
Arrhythmic Manifestations of Cardiac Amyloidosis: Challenges in Risk Stratification and Clinical Management

 , , and
, , and
Abstract
:1. Introduction
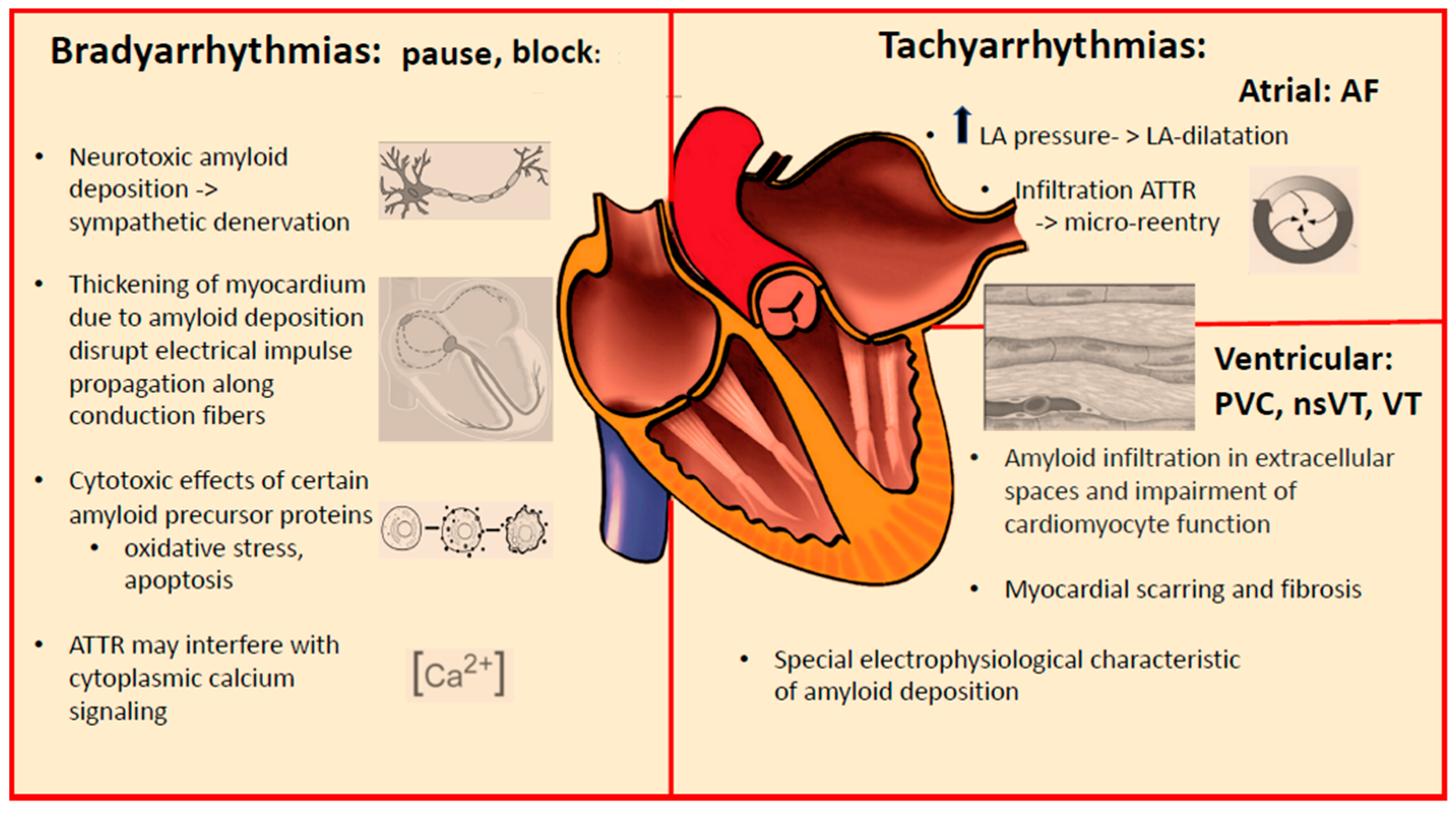
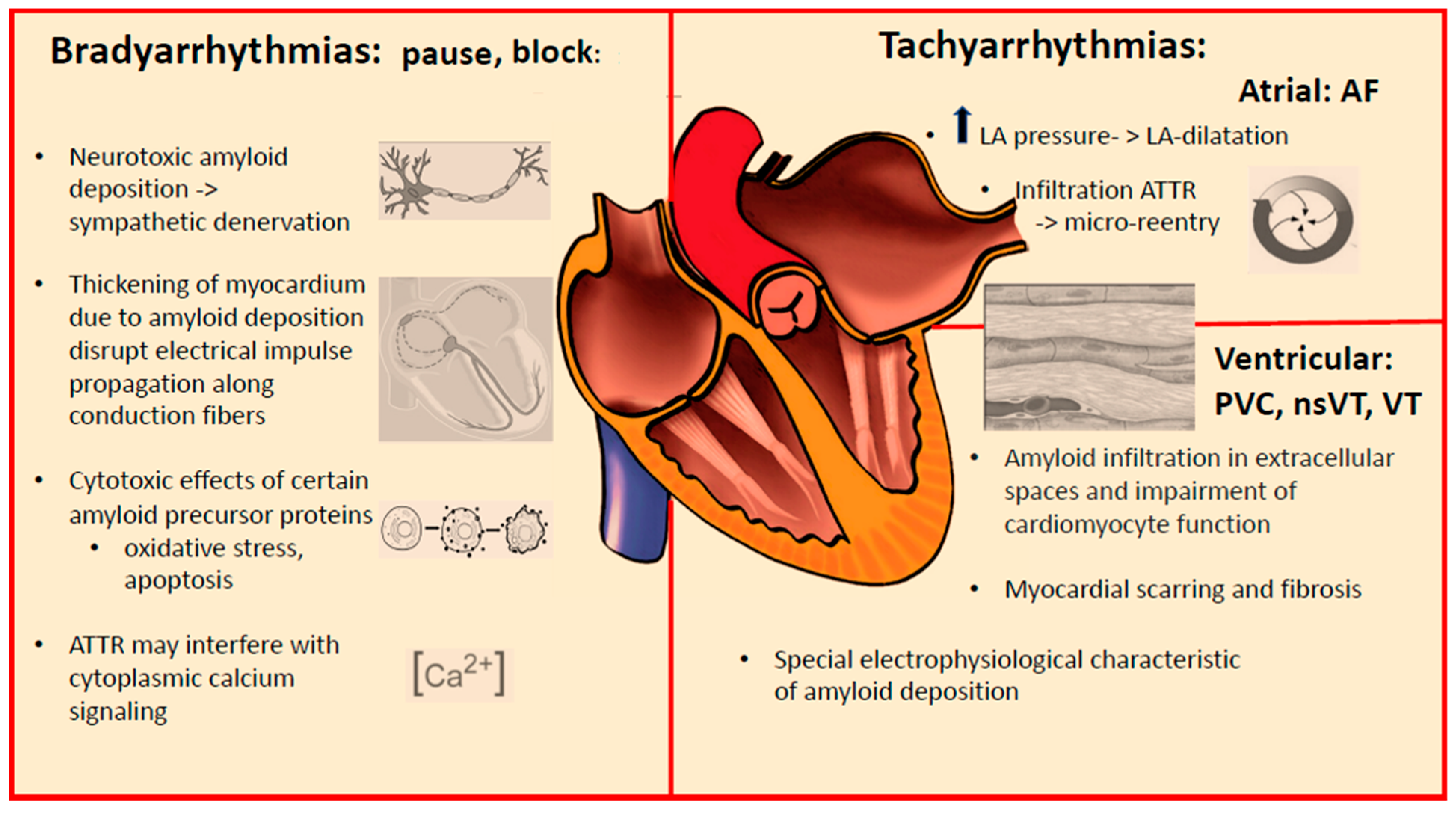
2. Arrhythmic Manifestations of Cardiac Amyloidosis
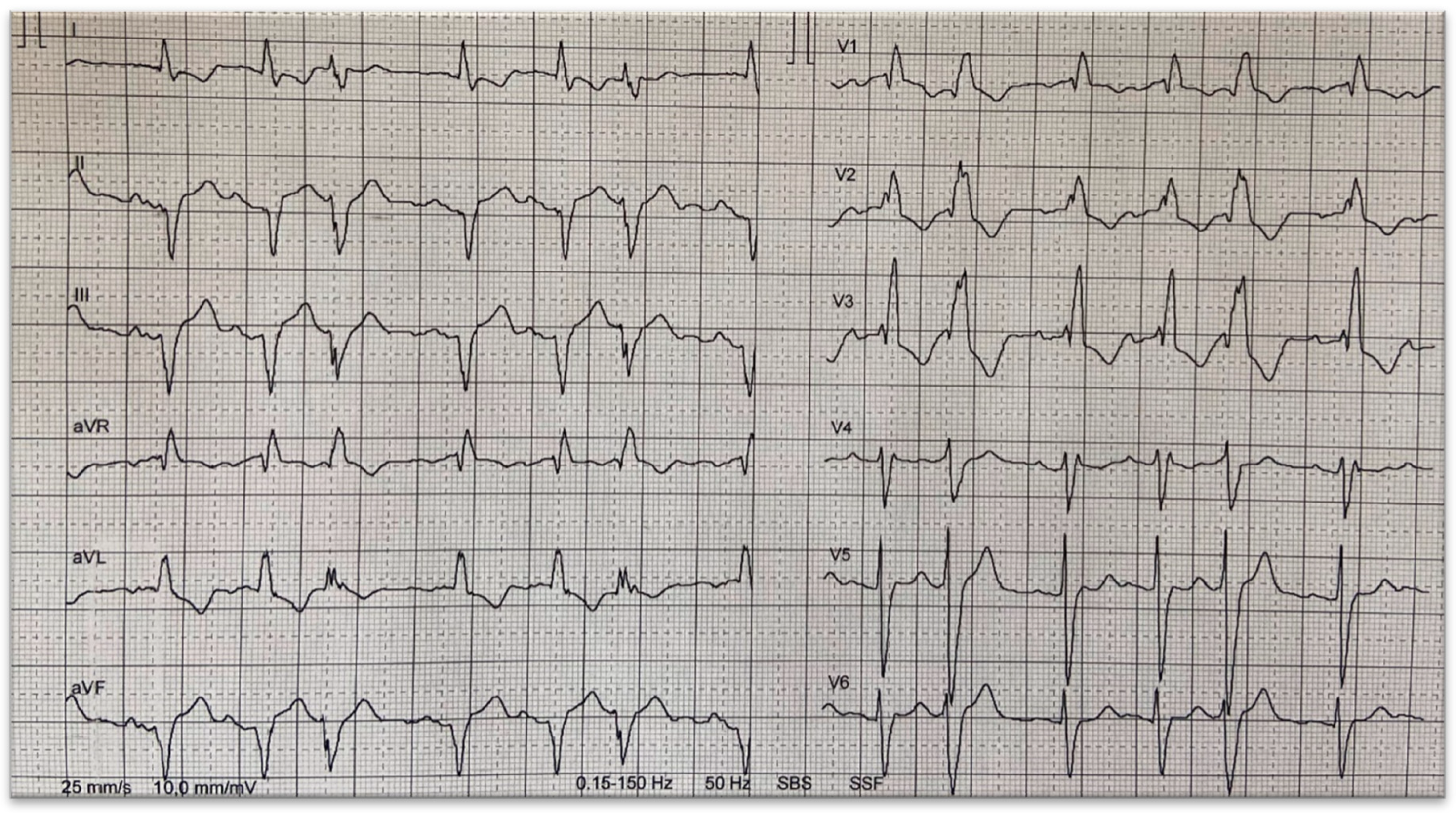
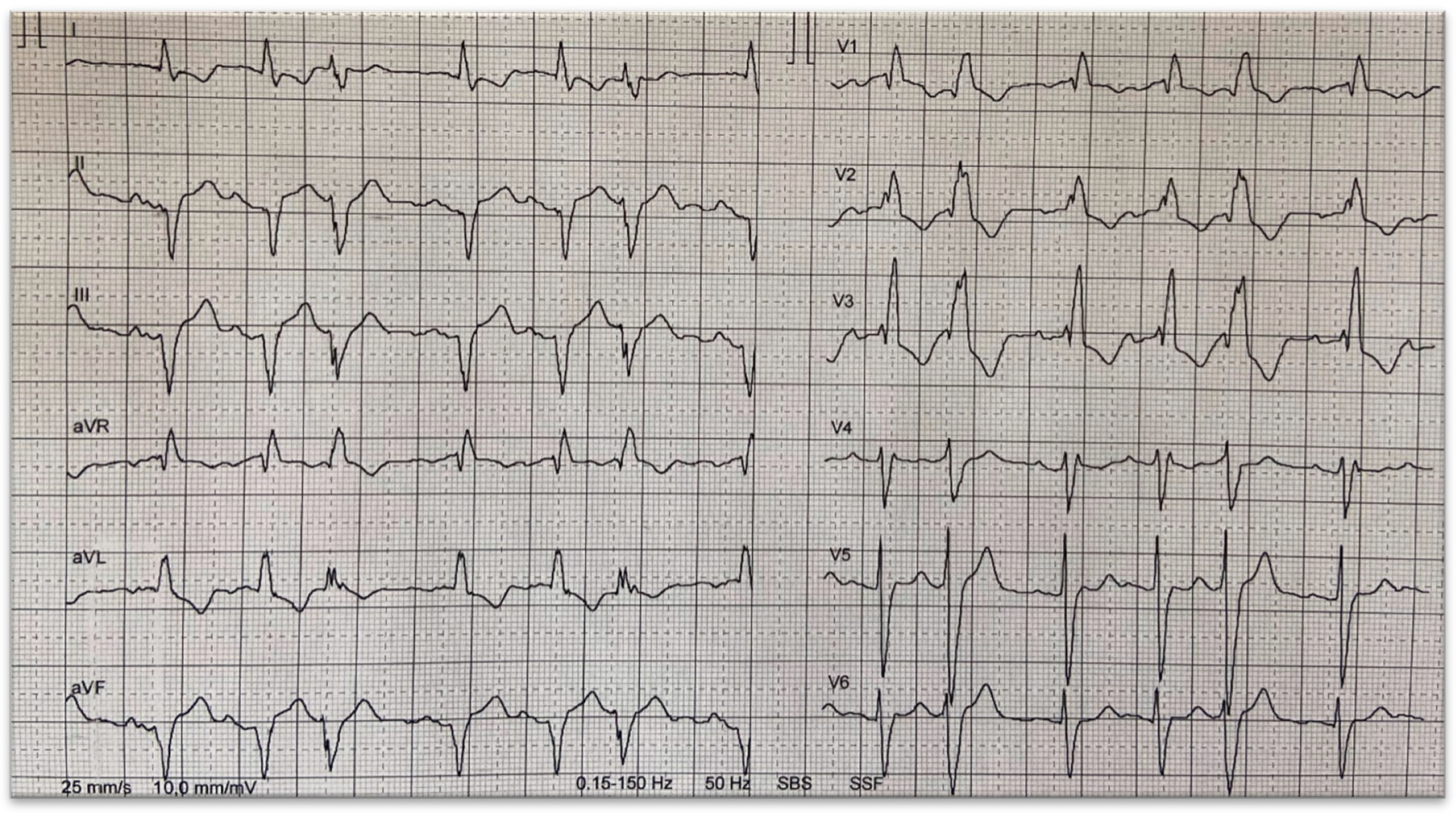
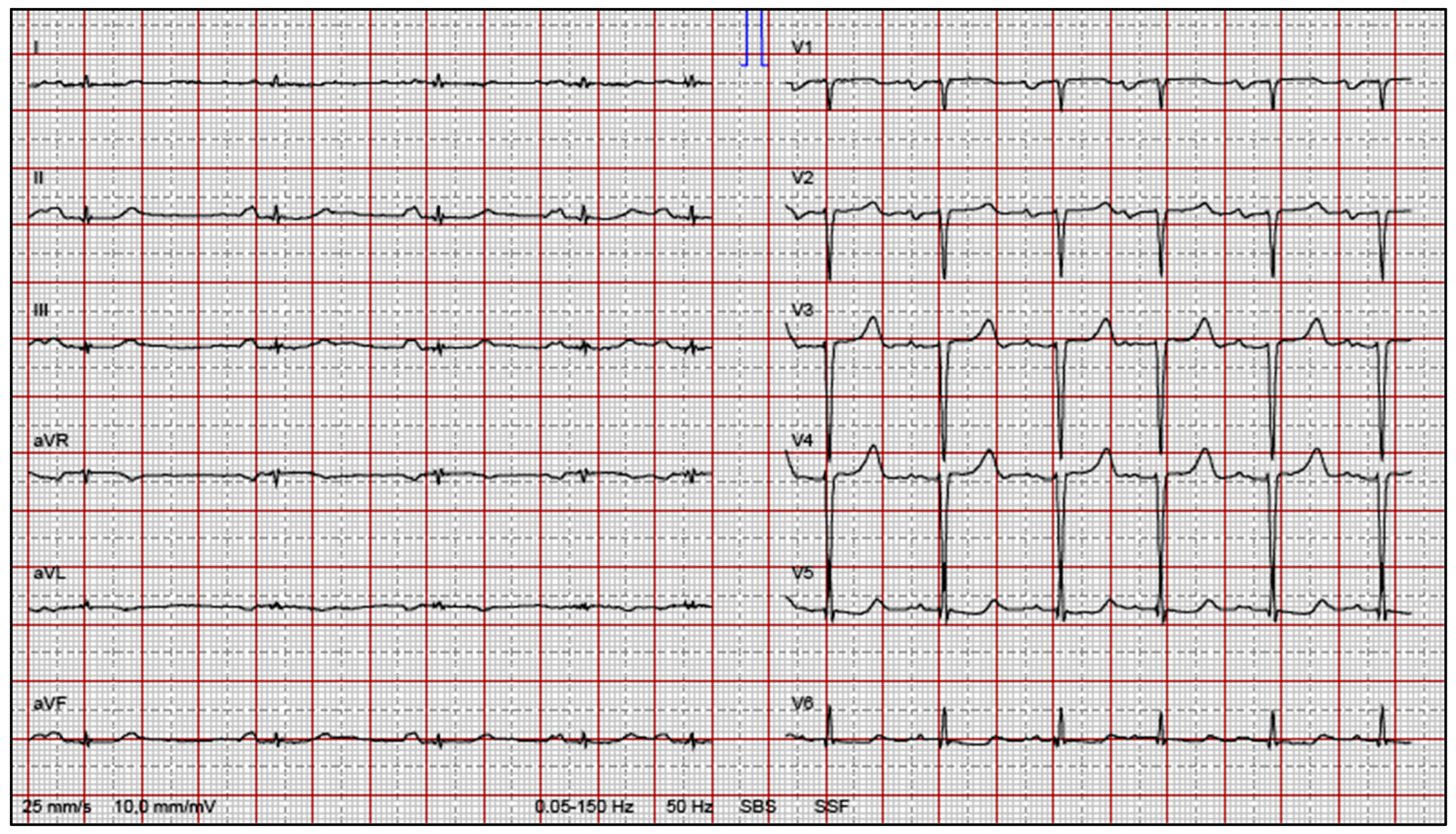
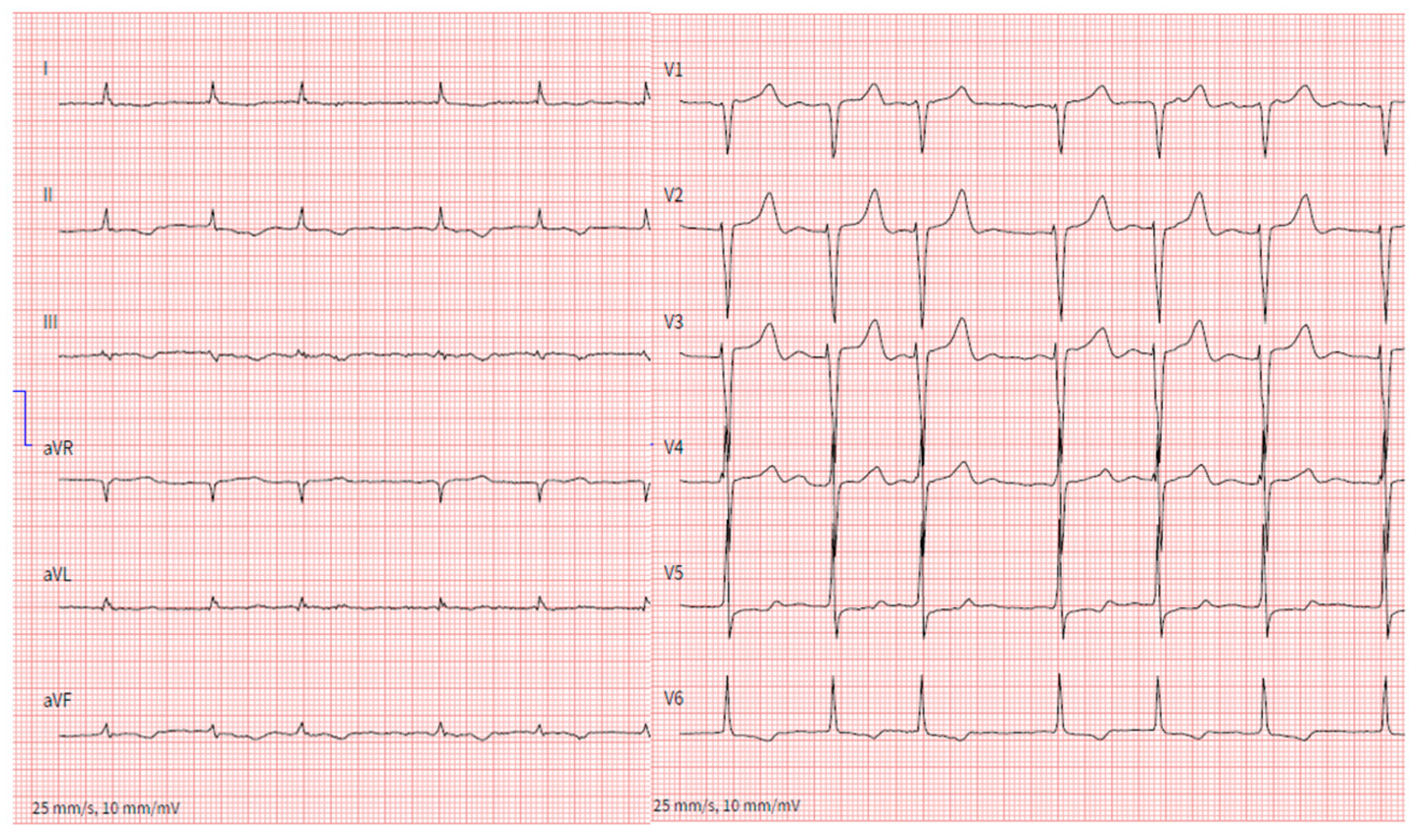
3. Conduction System Disturbances
Pacemaker Therapy in Cardiac Amyloidosis
4. Supraventricular Tachyarrhythmias
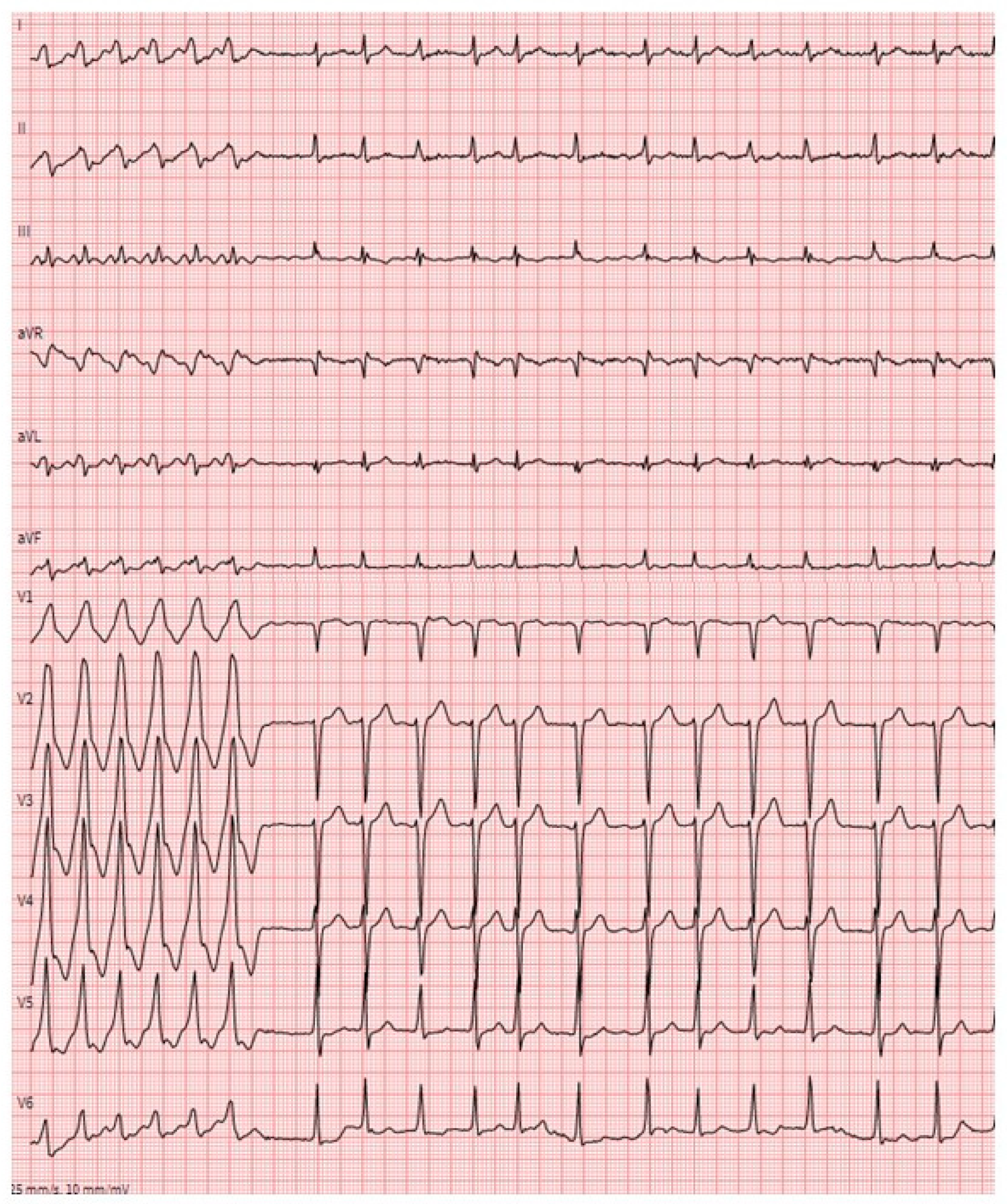
5. Ventricular Tachyarrhythmias
Implantable Cardioverter-Defibrillators in Cardiac Amyloidosis
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wechalekar, A.D.; Gillmore, J.D.; Hawkins, P.N. Systemic amyloidosis. Lancet 2016, 387, 2641–2654. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Naharro, A.; Hawkins, P.N.; Fontana, M. Cardiac amyloidosis. Clin. Med. 2018, 18 (Suppl. 2), s30–s35. [Google Scholar] [CrossRef] [PubMed]
- Gertz, M.A. Cardiac Amyloidosis. Heart Fail. Clin. 2022, 18, 479–488. [Google Scholar] [CrossRef] [PubMed]
- Stelmach-Gołdyś, A.; Zaborek-Łyczba, M.; Łyczba, J.; Garus, B.; Pasiarski, M.; Mertowska, P.; Małkowska, P.; Hrynkiewicz, R.; Niedźwiedzka-Rystwej, P.; Grywalska, E. Physiology, Diagnosis and Treatment of Cardiac Light Chain Amyloidosis. J. Clin. Med. 2022, 11, 911. [Google Scholar] [CrossRef]
- Quarta, C.C.; Kruger, J.L.; Falk, R.H. Cardiac amyloidosis. Circulation 2012, 126, e178–e182. [Google Scholar] [CrossRef]
- Schwotzer, R.; Flammer, A.J.; Gerull, S.; Pabst, T.; Arosio, P.; Averaimo, M.; Bacher, V.U.; Bode, P.; Cavalli, A.; Condoluci, A.; et al. Expert recommendation from the Swiss Amyloidosis Network (SAN) for systemic AL-amyloidosis. Swiss Med. Wkly. 2020, 150, w20364. [Google Scholar] [CrossRef]
- Condoluci, A.; Théaudin, M.; Schwotzer, R.; Pazhenkottil, A.P.; Arosio, P.; Averaimo, M.; Bacher, U.; Bode, P.; Cavalli, A.; Dirnhofer, S.; et al. Management of transthyretin amyloidosis. Swiss Med. Wkly. 2021, 151, w30053. [Google Scholar] [CrossRef]
