Abstract
For many years, the failure of randomized controlled trials (RCTs) has prevented patients with systemic lupus erythematosus (SLE) from benefiting from biological drugs that have proved to be effective in other rheumatological diseases. Only two biologics are approved for SLE, however they can only be administered to a restricted proportion of patients. Recently, several phase II RCTs have evaluated the efficacy and safety of new biologics in extra-renal SLE and lupus nephritis. Six drug trials have reported encouraging results, with an improvement in multiple clinical and serological outcome measures. The possibility of combining B-cell depletion and anti-BLyS treatment has also been successfully explored.
1. Introduction
Systemic lupus erythematosus (SLE) is an autoimmune rheumatic disease characterized by an unpredictable course and highly variable manifestations of differing severity. The main pathogenic feature of the disease is the presence of antibodies directed against autoantigens. The autoantibodies form immune complexes, which precipitate in different tissues and cause progressive organ damage, which has been associated with a higher risk of future mortality, with at least two-fold higher than the general population [1,2].
In the 1950s, the four-year survival for patients with systemic lupus erythematosus was 50% [3]. It is currently in the order of 85% fifteen-year survival [4,5]. This clearly represents a major improvement over the past seventy years, but also implies that the best of the conventional drugs (hydroxychloroquine, prednisolone, immunosuppressives) struggle to contain the disease and clearly some patients with SLE continue to die far too young. Moreover, drug toxicity of conventional therapies has been implicated in the progression of irreversible organ damage [6,7]. In a multi-ethnic study cohort, patients with damage were significantly more likely to have been treated with some immunosuppressants and biologics that are commonly used in clinical practice (azathioprine, mycophenolate, and rituximab) when compared to patients who did not develop damage [8]. This highlights the urgent need to find alternative therapies [9].
The remarkable development of the biologic drugs to treat patients with the autoimmune rheumatic diseases has radically improved the outcome and quality of life for many patients with rheumatoid arthritis, psoriatic arthritis, and ankylosing spondylitis [10]. In stark contrast, relatively few biologic drugs have “made the grade” in the routine management of lupus. B-cell blocking therapeutics currently approved for the use in SLE have proved to be effective in controlling disease activity, reducing the steroid dose [11], and decreasing damage progression in the open-label extension of randomized controlled trials [12]. Unfortunately, not all patients respond to these drugs, with rituximab having a response rate of approximately 70 to 80% [13,14] and belimumab showing at least 50% improvement in approximately half of the patients treated in the OBSErve Study [15]. In addition, some patients with an initial good response to rituximab experience relapse [16], while others develop allergic reactions [17].
However, there are now more encouraging signs of both short and longer-term successes with genuine hope that a number of biologic drugs will become part of the standard repertoire when treating lupus patients. There is an increasing expectation that there will be a much broader selection of biologic drugs to choose from in the next five to ten years.
In this article, we will review the approved biologics for SLE patients, those that were effective in phase II trials and are currently in phase III trials, and future perspectives.
2. Conventional Lupus Treatment
Conventional drugs for the treatment of lupus patients have been available for the past twenty to fifty years. Although there is some individual variation (e.g., methotrexate is particularly helpful in inflammatory arthritis), corticosteroids and immunosuppressive drugs are used across an array of common lupus features (Figure 1) [18,19]. Hydroxychloroquine is recommended for all patients, given its beneficial effects on disease control, organ damage, and thrombotic risk [20]. Patients with antiphospholipid syndrome in the context of SLE will invariably require anticoagulation in addition to immunosuppressive therapies.
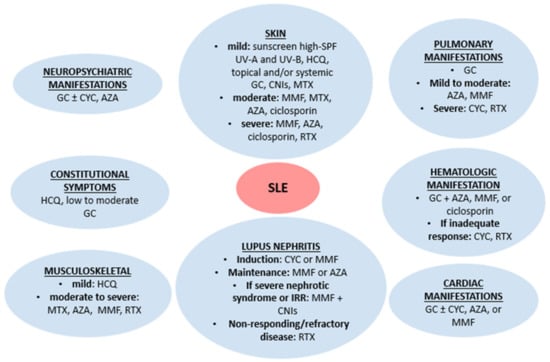
Figure 1.
Conventional lupus therapies. AZA: azathioprine. CNIs: calcineurin inhibitors. CYC: cyclophosphamide. GC: glucocorticoids. HCQ: hydroxychloroquine. IRR: incomplete renal response. MMF: mycophenolate mofetil. RTX: rituximab. SLE: systemic lupus erythematosus. SPF: sun protection factor.
3. Brief Overview of the Immunopathology of SLE and How This Guides the Development of Relevant Biologic Drugs
In the last past twenty years, the growing understanding of the cellular interactions and molecular mechanisms involved in the immunopathogenesis of SLE has allowed the development of several drugs with different targets (Figure 2).
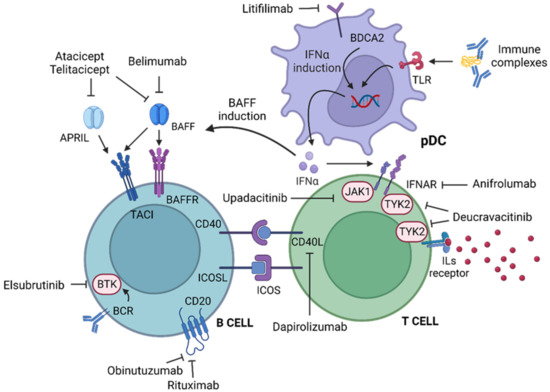
Figure 2.
Created with BioRender.com (accessed on 19 April 2023). Molecules and cells involved in the immunopathogenesis of SLE. The mechanism of the drugs discussed in the review is shown. APRIL: a proliferation-inducing ligand. BAFF: B-cell-activating factor. BAFFR: BAFF receptor. BCR: B-cell receptor. BDCA2: blood dendritic cell antigen 2. BTK: Bruton tyrosine protein kinase. CD40L: CD40 ligand. ICOS: inducible T-cell co-stimulator. ICOSL: ICOS ligand. IFNα: interferon α. IFNAR: type 1 interferon receptor. ILs: interleukins. JAK1: Janus kinase 1. pDC: plasmacytoid dendritic cell. TACI: Transmembrane Activator and Calcium-modulator and cyclophilin ligand Interaction. TLR: Toll-like receptor. TYK2: Tyrosine kinase 2.
The central event in SLE pathogenesis is the loss of tolerance, which results in the survival of autoreactive B-cells. Different phenomena are thought to contribute to the breakdown of self-tolerance, including the defective clearance of apoptotic debris [21], the inability to degrade neutrophil extracellular traps (NETs) [22], and abnormalities in B-cell receptor (BCR) signaling [23]. After recognizing the antigens on the surface of antigen presenting cells (APCs), BCRs on naïve B-cells initiate a cytoplasmatic signal transduction pathway leading to antigen presentation to T cells [24]. Several molecules are involved in the transduction, notably CD19, CD20, and CD22 [25,26], and many have been reported to be altered or defective in lupus patients and lupus animal models [27,28]. BCR function is also regulated by FcγRIIB, which increases the threshold for B-cell activation [29], and Bruton’s tyrosine kinase (BTK), whose inhibition has improved the manifestations of autoimmune diseases in studies on arthritis and lupus mouse models, suggesting that BTK might be a therapeutic target in SLE [30]. Since rituximab, a chimeric anti-CD20 antibody, first used in SLE patients in 2002 [31], the membrane protein CD20 has increasingly gained attention among researchers because of the possibility of using humanized monoclonal antibodies without the risk of the allergic reactions observed with rituximab. The rationale behind the administration of anti-CD20 antibodies is that their use has been associated with a significant reduction in plasma cell population. This is linked to a reduction in many lupus antibodies including anti-dsDNA, anti-nucleosome and anti-cardiolipin [32,33]. Furthermore, it has been observed that activated CD20+ B lymphocytes are able to activate T cells and can lead to an increased production of cytokines [34].
In SLE, altered expression of costimulatory molecules has also been described. In 2016, Menard et al. analyzed B-cell subsets and T cells in a cohort of SLE patients of African American origin. Among the several molecules studied, they noted the expression of CD40 and CD40 ligand (CD40L), which provide an essential signal for the maturation of B-cells and the isotype switching [35,36]. The results described alterations in the levels of CD40 and CD40L on B and T cells surface, suggesting that this pathway was involved in the more severe course of the disease that is often observed in African American/Afro-Caribbean patients [37]. Closely linked to CD40-CD40L, inducible co-stimulatory molecules (ICOS) and its ligand (ICOSL) are critically involved in the differentiation of B-cells. In SLE mouse models, ICOS can support self-reactive T cell survival [38].
One of the most important factors in the survival of peripheral self-reactive B-cells is the B-cell-activating factor (BAFF), also known as BLyS, a member of the tumor necrosis factor (TNF) family. Thien et al. found that BAFF overexpression rescued self-reactive B-cells from depletion [39]. Moreover, serum BAFF levels have been found to be associated with anti-dsDNA antibody levels and a higher rate of relapse following B-cell depletion therapy with rituximab [40]. In addition to BAFF, another B-cell activating factor, the proliferation-induced ligand APRIL, and TWEAK (TNF-like weak inducer of apoptosis) contribute to the homeostasis of B-cells and have been reported to be increased in SLE patients with lupus nephritis compared to controls [41,42,43].
T cells are thought to play an important role in the pathogenesis of lupus nephritis. In kidney biopsies of SLE patients with renal involvement, T cells are one of the dominant cell populations [44]. In 2020, Zhou et al. found that the administration of tofacitinib, a Janus kinase (JAK) inhibitor, into lupus-prone mouse models led to the prevention of lupus nephritis and to the reduction of the number of CD8+ renal resident memory T cells [45]. With previous evidence of efficacy of JAK inhibitors in lupus animal models [46], this finding supported further investigations in humans.
