Google Scholar] [CrossRef]
- Van Houdt, W.J.; Griffin, A.M.; Wunder, J.S.; Ferguson, P.C. Oncologic Outcome and Quality of Life After Hindquarter Amputation for Sarcoma: Is It Worth It? Ann. Surg. Oncol. 2018, 25, 378–386. [Google Scholar] [CrossRef]
- Hickey, M.; Farrokhyar, F.; Deheshi, B.; Turcotte, R.; Ghert, M. A Systematic Review and Meta-Analysis of Intralesional versus Wide Resection for Intramedullary Grade I Chondrosarcoma of the Extremities. Ann. Surg. Oncol. 2011, 18, 1705–1709. [Google Scholar] [CrossRef] [PubMed]
- Dierselhuis, E.F.; Goulding, K.A.; Stevens, M.; Jutte, P.C. Intralesional Treatment versus Wide Resection for Central Low-Grade Chondrosarcoma of the Long Bones. Cochrane Database Syst. Rev. 2019, 3, CD010778. [Google Scholar] [CrossRef]
- Schwab, J.H.; Wenger, D.; Unni, K.; Sim, F.H. Does Local Recurrence Impact Survival in Low-Grade Chondrosarcoma of the Long Bones? Clin. Orthop. Relat. Res. 2007, 462, 175–180. [Google Scholar] [CrossRef]
- Song, K.; Song, J.; Chen, F.; Lin, K.; Ma, X.; Jiang, J. Does Resection of the Primary Tumor Improve Survival in Patients with Metastatic Chondrosarcoma? Clin. Orthop. Relat. Res. 2019, 477, 573–583. [Google Scholar] [CrossRef]
- Italiano, A.; Mir, O.; Cioffi, A.; Palmerini, E.; Piperno-Neumann, S.; Perrin, C.; Chaigneau, L.; Penel, N.; Duffaud, F.; Kurtz, J.E.; et al. Advanced Chondrosarcomas: Role of Chemotherapy and Survival. Ann. Oncol. 2013, 24, 2916–2922. [Google Scholar] [CrossRef] [PubMed]
- Van Maldegem, A.M.; Gelderblom, H.; Palmerini, E.; Dijkstra, S.D.; Gambarotti, M.; Ruggieri, P.; Nout, R.A.; Van De Sande, M.A.; Ferrari, C.; Ferrari, S. Outcome of Advanced, Unresectable Conventional Central Chondrosarcoma. Cancer 2014, 120, 3159–3164. [Google Scholar] [CrossRef] [Green Version]
- Frezza, A.M.; Cesari, M.; Baumhoer, D.; Biau, D.; Bielack, S.; Campanacci, D.A.; Casanova, J.; Esler, C.; Ferrari, S.; Funovics, P.T. Mesenchymal Chondrosarcoma: Prognostic Factors and Outcome in 113 Patients. A European Musculoskeletal Oncology Society Study. Eur. J. Cancer 2015, 51, 374–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsuda, Y.; Ogura, K.; Hakozaki, M.; Kikuta, K.; Ae, K.; Tsuchiya, H.; Iwata, S.; Ueda, T.; Kawano, H.; Kawai, A. Mesenchymal Chondrosarcoma: A Japanese Musculoskeletal Oncology Group (JMOG) Study on 57 Patients. J. Surg. Oncol. 2017, 115, 760–767. [Google Scholar] [CrossRef] [Green Version]
- Kawaguchi, S.; Sun, T.; Lin, P.P.; Deavers, M.; Harun, N.; Lewis, V.O. Does Ifosfamide Therapy Improve Survival of Patients with Dedifferentiated Chondrosarcoma? Clin. Orthop. Relat. Res. 2014, 472, 983–989. [Google Scholar] [CrossRef] [Green Version]
- Kostine, M.; Cleven, A.H.; de Miranda, N.F.C.C.; Italiano, A.; Cleton-Jansen, A.-M.; Bovée, J.V.M.G. Analysis of PD-L1, T-Cell Infiltrate and HLA Expression in Chondrosarcoma Indicates Potential for Response to Immunotherapy Specifically in the Dedifferentiated Subtype. Mod. Pathol. 2016, 29, 1028–1037. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, A.R.; Tan, T.-S.; Unni, K.K.; Collins, M.S.; Wenger, D.E.; Sim, F.H. Secondary Chondrosarcoma in Osteochondroma: Report of 107 Patients. Clin. Orthop. Relat. Res. 2003, 411, 193–206. [Google Scholar] [CrossRef]