- Liżewska-Springer, A.; Sławiński, G.; Lewicka, E. Arrhythmic Sudden Cardiac Death and the Role of Implantable Cardioverter-Defibrillator in Patients with Cardiac Amyloidosis—A Narrative Literature Review. J. Clin. Med. 2021, 10, 1858. [Google Scholar] [CrossRef]
- Pattanshetty, D.J.; Bhat, P.K.; Chamberlain, W.A.; Lyons, M.R. Isolated cardiac involvement in primary amyloidosis: Presenting as sick sinus syndrome and heart failure. Tex. Heart Inst. J. 2013, 40, 615. [Google Scholar]
- Abdelazeem, B.; Malik, B.; Baral, N.; Gjeka, R.; Kunadi, A. A Case Report of Sick Sinus Syndrome as an Initial Presentation of Primary Amyloidosis. Cureus 2021, 13, e13922. [Google Scholar] [CrossRef]
- Lindow, T.; Lindqvist, P. The Prevalence of Advanced Interatrial Block and Its Relationship to Left Atrial Function in Patients with Transthyretin Cardiac Amyloidosis. J. Clin. Med. 2021, 10, 2764. [Google Scholar] [CrossRef]
- Barbhaiya, C.R.; Kumar, S.; Baldinger, S.; Michaud, G.F.; Stevenson, W.G.; Falk, R.; John, R.M. Electrophysiologic assessment of conduction abnormalities and atrial arrhythmias associated with amyloid cardiomyopathy. Heart Rhythm 2016, 13, 383–390. [Google Scholar] [CrossRef]
- Rapezzi, C.; Merlini, G.; Quarta, C.C.; Riva, L.; Longhi, S.; Leone, O.; Salvi, F.; Ciliberti, P.; Pastorelli, F.; Biagini, E.; et al. Systemic cardiac amyloidoses: Disease profiles and clinical courses of the 3 main types. Circulation 2009, 120, 1203–1212. [Google Scholar] [CrossRef] [Green Version]
- Hartnett, J.; Jaber, W.; Maurer, M.; Sperry, B.; Hanna, M.; Collier, P.; Patel, D.R.; Wazni, O.M.; Donnellan, E. Electrophysiological Manifestations of Cardiac Amyloidosis: JACC: CardioOncology State-of-the-Art Review. JACC CardioOncol. 2021, 3, 506–515. [Google Scholar] [CrossRef]
- Porcari, A.; Rossi, M.; Cappelli, F.; Canepa, M.; Musumeci, B.; Cipriani, A.; Tini, G.; Barbati, G.; Varrà, G.G.; Morelli, C.; et al. Incidence and risk factors for pacemaker implantation in light-chain and transthyretin cardiac amyloidosis. Eur. J. Heart Fail. 2022, 24, 1227–1236. [Google Scholar] [CrossRef]