The interferon (IFN) pathway has also been extensively studied in the context of lupus pathogenesis. In 2005, a French study group demonstrated that IFN-α could induce BAFF mRNA expression in salivary gland epithelial cells of patients with primary Sjogren’s syndrome [47]. This discovery shone a light on the complex interplay between innate and adaptive immune responses; it is thought that self-nucleic acids in autoimmune complexes can increase the secretion of IFN-α by plasmacytoid dendritic cells via Toll-like receptors [48,49]. This rise in IFN-α levels will eventually support B-cells survival and differentiation by inducing BAFF. The role of IFN-α in SLE has been emphasized by several groups, who have reported a dysregulation of interferon gene signatures in different tissues [50], and have shown that type I IFN activity correlated with activity measures [51].
4. Development and Current Usage of Biological Drugs Approved for the Treatment of SLE
Two biologic agents, belimumab and anifrolumab, have been approved for use in patients with SLE by the Food and Drug Administration (FDA). The use of rituximab is approved in the UK by the NHS (National Health Service) England and recommended by the European Alliance of Associations for Rheumatology (EULAR) and the American College of Rheumatology (ACR) in severe refractory cases of nephritis. It is also widely used in patients who fail to respond or develop major side effects with conventional immunosuppressants [18].
4.1. Rituximab
Rituximab is a chimeric anti-CD20 monoclonal antibody, whose use in SLE was first reported in an open study of six patients in 2002 [31]. It had already been administered successfully to patients with rheumatoid arthritis [52], following the discovery of B-cells infiltrating the synovial membrane of affected joints [53]. In subsequent cohort and open-label studies, both lupus patients with lupus nephritis and those with extra-renal manifestations, notably articular involvement and autoimmune thrombocytopenic purpura, benefited from rituximab or a biosimilar [54,55,56,57]. However, two major trials failed to demonstrate the superiority of rituximab as an add-on therapy over the standard of care in achieving clinical response in lupus nephritis and extra-renal disease [58,59]. The Exploratory Phase II/III SLE Evaluation of Rituximab (EXPLORER) trial was a randomized, double-blind, placebo-controlled trial based in North America studying the efficacy and safety of rituximab in moderate to severe extra-renal SLE manifestations. The primary endpoint was defined as the percentage of clinical responses measured by the British Isles Lupus Assessment Group index (BILAG) at week 52. The results showed no difference in the proportion of patients achieving a complete or partial response between the rituximab group and the placebo group [58].
With regard to proliferative lupus nephritis (LN), a phase III randomized controlled trial (RCT) of rituximab as an add-on therapy (LUNAR trial) failed to demonstrate superiority over mycophenolate and corticosteroids, compared to placebo in achieving complete or partial renal response [59]. The possible reasons why these trials failed have been carefully considered [60]. It is thought that some clinical features might have been misclassified as activity, leading to biased results that did not show any improvement because the features were actually due to damage. The extreme heterogeneity of SLE among different groups of patients might be implicated. Indeed, secondary analysis of the population enrolled in the EXPLORER trial highlighted the potential benefit of rituximab in African American patients [58]. Other reasons why the trials were not able to detect any significant benefit of rituximab may be the substantial background medication (steroids and conventional immunosuppressants) prescribed for the population in the trial and the inadequate sensitivity of the outcome measures. In 2016, the BAFF surge observed in some patients after the administration of rituximab has been hypothesized to be linked to the unsuccessful RCTs of rituximab in lupus [61]. This observation has led to further trials studying the efficacy of treatment schemes combining rituximab and belimumab, that will be discussed later.
Despite the disappointing clinical results of the RCTs, in 2019 EULAR recommended rituximab as an induction regimen for patients with extra-renal disease after the failure of multiple therapeutic options, or in case of contraindications to conventional immunosuppressants [18]. It also recommended rituximab alone or added to mycophenolate mofetil or cyclophosphamide in non-responding patients with lupus nephritis [62].
In 2022, Chen et al. investigated the potential role of rituximab as maintenance therapy in a multicenter prospective observational cohort study. Eighty-two patients were enrolled and were all treated with one course of rituximab to induce remission. Subsequently, those who responded clinically within 6 months were divided into two different groups, those receiving the maintenance therapy with rituximab in a single dose or conventional immunosuppressants. The relapse rate in the group treated with rituximab was statistically significantly reduced compared to the group on immunosuppressive agents. This suggests that rituximab might be a valuable treatment to maintain a sustained remission in SLE [63]. The doses and frequency of rituximab in the maintenance setting are not yet well defined. A randomized controlled trial (NCT04127747) has recently focused on two different administration schemes to determine whether an individualized scheme based on CD19+ B-cells, dsDNA, and C3 can control the disease better than a standard one. The group on standard dose will receive an infusion of rituximab 500 mg on the first day, on the 6th, 12th, 18th and 24th month, while the patients on individualized dose will receive rituximab 500 mg on the first day and then will be followed-up every 3 months and will receive another infusion of rituximab 500 mg, if CD19+ B-cell count ≥1%, or anti-dsDNA antibodies titre increased, or complement C3 level decreased. The results are awaited.
4.2. Belimumab
Belimumab (Benlysta) is a human monoclonal antibody that neutralizes the soluble form of BLyS. It was the first biological drug approved by the FDA specifically for the treatment of SLE. Advances in biotechnology turned out to be vital for the development of belimumab [64]. In the 1990s, an analysis of human neutrophil-derived cDNA led to the identification of the gene encoding BLyS. After the observation that BLyS-transgenic mice had developed clinical and serological features similar to lupus [65] in 2001, the levels of BAFF were measured in lupus patients and were found to be elevated compared to healthy controls. In this study, BAFF correlated with IgG levels and with anti-dsDNA antibodies [66]. This discovery supported the idea that BAFF could be a candidate therapeutic target in SLE. Belimumab was developed using the technology of human single chain variable fragment (scFv) phage display, which allowed the selection of a limited number of scFvs, those with highest binding and inhibitory activity against BLyS. Then, the selected scFvs underwent a phase of conversion to obtain full-length immunoglobulins and were further processed. The result was a fully human monoclonal antibody, eventually named “belimumab” [67].
In 2009, the results of a phase II trial of belimumab were published. The study did not meet its co-primary endpoints of change using the Safety of Estrogens in Lupus National Assessment-Systemic Lupus Erythematosus Disease Activity Index (SELENA-SLEDAI) and time to first flare. It appeared that belimumab was not better than placebo in controlling disease activity in lupus patients. However, a secondary analysis suggested that it could stabilize the disease [68].
More encouragingly, two phase III trials (BLISS-52 and BLISS-76) reported the superiority of belimumab over placebo in SLE patients (especially those with high anti-dsDNA antibodies and low C3 levels) without severe renal or central nervous system (CNS) involvement. Belimumab was added to the background therapy with conventional immunosuppressants and steroids. In both BLISS-52 and BLISS-76, two different doses of intravenous belimumab were tested (1 mg/kg and 10 mg/kg). BLISS-52 reported higher proportions of response rate, measured with the SRI-4 (Systemic lupus erythematosus Response Index), in belimumab-treated groups regardless of the dose (51% for 1 mg/kg, 58% for 10 mg/kg, 48% for placebo). In BLISS-76, the benefit was significant only in patients given 10 mg/kg of belimumab (43.2% of SRI responders versus 33.5% in the placebo group, p = 0.017), whereas the risk of severe SELENA-SLEDAI flare over 76 weeks was significantly reduced only in those given 1 mg/kg of belimumab (18.5% versus 26.5% in placebo group, p = 0.023). In both studies, no increase in the rates of adverse events was noted in the groups on active treatment [69,70]. In BLISS-52 and BLISS-76 patients of Black/African American ancestry and Asian ethnicity were underrepresented, accounting for approximately 25% and less than 5% in BLISS-76, respectively [70]. Further studies were conducted to assess the efficacy of belimumab in these minorities. In Asian patients enrolled in the BLISS-NEA trial, a significant improvement in disease activity and a reduction of the cumulative steroid dose in patients given belimumab was noted [71]. In contrast, the EMBRACE study in African Americans failed to meet the primary endpoint of superiority of belimumab over standard of care, although a higher but insignificant response rate was observed in patients on active treatment compared to placebo [72]. Intravenous belimumab at the dose of 10 mg/kg every 4 weeks as an add-on therapy over standard of care was also assessed in pediatric patients aged >4 years, with results similar to those reported in RCTs on adults [73].
In 2011, the US FDA and the European Medicines Agency (EMA) approved intravenous belimumab at the dose of 10 mg/kg for the treatment of active SLE in adult patients with positive serology who were receiving standard of care. In the UK, the National Institute for Health and Care Excellence recommended the use of belimumab for SLE only in 2016 under strict restrictions (e.g., the patients had to have a SLEDAI-2K score of >10 and could only have skin and/or joint disease). Two years later, the FDA approval was extended to pediatric patients aged five years and above. However, the route of administration impacts heavily on the costs the Health Service has to bear though, by forcing patients to go to the hospital for the infusion regularly, uncertainty about patients compliance is abolished. In terms of treatment adherence, this is beneficial. The BLISS-SC trial demonstrated the equivalent safety and efficacy of subcutaneous belimumab at a fixed dose of 200 mg administered once a week when given in addition to standard of care. The beneficial effect of belimumab was clear early, with significant higher response rates detected at the end of the second month of treatment, which was maintained throughout the study [74].
Although severe active lupus nephritis was an exclusion criterion in BLISS-52 and BLISS-76, a post-hoc analysis underlined the potential improvement in renal parameters in those patients enrolled with mild to moderate renal involvement who were assigned randomly to belimumab treatment. A higher remission rate, shorter time to achieve the remission status, lower risk of flare, and more marked reduction in proteinuria than patients on standard therapy alone was noted [75]. Based on these promising data, The Belimumab International Study in Lupus Nephritis (BLISS-LN) assessed the addition of intravenous belimumab to standard therapy with cyclophosphamide or mycophenolate mofetil. A significantly greater proportion of belimumab-treated patients compared to placebo-treated patients achieved the primary efficacy renal response at week 104, 43%, and 32%, respectively (p = 0.03). The primary efficacy renal response was determined by a combination of urine protein creatinine ratio ≤0.7, an estimated glomerular filtration rate no worse than 20% compared to the value recorded before the flare or at least 60 mL/min/1.73 m2, and without the need for rescue therapy [76]. These results led to the FDA approving belimumab for lupus nephritis. However, it must be acknowledged that, despite the approval of belimumab in LN, there are some reports of patients who developed renal involvement while taking belimumab [77,78,79].