- Altay, M.; Bayrakci, K.; Yildiz, Y.; Erekul, S.; Saglik, Y. Secondary Chondrosarcoma in Cartilage Bone Tumors: Report of 32 Patients. J. Orthop. Sci. 2007, 12, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Chen, G.; Chen, X.; Huang, X.; Liu, M.; Pan, W.; Yan, X.; Lin, N.; Ye, Z. Predictors of the Survival of Patients with Chondrosarcoma of Bone and Metastatic Disease at Diagnosis. J. Cancer 2019, 10, 2457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strotman, P.K.; Reif, T.J.; Kliethermes, S.A.; Sandhu, J.K.; Nystrom, L.M. Dedifferentiated Chondrosarcoma: A Survival Analysis of 159 Cases from the SEER Database (2001–2011). J. Surg. Oncol. 2017, 116, 252–257. [Google Scholar] [CrossRef] [PubMed]
- Schneiderman, B.A.; Kliethermes, S.A.; Nystrom, L.M. Survival in Mesenchymal Chondrosarcoma Varies Based on Age and Tumor Location: A Survival Analysis of the SEER Database. Clin. Orthop. Relat. Res. 2017, 475, 799–805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakayama, R.; Hayakawa, K.; Kobayashi, E.; Endo, M.; Asano, N.; Yonemoto, T.; Kawashima, H.; Hamada, K.; Watanabe, I.; Futani, H. What Factors Are Associated with Treatment Outcomes of Japanese Patients with Clear Cell Chondrosarcoma? Clin. Orthop. Relat. Res. 2020, 478, 2537–2547. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SUBTYPE | Age | Location | Radiology | Histopathology | |
|---|---|---|---|---|---|
| Dedifferentiated | 60–80 | Femur, pelvis, humerus | Aggressive, destructive cartilaginous tumor | Low-grade chondrosarcoma with abrupt transition to high-grade non-cartilaginous sarcoma | + PD-L1 + IDH1 |
| Periosteal | 20–40 | Metaphyseal of long bones (femur and humerus) | Large lesions (>5 cm), arise from periosteum. Rarely medullary canal involvement | Resemble grade I–II conventional chondrosarcoma | + IDH1 |
| Mesenchymal | 20–30 | Wide variation, including extraskeletal soft tissue involvement | Primarily lytic, aggressive with wide zone of transition | Biphasic; portions of poorly differentiated small round or spindled mesenchymal cells mixed with islands of hyaline cartilage | + S100 + SOX9 + Bcl-2 − IDH |
| Clear Cell | 25–50 | Epiphyseal, primarily proximal femur and humerus | Well defined, lytic, epiphyseal-based lesions | Sheets of cells with large round nuclei. Cells have a distinct pale or clear cytoplasm | + S100 + SOX9 + Bcl-2 − IDH |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gazendam, A.; Popovic, S.; Parasu, N.; Ghert, M. Chondrosarcoma: A Clinical Review. J. Clin. Med. 2023, 12, 2506. https://doi.org/10.3390/jcm12072506
Gazendam A, Popovic S, Parasu N, Ghert M. Chondrosarcoma: A Clinical Review. Journal of Clinical Medicine. 2023; 12(7):2506. https://doi.org/10.3390/jcm12072506
Chicago/Turabian StyleGazendam, Aaron, Snezana Popovic, Naveen Parasu, and Michelle Ghert. 2023. "Chondrosarcoma: A Clinical Review" Journal of Clinical Medicine 12, no. 7: 2506. https://doi.org/10.3390/jcm12072506
APA StyleGazendam, A., Popovic, S., Parasu, N., & Ghert, M. (2023). Chondrosarcoma: A Clinical Review. Journal of Clinical Medicine, 12(7), 2506. https://doi.org/10.3390/jcm12072506