- Cappelli, F.; Vignini, E.; Martone, R.; Perlini, S.; Mussinelli, R.; Sabena, A.; Morini, S.; Gabriele, M.; Taborchi, G.; Bartolini, S.; et al. Baseline ECG Features and Arrhythmic Profile in Transthyretin Versus Light Chain Cardiac Amyloidosis. Circ. Heart Fail. 2020, 13, e006619. [Google Scholar] [CrossRef]
- Rehorn, M.R.; Loungani, R.S.; Black-Maier, E.; Coniglio, A.C.; Karra, R.; Pokorney, S.D.; Khouri, M.G. Cardiac Implantable Electronic Devices: A Window into the Evolution of Conduction Disease in Cardiac Amyloidosis. JACC Clin. Electrophysiol. 2020, 6, 1144–1154. [Google Scholar] [CrossRef]
- Donnellan, E.; Wazni, O.M.; Saliba, W.I.; Baranowski, B.; Hanna, M.; Martyn, M.; Patel, D.; Trulock, K.; Menon, V.; Hussein, A.; et al. Cardiac devices in patients with transthyretin amyloidosis: Impact on functional class, left ventricular function, mitral regurgitation, and mortality. J. Cardiovasc. Electrophysiol. 2019, 30, 2427–2432. [Google Scholar] [CrossRef]
- Feng, D.; Edwards, W.D.; Oh, J.K.; Chandrasekaran, K.; Grogan, M.; Martinez, M.W.; Syed, I.I.; Hughes, D.A.; Lust, J.A.; Jaffe, A.S.; et al. Intracardiac Thrombosis and Embolism in Patients with Cardiac Amyloidosis. Circulation 2007, 116, 2420–2426. [Google Scholar] [CrossRef] [Green Version]
- Feng, D.; Syed, I.S.; Martinez, M.; Oh, J.K.; Jaffe, A.S.; Grogan, M.; Edwards, W.D.; Gertz, M.A.; Klarich, K.W. Intracardiac Thrombosis and Anticoagulation Therapy in Cardiac Amyloidosis. Circulation 2009, 119, 2490–2497. [Google Scholar] [CrossRef] [Green Version]
- Donnellan, E.; Wazni, O.M.; Hanna, M.; Elshazly, M.B.; Puri, R.; Saliba, W.; Kanj, M.; Vakamudi, S.; Patel, D.R.; Baranowski, B.; et al. Atrial Fibrillation in Transthyretin Cardiac Amyloidosis: Predictors, Prevalence, and Efficacy of Rhythm Control Strategies. JACC Clin. Electrophysiol. 2020, 6, 1118–1127. [Google Scholar] [CrossRef] [PubMed]
- Vilches, S.; Fontana, M.; Gonzalez-Lopez, E.; Mitrani, L.; Saturi, G.; Renju, M.; Griffin, J.M.; Caponetti, A.; Gnanasampanthan, S.; Santos, J.D.L.; et al. Systemic embolism in amyloid transthyretin cardiomyopathy. Eur. J. Heart Fail. 2022, 24, 1387–1396. [Google Scholar] [CrossRef] [PubMed]