The Belimumab Assessment of Safety in SLE (BASE) trial showed, in 2019, an increased risk of psychiatric issues in patients on belimumab [80], leading the Medicines and Healthcare products Regulatory Agency to express concern about the safety profile of the drug. However, a subsequent meta-analysis of eleven randomized controlled trials (eight thousand eight hundred and twenty-four patients) did not confirm the excess risk of psychiatric events [81].
Currently, belimumab is approved for extra-renal and renal SLE in both adult and pediatric SLE patients with high disease activity and positive serology (high anti-dsDNA and low complement). Its use in severe active central nervous system involvement in SLE is not recommended, although not on the basis of any compelling data.
4.3. Anifrolumab
The increasing amount of evidence about the role of IFN-α in lupus, reviewed by Ramaswamy et al. [82], confirmed the efficacy and safety of anifrolumab, a fully human antibody IgG1k against the type I IFN-α/β/ω receptor, in randomized controlled trials. In the phase IIb MUSE (MEDI-546 in Uncontrolled Systemic lupus Erythematosus) trial, a significantly greater proportion of patients treated with anifrolumab plus standard therapy achieved an SRI-4 response with sustained reduction of steroids at week 24 (34.3% for the group treated with anifrolumab 300 mg, 28.8% for 1000 mg) compared to placebo-treated patients (17.6%). When stratifying the patients according to the IFN signature, a greater effect was found in those with a high type I IFN signature (36% for anifrolumab 300 mg, 28.2% for anifrolumab 1000 mg, 13.2% for placebo patients). This result suggested the possibility of utilizing a more precision mode of treatment for some SLE patients. A higher incidence of herpes zoster was reported in patients given anifrolumab [83].
The results of the first phase III RCT, TULIP-1 (Treatment of the Uncontrolled Lupus via the Interferon Pathway-1), of anifrolumab in extra-renal SLE were published in 2019. Two doses of anifrolumab were evaluated (300 mg and 150 mg) over a 48-week period, followed by 4 weeks of follow-up. At week 52 there was no difference in the SRI-4 response between anifrolumab and the placebo group (primary endpoint). However, the BICLA (BILAG-based Composite Lupus Assessment) responses were higher in anifrolumab-treated patients [84]. The authors of the study suspected that the primary endpoint was not reached because the design was too strict and because the SRI was not able to detect partial improvements. As with belimumab RCTs, a careful choice of the outcome measure was vital. Interestingly, a post-hoc analysis of BICLA and SRI-4 discordant responder status highlighted that, in TULIP-1, the subgroup of placebo-treated patients, who achieved an SRI-4 response but not BICLA response, had lower baseline joint count scores and took higher steroid dose compared to anifrolumab group. These factors might have prevented the authors from detecting significant differences between treatment groups [85]. In the second phase III RCT, TULIP-2, SRI-4 was replaced by BICLA as the primary outcome measure. Patients with active severe lupus nephritis or neuropsychiatric SLE were again excluded from the study. TULIP-2 demonstrated the superiority of intravenous 300 mg of anifrolumab administered monthly in active moderate to severe SLE over placebo. A greater proportion of patients treated with anifrolumab achieved a BICLA response compared to placebo at week 52 (47.8% versus 31.5%, p = 0.001). Ironically, the SRI-4, a secondary outcome measure in this study, was also met. When the population was stratified according to the interferon signature, only in the subpopulation characterized by a high interferon signature was a significantly higher percentage of BICLA-responders noted (48.0% versus 30.7%, p = 0.002) [86].
In 2021, the FDA approved the use of anifrolumab over standard treatments in adult lupus patients with moderate to severe manifestations without severe active renal or neuropsychiatric involvement. Currently, a 116-week phase 3 randomized controlled trial (IRIS) is recruiting lupus patients to evaluate the efficacy and safety of intravenous infusion of anifrolumab plus standard of care (mycophenolate mofetil and corticosteroids) in active class III or IV lupus nephritis (NCT05138133).
5. Reviewing Those Drugs That Have Achieved Phase II Drug Trial Endpoints and Are in or about to Start Phase III Trials
5.1. Obinutuzumab
Obinutuzumab is a fully humanized anti-CD20 monoclonal antibody which causes a more complete B-cell depletion than rituximab [87].
A phase II trial (NOBILITY, Table 1) [88] of obinutuzumab added to mycophenolate and steroids studied 242 SLE patients aged 18–75 years with histologic evidence of class III or IV lupus nephritis, a urine protein to creatinine ratio (UPCR) >1 and an estimated glomerular filtration rate ≥30 mL/min/1.73 m2. Sixty-three patients were randomly assigned to the obinutuzumab group and followed until week 104. Obinutuzumab 1000 mg or placebo was administered intravenously on day 1, week 2, 24, and 26. The primary endpoint, evaluated at week 52, was the proportion of complete renal responses (CRR), defined by UPCR <0.5, serum creatinine within normal range, absence of worsening of serum creatinine by >15%, and <10 red blood cells per high-power field in urinary sediment without red blood cells casts. The trial reached its primary objective, with a greater proportion of patients in the obinutuzumab group who achieved CRR at week 52 compared to the placebo, 35% and 23%, respectively (p = 0.115 with a prespecified alpha level of 0.2). In addition to CRR, partial renal responses (PRR) were also considered as a component of the overall renal responses (ORR), including both CRR and PRR. The requirements to achieve PRR were the UPCR reduced by at least 50% from baseline down to values <1 or <3 if the baseline UPCR was ≥3, a worsening in the serum creatinine of less than 15%, <10 red blood cells per high-power field in the urinary sediment or less than 50% increase from baseline. At week 52, a higher proportion of patients in the obinutuzumab group achieved ORR than in the placebo group (approximately 55% versus around 35%, p < 0.05). Notably, this advantage was maintained throughout the study (p < 0.01 at week 104), despite the last dose of the drug having been administered at week 26. The benefit of obinutuzumab was particularly evident in class IV lupus nephritis and in patients with baseline UPCR ≥ 3. No improvement in outcome was observed in those patients with LN class V.

Table 1.
Phase II trial of obinutuzumab.
Although the proportion of patients with B-cell depletion (CD19+ ≤5 cells/µL) was similar between groups at week 104, the CD19+ B-cells in patients taking obinutuzumab were depleted more rapidly than in patients given the placebo. Indeed, at week 2, the levels of CD19+ cells were <5 cells/µL in 98% of patients treated with obinutuzumab, compared to the placebo group in which only 2% of patients achieved B-cell depletion. Treatment with obinutuzumab was associated with low IgM levels at week 104 (33% of patients versus 8% in the placebo group) and with early significant improvement in anti-dsDNA antibodies and complement levels. Considering safety, the rate of serious infections in the obinutuzumab group (8%) was not higher than in the placebo group (18%). Obinutuzumab-treated patients had a slightly higher incidence of non-severe infusion-related reactions (notably headache, tachycardia, nausea, and hypertension) than placebo-treated patients, 16% and 10%, respectively.
These data have led to the development of a phase III trial (REGENCY) of obinutuzumab in class III or IV lupus nephritis. For this study, investigators are asked to recruit patients aged 18–75, with an estimated enrollment of two hundred fifty-two patients, who will be randomized into three different treatment groups. Those in group 1 will receive 1000 mg of intravenous obinutuzumab at baseline and weeks 2, 24, 26, 50, and 52, in addition to mycophenolate mofetil and oral prednisone; patients in group 2 will receive the same treatment except for the infusion at week 50, that will be replaced by placebo, and those in group 3 will receive placebo infusions plus mycophenolate mofetil and oral steroids. The primary endpoint will be the percentage of complete renal responses at week 76.
Another phase III trial (OBILUP, NCT04702256) will explore the efficacy of obinutuzumab in proliferative lupus nephritis (class III or IV). The OBILUP study is a randomized, open-label, controlled, non-inferiority trial investigating the association of obinutuzumab and mycophenolate mofetil compared to mycophenolate mofetil plus steroids in achieving complete renal response in adolescents (aged 14 years and above) and adults. The patients in the obinutuzumab group will not be given oral steroids unless they have extrarenal involvement. In those with extrarenal manifestations, the steroid dose will be kept under 10 mg/day at any time, <7.5 mg/day after 6 months and <5 mg/day after 9 months.
The potential role of obinutuzumab in extra-renal SLE will be evaluated in a phase III trial (ALLEGORY, NCT04963296) including only patients with anti-nuclear antibody (ANA) ≥ 1:80, or elevated anti-dsDNA and/or anti-Sm antibodies, hypocomplementemia C3 and/or C4 and/or low CH50. Other key inclusion criteria will be high disease activity at screening, defined according to the BILAG-2004, the Systemic Lupus Erythematosus Disease Activity Index 2000 (SLEDAI-2K) and Physician’s Global Assessment (PGA). The therapy will be administered with the same scheme used in the NOBILITY trial. The primary objective will be the proportion of SRI-4 responders at week 52. The response rates according to SRI-6, SRI-8 and BICLA will be also assessed as secondary endpoints at week 52.
5.2. Dapirolizumab
Dapirolizumab is a polyethylene glycol (PEG)-conjugated antigen-binding fragment (Fab’) targeting CD40L. It lacks the functional fragment crystallizable domain, that has been implicated in the thromboembolic events reported with previous anti-CD40 drugs [89]. In early clinical trials, dapirolizumab was well tolerated and the rate of adverse events was similar between dapirolizumab and placebo groups [90,91].