- Isath, A.; Correa, A.; Siroky, G.P.; Perimbeti, S.; Mohammed, S.; Chahal, C.A.A.; Padmanabhan, D.; Mehta, D. Trends, burden, and impact of arrhythmia on cardiac amyloid patients: A 16-year nationwide study from 1999 to 2014. J. Arrhythmia 2020, 36, 727–734. [Google Scholar] [CrossRef] [PubMed]
- Tschöpe, C.; Elsanhoury, A. Treatment of Transthyretin Amyloid Cardiomyopathy: The Current Options, the Future, and the Challenges. J. Clin. Med. 2022, 11, 2148. [Google Scholar] [CrossRef] [PubMed]
- Pollak, A.; Falk, R.H. Left ventricular systolic dysfunction precipitated by verapamilin cardiac amyloidosis. Chest 1993, 104, 618–620. [Google Scholar] [CrossRef]
- Griffiths, B.E.; Hughes, P.; Dowdle, R.; Stephens, M.R. Cardiac amyloidosis with asymmetrical septal hypertrophy and deterioration after nifedipine. Thorax 1982, 37, 711–712. [Google Scholar] [CrossRef] [Green Version]
- Pour-Ghaz, I.; Bath, A.; Kayali, S.; Alkhatib, D.; Yedlapati, N.; Rhea, I.; Khouzam, R.N.; Jefferies, J.L.; Nayyar, M. A Review of Cardiac Amyloidosis: Presentation, Diagnosis, and Treatment. Curr. Probl. Cardiol. 2022, 47, 101366. [Google Scholar] [CrossRef]
- Rubinow, A.; Skinner, M.; Cohen, A.S. Digoxin sensitivity in amyloid cardiomyopathy. Circulation 1981, 63, 1285–1288. [Google Scholar] [CrossRef] [Green Version]
- Ibrahim, R.; Takamatsu, C. Digoxin in amyloidosis: Is it associated with a greater incidence of arrhythmogenic potential? J. Arrhythmia 2022, 38, 831. [Google Scholar] [CrossRef]
- Donnelly, J.P.; Sperry, B.W.; Gabrovsek, A.; Ikram, A.; Tang, W.W.; Estep, J.; Hanna, M. Digoxin Use in Cardiac Amyloidosis. Am. J. Cardiol. 2020, 133, 134–138. [Google Scholar] [CrossRef]
- Muchtar, E.; Gertz, M.A.; Kumar, S.K.; Lin, G.; Boilson, B.; Clavell, A.; Lacy, M.Q.; Buadi, F.K.; Hayman, S.R.; Kapoor, P.; et al. Digoxin use in systemic light-chain (AL) amyloidosis: Contra-indicated or cautious use? Amyloid 2018, 25, 86–92. [Google Scholar] [CrossRef]
- Mints, Y.Y.; Doros, G.; Berk, J.L.; Connors, L.; Ruberg, F.L. Features of atrial fibrillation in wild-type transthyretin cardiac amyloidosis: A systematic review and clinical experience. ESC Heart Fail. 2018, 5, 772–779. [Google Scholar] [CrossRef]
- Ruberg, F.L.; Grogan, M.; Hanna, M.; Kelly, J.W.; Maurer, M.S. Transthyretin Amyloid Cardiomyopathy: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2019, 73, 2872–2891. [Google Scholar] [CrossRef]
- Dale, Z.; Chandrashekar, P.; Al-Rashdan, L.; Kim, M.; Masri, A.; Nazer, B. Management Strategies for Atrial Fibrillation and Flutter in Patients with Transthyretin Cardiac Amyloidosis. Am. J. Cardiol. 2021, 157, 107–114. [Google Scholar] [CrossRef]
- Reisinger, J.; Dubrey, S.W.; Lavalley, M.; Skinner, M.; Falk, R.H. Electrophysiologic Abnormalities in AL (Primary) Amyloidosis With Cardiac Involvement. J. Am. Coll. Cardiol. 1997, 30, 1046–1051. [Google Scholar] [CrossRef] [Green Version]
- D’Errico, S.; Mazzanti, A.; Baldari, B.; Maiese, A.; Frati, P.; Fineschi, V. Sudden death in lambda light chain AL cardiac amyloidosis: A review of literature and update for clinicians and pathologists. Int. J. Clin. Exp. Pathol. 2020, 13, 1474–1482. [Google Scholar]
- Kumar, S.; Dispenzieri, A.; Lacy, M.Q.; Hayman, S.R.; Buadi, F.K.; Colby, C.; Laumann, K.; Zeldenrust, S.R.; Leung, N.; Dingli, D.; et al. Revised Prognostic Staging System for Light Chain Amyloidosis Incorporating Cardiac Biomarkers and Serum Free Light Chain Measurements. J. Clin. Oncol. 2012, 30, 989–995. [Google Scholar] [CrossRef] [Green Version]
- Lilleness, B.; Ruberg, F.L.; Mussinelli, R.; Doros, G.; Sanchorawala, V. Development and validation of a survival staging system incorporating BNP in patients with light chain amyloidosis. Blood 2019, 133, 215–223. [Google Scholar] [CrossRef] [Green Version]
- Grogan, M.; Scott, C.G.; Kyle, R.A.; Zeldenrust, S.R.; Gertz, M.A.; Lin, G.; Klarich, K.W.; Miller, W.L.; Maleszewski, J.J.; Dispenzieri, A. Natural History of Wild-Type Transthyretin Cardiac Amyloidosis and Risk Stratification Using a Novel Staging System. J. Am. Coll. Cardiol. 2016, 68, 1014–1020. [Google Scholar] [CrossRef]
- Gillmore, J.D.; Damy, T.; FontAna, M.; Hutchinson, M.; Lachmann, H.J.; Martinez-Naharro, A.; Quarta, C.C.; Rezk, T.; Whelan, C.J.; Gonzalez-Lopez, E.; et al. A new staging system for cardiac transthyretin amyloidosis. Eur. Heart J. 2018, 39, 2799–2806. [Google Scholar] [CrossRef]