A phase IIb, randomized, double-blind, placebo-controlled trial (Table 2) [92] aimed to establish a dose-response relationship and investigated the safety and efficacy of dapirolizumab administered intravenously in addition to standard of care in moderate to severe SLE. Patients with a new diagnosis of lupus nephritis (class III or IV), worsening pre-existing LN, and neuropsychiatric SLE were excluded from the trial. One hundred eighty-two patients, aged 18 years or above, were enrolled and randomly assigned to one of four groups receiving intravenous dapirolizumab at three different doses (6 mg/kg, 24 mg/kg, or 45 mg/kg) or intravenous placebo monthly up to week 20. All patients, then, underwent an observational period, lasting 24 weeks. One hundred sixty-eight(90.1%) patients completed the study. The trial was unable to identify a dose-response relationship (its primary endpoint) even when a stratification for baseline steroid dose was performed. For the primary objective the responses were determined using BICLA at week 24. An overall greater improvement in SRI-4, BICLA, SLEDAI-2K, PGA, BILAG, and Cutaneous Lupus Erythematosus Disease Area and Severity Index–Activity (CLASI-A) was observed throughout the study in dapirulizumab-treated patients when compared to placebo-treated patients, together with a greater reduction in the levels of anti-dsDNA and antiphospholipid antibodies and a rise in C3 and C4. There were also fewer flares in the group on dapirolizumab compared to the placebo (5 versus 7). After the discontinuation of the treatment at week 24 required by the study protocol, auto-antibodies and complement components tended to return to pre-treatment values. During this period of observation, a stabilization of BILAG, SLEDAI, and PGA was observed, where there was a reduction of the BICLA and SRI-4 response rates, probably due to the use of rescue therapy. Unlike earlier anti-CD40L monoclonal antibodies, dapirolizumab was not associated with a higher risk of thromboembolic events compared to placebo. Its safety profile was considered acceptable; severe treatment-associated adverse events (AEs) were similar in the treatment groups.
Table 2.
Phase II trial of dapirolizumab.
Importantly, this phase II study did not have as its primary endpoint the response rate. Thus, the failure to meet the primary objective cannot be considered as evidence of inefficacy. In contrast, it was felt that the overall improvement in the secondary endpoint supported further investigation and the subsequent design of a phase III, randomized, controlled trial (PHOENYCS GO), which will assess dapirolizumab in lupus patients with moderate or severe disease in spite of standard of care. Approximately four hundred fifty patients, aged 16 and above, will be enrolled. Those with neuropsychiatric manifestation or renal involvement causing estimated glomerular filtration rate <30 mL/min/1.73 m2, serum creatinine >2.5 mg/dL, proteinuria >3 g/day, or protein to creatinine ratio >340 mg/mmol, will be excluded. The primary outcome measure will be the proportion of BICLA responders at week 48. The study will have multiple secondary endpoints, notably the proportion of patients achieving SRI-4 response at week 48, BICLA response at week 12 and at week 24, and Lupus Low Disease Activity State (LLDAS) ≥50% of post-baseline visit. Other secondary outcome measures will be the rate of BILAG flare until the end of week 48, BILAG improvement without worsening at week 48, the time to first BILAG flare, the change in PGA from baseline at week 48, and the rate of adverse events from baseline up to week 54 (NCT04294667). The safety and tolerability of dapirolizumab will be also assessed in an ongoing phase III open label study (NCT04976322) evaluating the incidence of treatment-emergent adverse events up to week 110 in seven hundred sixty patients aged ≥16 years. The secondary endpoints will be the efficacy measures (proportion of BILAG flares at different time points, achievement of LLDAS, BICLA responses).
5.3. Deucravacitinib
Deucravacitinib is a highly selective Tyrosine Kinase 2 (TYK2) inhibitor whose activity is due to the binding to a regulatory site which results in allosteric inhibition [93].
The phase II trial (PAISLEY, Table 3) [94] of deucravacitinib in SLE evaluated the efficacy and safety of three different dosages (3 mg twice daily, 6 mg twice daily, or 12 mg once daily) in active SLE with at least one BILAG-2004 grade A or at least 2 BILAG-2004 grade B from the musculoskeletal or mucocutaneous domain and SLEDAI-2K ≥6. Three hundred sixty-three patients aged 18–75 years were included, of whom ninety-one received the lowest dose of the drug, ninety-three received 6 mg twice daily, and eighty-nine received 12 mg once daily. The remaining patients received placebo tablets. All patients were taking standard of care treatment. Deucravacitinib was administered orally for 48 weeks. Patients with severe, active lupus nephritis, neuropsychiatric SLE, recent herpes zoster, herpes simplex or influenza infection or history of disseminated or complicated herpes zoster were excluded. The primary endpoint of the proportion of SRI-4 responders at week 32 was met. In the patients taking 3 mg twice daily of deucravacitinib, there was a significantly higher percentage of SRI-4 responders than in the placebo group (58.2% versus 34.4%, p <0.001). A significantly higher proportion of SRI-4 responses (49.5%) was also noted among those taking 6 mg twice daily dose when compared with placebo (p = 0.02). No significant difference was observed between patients on deucravacitinib 12 mg daily and the placebo group.
Table 3.
Phase II trial of deucravacitinib.
With regard to the secondary endpoints evaluated at week 48, only the group taking deucravacitinib 3 mg twice daily differed significantly when compared with placebo. In fact, it showed higher proportions in SRI-4 responses (57.1% versus 34.4%, p < 0.001), BICLA responses (47.3% versus 25.6%, p = 0.001), CLASI-50 responses (69.6% versus 16.7%, p < 0.001), LLDAS (36.3% versus 13.3%, p < 0.001), and a significantly greater mean change from baseline in the joint count (–8.9 versus –7.6, p = 0.001). The patients taking deucravacitinib also showed an increasing improvement in the levels of anti-dsDNA antibodies and C3 and C4 levels throughout the study and a dramatic reduction in the IFN gene expression from week 4 was noted.
In terms of safety, AEs were mainly mild or moderate infections, with upper respiratory tract infections and nasopharyngitis being the most common. Encouragingly, the incidence of herpes zoster infection, a well-known complication of the treatment with JAK inhibitors [95], was similar between the deucravacitinib and the placebo groups. However, there was a higher frequency of cutaneous adverse events (acne and rash) in deucravacitinib-treated patients compared with placebo (16.5% in the group taking 3 mg twice daily of deucravacitinib, 34.4% in the group taking 6 mg twice daily, 33.7% in the group taking 12 mg once daily, 13.3% in the placebo group).
The promising results of the PAISLEY trial have led to the design of two-phase III trials (POETYC SLE-1—NCT05617677- and POETYC SLE-2 -NCT05620407-), which will evaluate the SRI-4 response rate at week 52 in patients with extra-renal SLE treated with deucravacitinib in addition to standard of care. These studies will have a long-term follow-up to detect adverse events up to week 156.
5.4. Litifilimab
Litifilimab is a humanized monoclonal antibody that targets the surface receptor blood dendritic cell antigen 2 (BDCA2). BDCA2 is expressed on plasmacytoid dendritic cells [96] and has been linked to the production of interferon α [97].
Litifilimab was evaluated in a two-part, phase II, multicenter, randomized, controlled trial (LILAC, Study to Evaluate the Efficacy of BIIB059 in Subjects with Systemic Lupus Erythematosus and Active Skin Manifestations and in Subjects with Cutaneous Lupus Erythematosus, Table 4) [98]. Here, we will only review the results of part A of the study. Part B involved patients with cutaneous lupus erythematosus, some of whom did not have systemic involvement.
Table 4.
Phase II trial of litifilimab.
Part A of the LILIAC study enrolled adults with a positive antinuclear antibody test ≥1:80 and/or anti-dsDNA antibodies ≥30 UI/mL, active skin disease defined as a score of at least 8 on the CLASI-A, a score of at least 4 on the SLEDAI-2K. According to the second version of the protocol, a minimum score on the CLASI-A was no longer required, but patients had to have at least one active skin lesion and at least four tender and swollen joints in the sites evaluated with the twenty-eight joint assessment to be considered eligible. The study met its primary endpoint and demonstrated the superiority of litifilimab 450 mg over standard of care in controlling joint involvement in SLE. The least-squares mean (LSM) difference in the number of active joints between litifilimab and placebo at week 24 was −3.4 (p = 0.04). When compared to the placebo, the group treated with litifilimab showed a significantly higher proportion of patients who experienced a decrease of at least 7 points on CLASI-A score (56% versus 34%, with LSM difference [95% confidence interval -CI-]: 21.6 [0.1 to 43.1]), a greater number of SRI-4 responders (56% versus 29%, with LSM difference [95% CI]: 26.4 [9.5 to 43.2]), and a greater absolute change in SLEDAI-2K (LSM –4.4 ± 0.5 versus –2.6 ± 0.5, with LSM difference [95% CI]: –1.7 [–3.0 to –0.5]). All these secondary outcome measures were evaluated at week 24. The serologic analysis of patients in the litifilimab arm did not identify any improvement in the anti-dsDNA antibodies, C3, C4, or immunoglobulins levels. The placebo group reported more AEs than the litifilimab group (68% versus 59%). AEs were mostly mild to moderate, with only 5% of litifilimab-treated patients and 11% of placebo-treated patients experiencing serious AEs. The most common AEs in the litifilimab group were diarrhea, nasopharyngitis, and urinary tract infections, each occurring in seven patients (5%).
Based on the promising results of the LILIAC trial, two phase III trials (TOPAZ-1 -NCT04895241- and TOPAZ-2—NCT04961567-) of low and high doses of litifilimab as an add-on therapy over standard of care in lupus patients have been designed and are ongoing. Both will measure the proportion of SRI-4 responders at week 24 as primary objective measure. Multiple secondary endpoints will be considered, including the joint count assessment, the change in CLASI-A score, and the reduction of steroid dose. The patients who will complete the TOPAZ-1 and TOPAZ-2 trials will be offered the possibility of entering the extension study (EMERALD, NCT05352919), lasting 180 weeks. The primary endpoint of the EMERALD trial will be the safety profile of litifilimab. The proportion of patients experiencing treatment-emergent and serious AEs, and the long-term impact of litifilimab on several efficacy outcome measures will be evaluated.
Another phase II/III trial of litifilimab (AMETHYST, NCT05531565) is currently re-cruiting patients with cutaneous SLE with or without systemic involvement who did not respond to antimalarial treatment. The primary endpoint will be the control of skin manifestations (achievement of Cutaneous Lupus Activity of Physician’s Global Assessment-Revised (CLA-IGA-R) Erythema Score of 0 or 1 and CLASI-70 responses).