- Varr, B.C.; Zarafshar, S.; Coakley, T.; Liedtke, M.; Lafayette, R.A.; Arai, S.; Schrier, S.L.; Witteles, R.M. Implantable cardioverter-defibrillator placement in patients with cardiac amyloidosis. Heart Rhythm 2014, 11, 158–162. [Google Scholar] [CrossRef] [PubMed]
- Frustaci, A.; Verardo, R.; Russo, M.A.; Caldarulo, M.; Alfarano, M.; Galea, N.; Miraldi, F.; Chimenti, C. Infiltration of Conduction Tissue Is a Major Cause of Electrical Instability in Cardiac Amyloidosis. J. Clin. Med. 2023, 12, 1798. [Google Scholar] [CrossRef] [PubMed]
- Fluechter, S.; Kuschyk, J.; Wolpert, C.; Doesch, C.; Veltmann, C.; Haghi, D.; Schoenberg, S.O.; Sueselbeck, T.; Germans, T.; Streitner, F.; et al. Extent of late gadolinium enhancement detected by cardiovascular magnetic resonance correlates with the inducibility of ventricular tachyarrhythmia in hypertrophic cardiomyopathy. J. Cardiovasc. Magn. Reson. 2010, 12, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fontana, M.; Pica, S.; Reant, P.; Abdel-Gadir, A.; Treibel, T.A.; Banypersad, S.M.; Maestrini, V.; Barcella, W.; Rosmini, S.; Bulluck, H.; et al. Prognostic Value of Late Gadolinium Enhancement Cardiovascular Magnetic Resonance in Cardiac Amyloidosis. Circulation 2015, 20, 1570–1579. [Google Scholar] [CrossRef]
- Orini, M.; Graham, A.J.; Martinez-Naharro, A.; Andrews, C.M.; de Marvao, A.; Statton, B.; Cook, S.A.; O’Regan, D.P.; Hawkins, P.N.; Rudy, Y.; et al. Noninvasive Mapping of the Electrophysiological Substrate in Cardiac Amyloidosis and Its Relationship to Structural Abnormalities. J. Am. Heart Assoc. 2019, 8, e012097. [Google Scholar] [CrossRef]
- Kristen, A.V.; Dengler, T.J.; Hegenbart, U.; Schönland, S.; Goldschmidt, H.; Sack, F.-U.; Voss, F.; Becker, R.; Katus, H.A.; Bauer, A. Prophylactic implantation of cardioverter-defibrillator in patients with severe cardiac amyloidosis and high risk for sudden cardiac death. Heart Rhythm 2008, 5, 235–240. [Google Scholar] [CrossRef]
- Brown, M.T.; Yalamanchili, S.; Evans, S.T.; Ram, P.; Blank, E.A.; Lyle, M.A.; Merchant, F.M.; Bhatt, K.N. Ventricular arrhythmia burden and implantable cardioverter-defibrillator outcomes in transthyretin cardiac amyloidosis. Pacing Clin. Electrophysiol. 2022, 45, 443–451. [Google Scholar] [CrossRef]
- Halawa, A.; Woldu, H.G.; Kacey, K.G.; Alpert, M.A. Effect of ICD implantation on cardiovascular outcomes in patients with cardiac amyloidosis: A systematic review and meta-anaylsis. J. Cardiovasc. Electrophysiol. 2020, 31, 1749–1758. [Google Scholar] [CrossRef]
- Towbin, J.A.; McKenna, W.J.; Abrams, D.J.; Ackerman, M.J.; Calkins, H.; Darrieux, F.C.C.; Daubert, J.; de Chillou, C.; DePasquale, E.C.; Desai, M.Y.; et al. 2019 HRS expert consensus statement on evaluation, risk stratification, and management of arrhythmogenic cardiomyopathy. Heart Rhythm 2019, 16, e301–e372. [Google Scholar] [CrossRef] [Green Version]
- Hamon, D.; Algalarrondo, V.; Gandjbakhch, E.; Extramiana, F.; Marijon, E.; Elbaz, N.; Selhane, D.; Dubois-Rande, J.-L.; Teiger, E.; Plante-Bordeneuve, V.; et al. Outcome and incidence of appropriate implantable cardioverter-defibrillator therapy in patients with cardiac amyloidosis. Int. J. Cardiol. 2016, 222, 562–568. [Google Scholar] [CrossRef]
- Kono, A.K.; Yamada, N.; Higashi, M.; Kanzaki, S.; Hashimura, H.; Morita, Y.; Sakuma, T.; Noguchi, T.; Naito, H.; Sugimura, K. Dynamic late gadolinium enhancement simply quantified using myocardium to lumen signal ratio: Normal range of ratio and diffuse abnormal enhancement of cardiac amyloidosis. J. Magn. Reson. Imaging 2011, 34, 50–55. [Google Scholar] [CrossRef]
- Hashimura, H.; Ishibashi-Ueda, H.; Yonemoto, Y.; Ohta-Ogo, K.; Matsuyama, T.-A.; Ikeda, Y.; Morita, Y.; Yamada, N.; Yasui, H.; Naito, H. Late gadolinium enhancement in cardiac amyloidosis: Attributable both to interstitial amyloid deposition and subendocardial fibrosis caused by ischemia. Heart Vessel. 2016, 31, 990–995. [Google Scholar] [CrossRef]
- Lin, G.; Dispenzieri, A.; Kyle, R.; Grogan, M.; Brady, P.A. Implantable Cardioverter Defibrillators in Patients with Cardiac Amyloidosis. J. Cardiovasc. Electrophysiol. 2013, 24, 793–798. [Google Scholar] [CrossRef]