5.5. Atacicept
Atacicept is a recombinant fusion protein constituted by the extracellular domain of TACI (Transmembrane Activator and Calcium-modulator and cyclophilin ligand Interaction) receptor and a modified portion of human IgG [99]. TACI is the receptor of two B- cell activating factors, BAFF and APRIL, and is normally expressed on B-cells [100]. Thus, atacicept binds BAFF and APRIL preventing them from activating the downstream signaling pathway. The two major phase II trials that investigated atacicept in extra-renal SLE did not meet their primary endpoints [101,102] and the phase II RCT of atacicept plus mycophenolate in lupus nephritis was terminated due to the occurrence of serious infections thought to be linked to hypogammaglobulinemia initially believed to be due to the atacicept, but in fact the concomitant mycophenolate is the more likely culprit [103]. Since the phase II trial of atacicept in lupus nephritis was interrupted after the enrollment of just six patients, the efficacy of the drug has not been adequately tested [103]. Recently Vera Therapeutics has acquired atacicept (from Merk Serono) and a phase III trial (COMPASS, NCT05609812) of atacicept in adult lupus patients with biopsy-proven active lupus nephritis is ongoing. It will last for 104 weeks, followed by 52 weeks of open-label treatment.
Here, we will summarize the design and main results of the phase II trials of atacicept in SLE (APRIL-SLE [101] and ADDRESS II [102], Table 5). The APRIL-SLE trial [101] was a 52-week, randomized, placebo-controlled, phase II/III trial investigating two different doses of atacicept (75 mg and 150 mg) after patients who had flared had been admitted into remission with steroids. The atacicept/placebo was given in addition to standard of care to prevent flares in patients with extra-renal moderate-to-severe SLE. The therapy was administered twice weekly for 4 weeks, then weekly up to week 48. Four hundred sixty-one patients aged 16 years or above were randomized. One hundred forty-five patients were administered atacicept 150 mg, one hundred fifty-nine were administered atacicept 75 mg and one hundred fifty-seven were assigned to the placebo group. The atacicept 150 mg arm was terminated before week 52 because of two fatal infections reported in this group (leptospirosis and pneumococcal pneumonia). This was an odd and unfortunate decision especially as other trials were not stopped in spite of many more deaths (e.g., the voclosporin phase II trial [104]). These events were not associated with hypogammaglobulinemia and the rate of serious infections in the APRIL-SLE study (6.9%) was not higher than the one reported in previous studies of belimumab [70] or rituximab [58], approximately 7% and 9.5%, respectively. With regard to the primary endpoint, atacicept 75 mg did not show superiority over the placebo in preventing BILAG A or B flares at week 52. However, a post-hoc analysis highlighted the potential benefit that atacicept 150 mg might have on flare prevention and on prolonging the time to first flare. When compared to the placebo, the flare rate in this group was significantly lower (37% versus 54%, p = 0.002) and the first flare was significantly delayed (Hazard Ratio, HR, 0.56, p = 0.009).
Table 5.
Phase II trials of atacicept.
Another phase II, randomized, placebo-controlled trial (ADDRESS II) [102] explored the potential role of two different doses of subcutaneous atacicept (75 mg or 150 mg) in adults with moderate to severe SLE (SLEDAI-2K ≥6) receiving standard therapy. Patients with severe lupus nephritis defined by UPCR >2.0 mg/mg and/or estimated glomerular filtration rate <40 mL/minute/1.73 m2) or with central nervous system manifestation were excluded. A total of three hundred and six patients were enrolled, of whom one hundred and two were assigned randomly to atacicept 75 mg once weekly, one hundred and four to atacicept 150 mg once weekly, and one hundred to the placebo group. The primary efficacy endpoint (SRI-4 responses at week 24) was not met. However, the exploratory analyses considering day 1 as baseline showed a significantly higher proportion of SRI-4 responders at week 24 in both atacicept groups when compared with the placebo group. Furthermore, the subgroup analyses highlighted a significantly higher SRI-4 response rate in atacicept-treated patients when only those with serologically active disease were considered (61.2% of those receiving atacicept 75 mg and 61.5% of those receiving atacicept 150 mg, with p equal to 0.003 and 0.002, respectively). In the subgroup treated with 150 mg of atacicept and with baseline high disease activity (HDA), defined by SLEDAI-2K score ≥10, 62.7% of patients achieved SRI-4 response at week 24. This percentage was significantly higher compared to placebo-treated patients with baseline HDA (42.3%, p = 0.029). A higher proportion of patients taking atacicept 150 mg also achieved SRI-6 response at week 24 compared to placebo (54.9% versus 28.8%, p = 0.005). The group taking atacicept 75 mg had less severe BILAG A flares than placebo, regardless of the severity of the disease at baseline (Hazard Ratio [HR] 0.24, p = 0.019). When only the subgroup with HDA was considered, a significant difference between the percentages of patients free from severe BILAG A flares was detected between the groups on atacicept 75 mg (HR 0.08, p = 0.002) or 150 mg (HR 0.32, p = 0.038) and the placebo group. Thus, it seemed that atacicept prevented flares, especially in patients with high disease activity.
In terms of the effect on biomarkers, atacicept was associated with an increase in the levels of complement components (C3 and C4) and with a reduction in the levels of anti-dsDNA antibodies. Significantly higher rates of upper respiratory tract infections (9.8% with atacicept 75 mg and 12.5% with atacicept 150 mg) and diarrhea (6.9% with atacicept 75 mg and 11.5% with atacicept 150 mg) were reported in patients on active treatment compared to the placebo group, in which 3.0% had an upper respiratory tract infection and 5.0% had diarrhea. Overall, the safety profile of atacicept was considered acceptable by the authors.
The long-term extension (LTE) study [105] confirmed the results of the ADDRESS II trial, without new safety concerns. The patients previously treated with placebo in the core study and who started atacicept 150 mg (PBO/atacicept group) showed an overall clinical improvement. However, the patients achieving the greatest proportion of SRI responses (SRI-4 58%, SRI-6 43.2%), low disease activity (47.7%), LLDAS (30.7%) and remission (26.1%) at 72 weeks were those taking 150 mg of atacicept continuously from the randomization in the core study. The percentages of patients in LDA and remission were significantly higher compared to those measured in the PBO/atacicept group.
5.6. Telitacicept
Telitacicept is a fusion protein consisting of a domain of TACI receptor and the crystallizable fragment (Fc) component of human IgG [106]. In China, telitacicept is approved in active SLE [107].
A phase IIb, randomized, placebo-controlled trial (Table 6) [108] studied telitacicept in combination with standard therapy compared to placebo in adults with SLE and a SELENA-SLEDAI score ≥8. Two hundred forty-nine patients were randomly assigned to four treatment groups. They received subcutaneous telitacicept 80 mg (n = 62), 160 mg (n = 63), 240 mg (n = 62), or placebo (n = 62) once a week as an add-on therapy. The primary efficacy endpoint was met. At week 48, in the groups treated with telitacicept, there were significantly greater proportions of SRI-4 responses than in the placebo group. The SRI-4 response was achieved by 71.0 % of the patients on telitacicept 80 mg (p < 0.0001), 68.3% of those on telitacicept 160 mg (p = 0.0001), 75.8% of those taking the highest dose (p < 0.0001). 33.9% of the patients in the placebo group were SRI-4 responders. Significantly higher proportions of telitacicept-treated patients experienced a reduction of ≥4 points in the SELENA-SLEDAI score compared to placebo-treated patients (75.8% with telitacicept 80 mg [p = 0.003], 77.8% with telitacicept 160 mg [p = 0.001], 79.0% with telitacicept 240 mg [p < 0.001], 50.0% with placebo). The frequency of AEs was similar between the groups.
Table 6.
Phase II trial of telitacicept.
A phase III trial (NCT04082416) [109] also based in China confirmed the efficacy and safety of telitacicept 160 mg. The study recruited adult SLE patients with a SELENA-SLEDAI score ≥8 who were given subcutaneous telitacicpet 160 mg once a week or placebo for 52 weeks. Three hundred thirty-five participants were randomized, of whom one hundred sixty-seven received telitacicept and one hundred sixty-eight received placebo. In the telitacicept group, 82.6% of patients achieved the SRI-4 response, compared to 38.1% of those treated with placebo (p <0.001). The higher response rate in telitacicept-treated patients was evident at week 4 and maintained throughout the study. Telitacicept was well tolerated, with only 7.2% of patients experiencing serious AEs versus 14.2% in the placebo group.
A phase III RCT (NCT05306574) of telitacicept is ongoing. Three hundred forty-one adolescent (≥12 years) and adult patients with moderate to severe SLE will be randomized to receive telitacicept 160 mg, telitacicept 240 mg, or placebo. The therapy will be administered once a week over the standard of care for 52 weeks. The primary endpoint will be the proportion of SRI-4 responders at 48 and 52 weeks.
An observational retrospective study of twenty Chinese patients [110], who were given telitacicept 160 mg weekly, provided early data on the efficacy and safety of telitacicept outside of the ideal setting of the RCTs. 80% of patients achieved the SRI-4 response, 90% succeeded in reducing the steroid dose by >25%, and none experienced severe flares. The therapy was well tolerated and only one patient discontinued the treatment because of AEs. An improvement in renal function was reported in patients with renal impairment, suggesting a potential role of telitacicept in lupus nephritis. Further investigations are needed.
5.7. Other Drugs
Tabalumab, an anti-BLyS monoclonal antibody, met its primary endpoint, achieving an SRI-5 response in one phase II trial (ILLUMINATE-2) [111] but not in a second trial (NCT01196091 [112]). Eli-Lilly decided not to take forward any further studies with this monoclonal. Blisibimod (anti-BLyS) and epratuzumab (anti-CD22) both gave encouraging phase II results [113,114] but did not meet their phase III endpoints [115,116] and no further trials with these monoclonals are planned.
6. Combination Therapies
The best studied combination therapy in SLE is rituximab and belimumab. Three phase II trials (Synbiose, CALIBRATE, and BEAT-LUPUS, Table 7) and the phase III BLISS-BELIEVE trial (Table 8) have explored the efficacy and safety of combining these two biologics.
Table 7.
Phase II trials of rituximab and belimumab (combination therapy).
Table 8.
Phase III trial of rituximab and belimumab (combination therapy).
The Synergic B-cell immunomodulation in SLE (Synbiose) trial [117] was a single-arm study investigating the immunological effects of sequential therapy with rituximab and belimumab. Sixteen patients with severe and refractory SLE were included. They were given intravenous rituximab 1 g at weeks 0 and 2, followed by intravenous belimumab at a dose of 10 mg/kg at weeks 4, 6, 8, and then monthly. The Synbiose trial demonstrated that belimumab, given after rituximab, inhibited the increasing levels of circulating BLyS detected in the serum of patients four weeks after the first infusion of rituximab. This effect was associated with a clinical improvement measured with disease activity indexes (SLEDAI and LLDAS). The median SLEDAI decreased significantly from 18 at baseline to 2 (p < 0.0001) at week 24 and thirteen patients achieved LLDAS at week 24.
A phase II, randomized, open-label (CALIBRATE) trial [118] assessed the safety of combining rituximab and belimumab in recurrent or refractory lupus nephritis. Forty-three adult patients, who previously had been treated with cyclophosphamide (CYC) or mycophenolate mofetil, were recruited and assigned to two groups. Every patient received CYC and intravenous rituximab at weeks 0 and 2. The patients of one group (RCB group) were given intravenous belimumab 10 mg/kg at weeks 4, 6, 8, and then monthly up to week 48. The patients in the other group (RC group) did not receive additional treatment beyond the CYC and rituximab. The study demonstrated that the combination of rituximab and belimumab was safe; the proportion of patients who experienced at least one severe AE did not differ significantly between the groups (23% in the RC group versus 9.5% in the RCB group, p > 0.05) at week 48. The combination of rituximab and belimumab was associated with a significantly lower B-cell count. This difference remained significant up to week 60 (p = 0.0012). Disappointingly, the RCB group did not have higher renal response rates compared to the RC group at all time points and the frequency of nonrenal flares was similar between the groups, suggesting that the addition of belimumab did not provide any clinical advantage. However, the trial was not powered to look for clinical change.
A 52 week, phase IIb, superiority RCT (BEAT-LUPUS) [119] studied the efficacy of belimumab given after rituximab infusions in patients with extrarenal and renal SLE. All fifty-two patients were given intravenous rituximab (1 g two weeks apart), after which they were randomly assigned to receive placebo or intravenous belimumab at the dose of 10 mg/kg at week 0, 2, 4, and then monthly up to week 52. The BEAT-LUPUS trial showed that, at 52 weeks, in the belimumab group there were significantly lower serum IgG anti-dsDNA antibody levels (primary endpoint of the study) than in the placebo group, 47 IU/mL versus 103 IU/mL of geometric mean, respectively (p < 0.001). The between-group difference in the serum anti-dsDNA levels was detectable since week 24 (p < 0.001). In contrast to the CALIBRATE trial, in the BEAT-LUPUS trial belimumab impacted significantly on the clinical outcome of the twenty-six belimumab-treated patients. Three had a severe flare compared to ten out of twenty-six in the placebo-treated group (p = 0.033). Considering safety, the frequency of adverse events was similar between the groups (two hundred forty-two AEs in the placebo group and two hundred forty-one AEs in the belimumab group). The BEAT-LUPUS trial thus demonstrated that the sequential therapy of belimumab given after rituximab is well tolerated and effective.
The BLISS-BELIEVE trial (NCT03312907) [120] was a 104-week, superiority, phase III, randomized controlled trial. It aimed to determine whether the combination of rituximab and belimumab was effective in achieving disease control and in reducing conventional therapy. Patients with severe kidney involvement or central nervous system lupus were excluded. Two hundred ninety-two patients were recruited and randomly assigned to three treatment arms. Patients in arm A (n = 72) received subcutaneous belimumab 200 mg weekly for 52 weeks and placebo infusions at weeks 4 and 6; in arm B (n = 144), instead of placebo infusions, patients received intravenous rituximab 1000 mg in addition to weekly subcutaneous belimumab injections. Those assigned to arm C (n = 76) were given subcutaneous belimumab 200 mg weekly plus standard of care up to 104 weeks. The primary endpoint was the proportion of patients who, at 52 weeks, achieved a state of disease control, defined as a SLEDAI-2K score ≤2, while keeping the steroid dose ≤5 mg/day and without immunosuppressants. The study did not meet its primary endpoint. There was no significant difference between the three arms in terms of achieving disease control. This was achieved by 16.7% of patients in arm A, 19.4% in arm B, and 25.5% in arm C.
A phase III open-label trial (Synbiose-2, NCT03747159) investigating the combination of belimumab and rituximab is ongoing. Seventy adults with severe extrarenal SLE and lupus nephritis will be enrolled and randomized to two treatment arms. One arm will receive subcutaneous belimumab at the dose of 200 mg weekly for two years, two intravenous infusion of rituximab 1000 mg at week 4 and 6, and the standard of care (steroids and mycophenolate mofetil). The other arm will receive only the standard of care. The primary endpoint of the study will be the proportion of treatment failures at 2 years. At 2 years the proportion of partial and complete renal responses will also be evaluated in patients with lupus nephritis. At frequent time points during the study the frequency of moderate to severe flares, the proportion of patients able to reduce concomitant immunosuppressants without flares, the disease activity captured by SLEDAI-2K, damage by using the System Lupus International Collaborating Clinics/American College of Rheumatology Damage Index for Systemic Lupus Erythematosus (SLICC), plus quality of life and safety will be assessed.
Apart from the sequential administration of rituximab and belimumab, elsubrutinib (a BTK inhibitor) and upadacitinib (a JAK inhibitor) have been investigated together as a combination therapy in a phase II trial (SLEek, NCT03978520), which has been recently completed. The SLEek study evaluated the proportion of SRI-4 responders who tapered the steroid dose to ≤10 mg of prednisone equivalent at week 24 and, as secondary endpoints, the achievement of SRI-4, BICLA, LLDAS, the number of flares, and the change in steroids dose. The results are awaited.
7. Pediatric SLE
The treatment of children with SLE is still challenging due to the small representation of this age group in clinical trials. Indeed, RCT eligibility criteria often require patients to be adults. As mentioned above, in pediatric lupus patients with renal and extra-renal involvement only intravenous belimumab has been approved by the FDA as a biological therapy. A phase II trial (PLUTO, NCT01649765) [73] showed that pediatric lupus patients (5–17 years) treated with intravenous belimumab had more favorable clinical outcomes compared to those treated with placebo, with 52.8% SRI-4 responders at 52 weeks versus 43.6%. The p-value was not reported, since the study did not enroll an adequate number of patients to detect a statistically significant difference. The subcutaneous formulation of belimumab has not yet been approved in children and adolescents. A phase II trial (NCT04179032) evaluating the pharmacokinetics and pharmacodynamics of subcutaneous belimumab in pediatric patients aged 5–17 years is currently ongoing. Among the phase II trials reported in this review, the only one that enrolled adolescents was the APRIL-SLE trial. However, a greater number of phase III trials will include adolescent patients, notably the studies evaluating dapirolizumab (PHOENYCS GO, NCT04294667, and NCT04976322: ≥16 years), telitacicept (NCT05306574, 12–70 years), and the OBILUP trial of obinutuzumab (NCT04702256), which will enroll patients aged at least 14 years. These trials will provide useful data on the efficacy of emerging drugs in adolescents. Given that none of these studies will enroll patients aged less than twelve years, the data on the efficacy in children will continue to be lacking.
8. Resume of Drugs in Phase I Clinical Trials
Several ongoing phase I clinical trials are evaluating the safety profile, pharmacokinetics, and pharmacodynamics of new potential drugs. We will briefly summarize the biological drugs that are in phase I trials: mosunetuzumab, itolizumab, CM313, and DS-7011a.
Mosunetuzumab, a CD20/CD3 bispecific antibody, engages T cells and redirects them to attack CD20+ B-cells [121]. It has been recently approved in Europe for the treatment of follicular lymphoma [122]. A phase I trial (NCT05155345) will evaluate the tolerance and the effect on the B-cell count of two different doses of mosunetuzumab in patients with active SLE (SLEDAI-2K ≥ 4).
Another biological drug targeting a molecule expressed on T cells is itolizumab. This is a humanized IgG1 monoclonal antibody which binds CD6 and downregulates T cell activation [123]. A phase I trial (EQUALISE, NCT04128579) will explore the safety profile of different doses of itolizumab in two patient cohorts, one with non-renal lupus and the other focusing on patients with proliferative lupus nephritis (class III or IV).
CM313 is a monoclonal antibody directed against CD38, a glycoprotein expressed mainly on plasma cells [124]. A phase Ib/II, randomized, double-blind, placebo-controlled trial (NCT05465707) will evaluate its safety, pharmacokinetics, and preliminary efficacy in patients with SLE who will receive different doses of CM313 (2 mg/kg, 4 mg/kg, 8 mg/kg, 16 mg/kg).
DS-7011a is a monoclonal antibody targeting the Toll-like receptor 7 (TLR7), which has been implicated in the promotion of autoimmune responses [125]. A phase Ib/II, randomized, double-blind, placebo-controlled trial (NCT05638802) will enroll twenty-four patients, to be randomized to receive placebo or intravenous infusions of DS-7011a at a dose of 20 mg/kg every 4 weeks. The proportion of AEs, multiple pharmacokinetic parameters, the change in disease activity, in the autoantibodies, and in complement component levels will be measured.
9. Discussion
In this review, the trials of biological therapies approved or under evaluation in lupus patients have been summarized. Many factors are thought to have limited the development of biologics in SLE, so that the treatment remains mostly based on conventional immunosuppressants, which have several adverse events and can increase the risk of damage accrual, especially in the case of corticosteroids.
Belimumab and anifrolumab are the two biologics approved by the FDA in lupus patients. However, few patients can benefit from these drugs, because of the limitations in their prescription. Rituximab has never been approved by regulatory agencies (except for NHS England in the UK), but it is recommended as an off-label treatment in case of severe or refractory SLE. B-cells have long represented an attractive target in SLE since they are thought to play a central role in the pathogenesis. Combining B-cell depletion with rituximab and anti-BLyS treatment with belimumab has proved effective in reducing the flare rate in the BEAT-LUPUS trial [119], without concerns regarding the safety. This supported the hypothesis that the EXPLORER [58] and LUNAR [59] trials failed because of the surge of BLyS observed after the infusions of rituximab [61]. Unfortunately, the BLISS-BELIEVE study was not successful so that the future for this form of combination therapy remains uncertain.