- Zeppenfeld, K.; Tfelt-Hansen, J.; de Riva, M.; Winkel, B.G.; Behr, E.R.; Blom, N.A.; Charron, P.; Corrado, D.; Dagres, N.; de Chillou, C.; et al. 2022 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death. Eur. Heart J. 2022, 43, 3997–4126. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
|
|
|
|
|
|
|
|
|
| AL | vATTR | wtATTR | |
|---|---|---|---|
| Arrhythmia risk | |||
| Sinus node disease | +/++ | ++ | ++ |
| AV conduction disease | ++ | +++ | +++ |
| Atrial fibrillation | ++/+++ | +++ | +++ |
| nsVT/VT | +++ | ++/+++ | ++/+++ |
| SCD risk | +++ | ++ | ++ |
| Device indication | |||
| PM | + | +++ | ++/+++ |
| CRT | +/++ | ++/+++ | ++ |
| ICD (primary prevention) | +/? | +/? | +/? |
| Bradyarrhythmias Potential candidates who may require cardiac pacing *
|
| Tachyarrhythmias Patients at risk for atrial fibrillation
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Laptseva, N.; Rossi, V.A.; Sudano, I.; Schwotzer, R.; Ruschitzka, F.; Flammer, A.J.; Duru, F. Arrhythmic Manifestations of Cardiac Amyloidosis: Challenges in Risk Stratification and Clinical Management. J. Clin. Med. 2023, 12, 2581. https://doi.org/10.3390/jcm12072581
Laptseva N, Rossi VA, Sudano I, Schwotzer R, Ruschitzka F, Flammer AJ, Duru F. Arrhythmic Manifestations of Cardiac Amyloidosis: Challenges in Risk Stratification and Clinical Management. Journal of Clinical Medicine. 2023; 12(7):2581. https://doi.org/10.3390/jcm12072581
Chicago/Turabian StyleLaptseva, Natallia, Valentina A. Rossi, Isabella Sudano, Rahel Schwotzer, Frank Ruschitzka, Andreas J. Flammer, and Firat Duru. 2023. "Arrhythmic Manifestations of Cardiac Amyloidosis: Challenges in Risk Stratification and Clinical Management" Journal of Clinical Medicine 12, no. 7: 2581. https://doi.org/10.3390/jcm12072581
APA StyleLaptseva, N., Rossi, V. A., Sudano, I., Schwotzer, R., Ruschitzka, F., Flammer, A. J., & Duru, F. (2023). Arrhythmic Manifestations of Cardiac Amyloidosis: Challenges in Risk Stratification and Clinical Management. Journal of Clinical Medicine, 12(7), 2581. https://doi.org/10.3390/jcm12072581