More encouragingly, six new biological drugs showed promising results in phase II trials and will be soon evaluated in phase III trials. The majority of the phase II trials included patients without lupus nephritis. Dapirolizumab, deucravacitinib, litifilimab, and telitacicept improved multiple clinical outcome measures in patients with extra-renal SLE and their safety profile was considered acceptable. An increase frequency of cutaneous AEs was noted during treatment with deucravacitinib, but these were not considered severe.
With regard to lupus nephritis, obinutuzumab and atacicept have been evaluated in phase II trials. In the NOBILITY trial [88], a rapid and sustained B-cell depletion and a higher proportion of renal responses were reported with obinutuzumab compared to placebo. If phase III trials confirm the beneficial effect reported, obinutuzumab could potentially replace rituximab in the treatment of refractory lupus nephritis. Indeed, fully humanized monoclonal antibodies compared to chimeric monoclonal antibodies have a lower risk of allergic reactions, a common issue that has often limited the use of rituximab. However, it must be acknowledged that the NOBILITY trial had some limitations, notably the limited sample size of one hundred twenty-five participants and the prespecified alpha level of 0.2.
The efficacy of atacicept in LN could not be evaluated in the phase II trial since it was discontinued prematurely due to the inadvertent concern about the infectious risk [103]. Two deaths due to infection occurred in patients on atacicept 150 mg in the APRIL-SLE trial with a subsequent termination of this treatment arm [101]. To note, the number of infections was not greater than the one observed in other previous lupus trials that were not discontinued and achieved significant responses. Furthermore, a subsequent integrated analysis of atacicept trials has provided reassurance that this monoclonal is not linked to an increased infection risk [126]. Despite the failure of the phase II trials in extra-renal SLE, it seemed that atacicept might be effective in a subgroup of patients with active serology and high disease activity.
10. Conclusions
The development of biologics in SLE clearly lags behind the therapeutic successes seen in other rheumatological diseases, with only two biologics approved in lupus patients. The analysis of the trials of rituximab, belimumab, and anifrolumab has taught the investigators that an accurate design of the studies and a careful choice of the outcome measures are vital to detect the potential benefit of new biological drugs in SLE. In addition, in such a heterogeneous disease as SLE, the beneficial effects of some drugs might not be evident in the whole population of lupus patients, but only in specific subgroups. The experience gained from the lupus trials should now allow better design of phase II and III studies which should eventually allow more effective biological drugs to emerge and take their place in the pantheon of lupus therapy.
11. Future directions
New effective biological drugs could at least partially replace conventional immunosuppressants, potentially leading to a reduction in organ damage and a subsequent improvement in the prognosis and patients’ quality of life. Moreover, with the expansion of the therapeutic armamentarium, different approaches to the disease would be feasible, notably combination and sequential therapies. Biological drugs targeting different molecules could also be the basis of a personalized medicine, tailored to the individual patient and their immunological alterations.
Funding
This research received no external funding.
Data Availability Statement
Data sharing is not applicable to this article.
Conflicts of Interest
D.A.I. has received honoraria from Amgen, Astra-Zeneca, UCB pharma, Vera Therapeutics, Merck Serono. These honoraria are passed onto a local arthritis charity. V.V. has declared no conflicts of interest.
References
- Bruce, I.N.; O’Keeffe, A.G.; Farewell, V.; Hanly, J.G.; Manzi, S.; Su, L.; Gladman, D.D.; Bae, S.-C.; Sanchez-Guerrero, J.; Romero-Diaz, J.; et al. Factors associated with damage accrual in patients with systemic lupus erythematosus: Results from the Systemic Lupus International Collaborating Clinics (SLICC) Inception Cohort. Ann. Rheum. Dis. 2015, 74, 1706–1713. [Google Scholar] [CrossRef] [PubMed]
- Moghaddam, B.; Marozoff, S.; Li, L.; Sayre, E.C.; Zubieta, J.A.A. All-cause and cause-specific mortality in systemic lupus erythematosus: A population-based study. Rheumatology 2021, 61, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Merrell, M.; Shulman, L.E. Determination of prognosis in chronic disease, illustrated by systemic lupus erythematosus. J. Chronic Dis. 1955, 1, 12–32. [Google Scholar] [CrossRef] [PubMed]
- Bernatsky, S.; Boivin, J.-F.; Joseph, L.; Manzi, S.; Ginzler, E.; Gladman, D.D.; Urowitz, M.; Fortin, P.R.; Petri, M.; Barr, S.; et al. Mortality in systemic lupus erythematosus. Arthritis Rheum. 2006, 54, 2550–2557. [Google Scholar] [CrossRef] [PubMed]
- Borchers, A.T.; Keen, C.L.; Shoenfeld, Y.; Gershwin, M. Surviving the butterfly and the wolf: Mortality trends in systemic lupus erythematosus. Autoimmun. Rev. 2004, 3, 423–453. [Google Scholar] [CrossRef]
- Gladman, D.D.; Urowitz, M.B.; Rahman, P.; Ibañez, M.; Tam, L.-S. Accrual of Organ Damage Over Time in Patients with Systemic Lupus Erythematosus. J. Rheumatol. 2003, 30, 1955–1959. [Google Scholar]
- Frodlund, M.; Reid, S.; Wetterö, J.; Dahlström; Sjöwall, C.; Leonard, D. The majority of Swedish systemic lupus erythematosus patients are still affected by irreversible organ impairment: Factors related to damage accrual in two regional cohorts. Lupus 2019, 28, 1261–1272. [Google Scholar] [CrossRef]
- Segura, B.T.; Bernstein, B.S.; McDonnell, T.; Wincup, C.; Ripoll, V.M.; Giles, I.; Isenberg, D.; Rahman, A. Damage accrual and mortality over long-term follow-up in 300 patients with systemic lupus erythematosus in a multi-ethnic British cohort. Rheumatology 2019, 59, 524–533. [Google Scholar] [CrossRef]
- Durcan, L.; O’Dwyer, T.; Petri, M. Management strategies and future directions for systemic lupus erythematosus in adults. Lancet 2019, 393, 2332–2343. [Google Scholar] [CrossRef]
- Burmester, G.R.; Bijlsma, J.W.J.; Cutolo, M.; McInnes, I.B. Managing rheumatic and musculoskeletal diseases — past, present and future. Nat. Rev. Rheumatol. 2017, 13, 443–448. [Google Scholar] [CrossRef]
- Fanouriakis, A.; Adamichou, C.; Koutsoviti, S.; Panopoulos, S.; Staveri, C.; Klagou, A.; Tsalapaki, C.; Pantazi, L.; Konsta, S.; Mavragani, C.P.; et al. Low disease activity—irrespective of serologic status at baseline—associated with reduction of corticosteroid dose and number of flares in patients with systemic lupus erythematosus treated with belimumab: A real-life observational study. Semin. Arthritis Rheum. 2018, 48, 467–474. [Google Scholar] [CrossRef]
- Agnihotri, P.; Fazel-Rezai, R.; Kaabouch, N. Comparative analysis of various brain imaging techniques. Annu. Int. Conf. IEEE Eng. Med. Biol. Soc. 2010, 2010, 3029–3032. [Google Scholar] [CrossRef] [PubMed]
- Aguiar, R.; Araújo, C.; Martins-Coelho, G.; Isenberg, D. Use of Rituximab in Systemic Lupus Erythematosus: A Single Center Experience Over 14 Years. Arthritis Care Res. 2017, 69, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Yusof, Y.M.; Shaw, D.; El-Sherbiny, Y.M.; Dunn, E.; Rawstron, A.C.; Emery, P.; Vital, E.M. Predicting and managing primary and secondary non-response to rituximab using B-cell biomarkers in systemic lupus erythematosus. Ann. Rheum. Dis. 2017, 76, 1829–1836. [Google Scholar] [CrossRef]
- Collins, C.E.; Cortes-Hernández, J.; Garcia, M.A.; Von Kempis, J.; Schwarting, A.; Touma, Z.; Kurtinecz, M.; Gairy, K. Real-World Effectiveness of Belimumab in the Treatment of Systemic Lupus Erythematosus: Pooled Analysis of Multi-Country Data from the OBSErve Studies. Rheumatol. Ther. 2020, 7, 949–965. [Google Scholar] [CrossRef] [PubMed]
- Freitas, S.; Ruiz, M.M.; Carneiro, A.C.; Isenberg, D.A. Why do some patients with systemic lupus erythematosus fail to respond to B-cell depletion using rituximab? Clin. Exp. Rheumatol. 2020, 38, 262–266. [Google Scholar] [CrossRef] [PubMed]
- Hennessey, A.; Lukawska, J.; Cambridge, G.; Isenberg, D.; Leandro, M. Adverse infusion reactions to rituximab in systemic lupus erythematosus: A retrospective analysis. BMC Rheumatol. 2019, 3, 32. [Google Scholar] [CrossRef]
- Fanouriakis, A.; Kostopoulou, M.; Alunno, A.; Aringer, M.; Bajema, I.; Boletis, J.N.; Cervera, R.; Doria, A.; Gordon, C.; Govoni, M.; et al. 2019 update of the EULAR recommendations for the management of systemic lupus erythematosus. Ann. Rheum. Dis. 2019, 78, 736–745. [Google Scholar] [CrossRef]
- Gordon, C.; Amissah-Arthur, M.-B.; Gayed, M.; Brown, S.; Bruce, I.N.; D’cruz, D.; Empson, B.; Griffiths, B.; Jayne, D.; Khamashta, M.; et al. The British Society for Rheumatology guideline for the management of systemic lupus erythematosus in adults. Rheumatology 2018, 57, e1–e45. [Google Scholar] [CrossRef]
- Ruiz-Irastorza, G.; Ramos-Casals, M.; Zeron, P.B.; Khamashta, M.A. Clinical efficacy and side effects of antimalarials in systemic lupus erythematosus: A systematic review. Ann. Rheum. Dis. 2010, 69, 20–28. [Google Scholar] [CrossRef]
- Poon, I.K.H.; Lucas, C.D.; Rossi, A.G.; Ravichandran, K.S. Apoptotic cell clearance: Basic biology and therapeutic potential. Nat. Rev. Immunol. 2014, 14, 166–180. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Kaplan, M.J. The role of neutrophils and NETosis in autoimmune and renal diseases. Nat. Rev. Nephrol. 2016, 12, 402–413. [Google Scholar] [CrossRef] [PubMed]
- Yap, D.Y.H.; Chan, T.M. B Cell Abnormalities in Systemic Lupus Erythematosus and Lupus Nephritis—Role in Pathogenesis and Effect of Immunosuppressive Treatments. Int. J. Mol. Sci. 2019, 20, 6231. [Google Scholar] [CrossRef] [PubMed]
- Robinson, R. Distinct B Cell Receptor Functions Are Determined by Phosphorylation. PLoS Biol. 2006, 4, e231. [Google Scholar] [CrossRef]
- Wang, K.; Wei, G.; Liu, D. CD19: A biomarker for B cell development, lymphoma diagnosis and therapy. Exp. Hematol. Oncol. 2012, 1, 36. [Google Scholar] [CrossRef]
- Polyak, M.J.; Li, H.; Shariat, N.; Deans, J.P. CD20 Homo-oligomers Physically Associate with the B Cell Antigen Receptor. J. Biol. Chem. 2008, 283, 18545–18552. [Google Scholar] [CrossRef]
- E Mei, H.; Schmidt, S.; Dörner, T. Rationale of anti-CD19 immunotherapy: An option to target autoreactive plasma cells in autoimmunity. Arthritis Res. Ther. 2012, 14, S1. [Google Scholar] [CrossRef]
- O’Keefe, T.L.; Williams, G.T.; Batista, F.D.; Neuberger, M.S. Deficiency in CD22, a B Cell–specific Inhibitory Receptor, Is Sufficient to Predispose to Development of High Affinity Autoantibodies. J. Exp. Med. 1999, 189, 1307–1313. [Google Scholar] [CrossRef]
- Smith, K.G.C.; Clatworthy, M.R. FcγRIIB in autoimmunity and infection: Evolutionary and therapeutic implications. Nat. Rev. Immunol. 2010, 10, 328–343. [Google Scholar] [CrossRef]
- Honigberg, L.A.; Smith, A.M.; Sirisawad, M.; Verner, E.; Loury, D.; Chang, B.; Li, S.; Pan, Z.; Thamm, D.H.; Miller, R.A.; et al. The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy. Proc. Natl. Acad. Sci. USA 2010, 107, 13075–13080. [Google Scholar] [CrossRef]
- Leandro, M.J.; Edwards, J.C.; Cambridge, G.; Ehrenstein, M.; Isenberg, D. An open study of B lymphocyte depletion in systemic lupus erythematosus. Arthritis Rheum. 2002, 46, 2673–2677. [Google Scholar] [CrossRef] [PubMed]
- Cambridge, G.; Leandro, M.J.; Teodorescu, M.; Manson, J.; Rahman, A.; Isenberg, D.; Edwards, J.C. B cell depletion therapy in systemic lupus erythematosus: Effect on autoantibody and antimicrobial antibody profiles. Arthritis Rheum. 2006, 54, 3612–3622. [Google Scholar] [CrossRef] [PubMed]
- Khattri, S.; Zandman-Goddard, G.; Peeva, E. B-cell directed therapies in antiphospholipid antibody syndrome — New directions based on murine and human data. Autoimmun. Rev. 2012, 11, 717–722. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.-X.; Fan, Y.-T.; Peng, B.-W. Distinct mechanisms underlying therapeutic potentials of CD20 in neurological and neuromuscular disease. Pharmacol. Ther. 2022, 238, 108180. [Google Scholar] [CrossRef] [PubMed]
- Karimi, M.H.; Pourfathollah, A.A. CD40 and Tolerance Induction. Iran. J. Allergy Asthma Immunol. 2012, 11, 1–13. [Google Scholar]
- Qamar, N.; Fuleihan, R.L. The Hyper IgM Syndromes. Clin. Rev. Allergy Immunol. 2014, 46, 120–130. [Google Scholar] [CrossRef]
- Menard, L.C.; Habte, S.; Gonsiorek, W.; Lee, D.; Banas, D.; Holloway, D.A.; Manjarrez-Orduno, N.; Cunningham, M.; Stetsko, D.; Casano, F.; et al. B cells from African American lupus patients exhibit an activated phenotype. JCI Insight 2016, 1, e87310. [Google Scholar] [CrossRef]
- Duarte, J.H. ICOS sustains pathogenic T-cell survival in SLE mouse model. Nat. Rev. Rheumatol. 2015, 11, 260. [Google Scholar] [CrossRef]
- Thien, M.; Phan, T.; Gardam, S.; Amesbury, M.; Basten, A.; Mackay, F.; Brink, R. Excess BAFF Rescues Self-Reactive B Cells from Peripheral Deletion and Allows Them to Enter Forbidden Follicular and Marginal Zone Niches. Immunity 2004, 20, 785–798. [Google Scholar] [CrossRef]
- Carter, L.M.; Isenberg, D.; Ehrenstein, M.R. Elevated serum B-cell activating factor (BAFF / BLyS) is associated with rising anti-dsDNA antibody levels and flare following B-cell depletion therapy in systemic lupus erythematosus: BAFF and SLE Relapse Following Rituximab. Arthritis Rheum. 2013, 65, 2672–2679. [Google Scholar] [CrossRef]
- Michaelson, J.S.; Wisniacki, N.; Burkly, L.C.; Putterman, C. Role of TWEAK in Lupus Nephritis: A bench-to-bedside review. J. Autoimmun. 2012, 39, 130–142. [Google Scholar] [CrossRef] [PubMed]
- Min, H.-K.; Kim, S.-M.; Park, J.-S.; Byun, J.-K.; Lee, J.; Kwok, S.-K.; Park, Y.-W.; Cho, M.-L.; Park, S.-H. Fn14-Fc suppresses germinal center formation and pathogenic B cells in a lupus mouse model via inhibition of the TWEAK/Fn14 Pathway. J. Transl. Med. 2016, 14, 98. [Google Scholar] [CrossRef] [PubMed]
- Parodis, I.; Zickert, A.; Sundelin, B.; Axelsson, M.; Gerhardsson, J.; Svenungsson, E.; Malmström, V.; Gunnarsson, I. Evaluation of B lymphocyte stimulator and a proliferation inducing ligand as candidate biomarkers in lupus nephritis based on clinical and histopathological outcome following induction therapy. Lupus Sci. Med. 2015, 2, e000061. [Google Scholar] [CrossRef] [PubMed]
- Tilstra, J.S.; Avery, L.; Menk, A.V.; Gordon, R.A.; Smita, S.; Kane, L.P.; Chikina, M.; Delgoffe, G.M.; Shlomchik, M.J. Kidney-infiltrating T cells in murine lupus nephritis are metabolically and functionally exhausted. J. Clin. Investig. 2018, 128, 4884–4897. [Google Scholar] [CrossRef]
- Zhou, M.; Guo, C.; Li, X.; Huang, Y.; Li, M.; Zhang, T.; Zhao, S.; Wang, S.; Zhang, H.; Yang, N. JAK/STAT signaling controls the fate of CD8+CD103+ tissue-resident memory T cell in lupus nephritis. J. Autoimmun. 2020, 109, 102424. [Google Scholar] [CrossRef]
- Ripoll; de Ramon, L.; Draibe, J.; Merino, A.; Bolaños, N.; Goma, M.; Cruzado, J.M.; Grinyó, J.M.; Torras, J. JAK3-STAT pathway blocking benefits in experimental lupus nephritis. Arthritis Res. Ther. 2016, 18, 134. [Google Scholar] [CrossRef]
- Ittah, M.; Miceli-Richard, C.; Gottenberg, J.E.; Lavie, F.; Lazure, T.; Ba, N.; Sellam, J.; Lepajolec, C.; Mariette, X. B cell-activating factor of the tumor necrosis factor family (BAFF) is expressed under stimulation by interferon in salivary gland epithelial cells in primary Sjögren’s syndrome. Arthritis Res. Ther. 2006, 8, R51. [Google Scholar] [CrossRef]
- Crow, M.K. Type I Interferon in the Pathogenesis of Lupus. J. Immunol. 2014, 192, 5459–5468. [Google Scholar] [CrossRef]
- Schiller, M.; Parcina, M.; Heyder, P.; Foermer, S.; Ostrop, J.; Leo, A.; Heeg, K.; Herrmann, M.; Lorenz, H.-M.; Bekeredjian-Ding, I. Induction of Type I IFN Is a Physiological Immune Reaction to Apoptotic Cell-Derived Membrane Microparticles. J. Immunol. 2012, 189, 1747–1756. [Google Scholar] [CrossRef]
- Higgs, B.W.; Liu, Z.; White, B.; Zhu, W.; I White, W.; Morehouse, C.; Brohawn, P.; A Kiener, P.; Richman, L.; Fiorentino, D.; et al. Patients with systemic lupus erythematosus, myositis, rheumatoid arthritis and scleroderma share activation of a common type I interferon pathway. Ann. Rheum. Dis. 2011, 70, 2029–2036. [Google Scholar] [CrossRef]
- Oke, V.; Gunnarsson, I.; Dorschner, J.; Eketjäll, S.; Zickert, A.; Niewold, T.B.; Svenungsson, E. High levels of circulating interferons type I, type II and type III associate with distinct clinical features of active systemic lupus erythematosus. Arthritis Res. Ther. 2019, 21, 107. [Google Scholar] [CrossRef] [PubMed]
- Edwards, J.C.W.; Cambridge, G. Sustained improvement in rheumatoid arthritis following a protocol designed to deplete B lymphocytes. Rheumatology 2001, 40, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Krenn, V.; Souto-Carneiro, M.M.; Kim, H.J.; Berek, C.; Starostik, P.; König, A.; Harms, H.; Müller-Hermelink, H.K. Histopathology and molecular pathology of synovial B-lymphocytes in rheumatoid arthritis. Histol. Histopathol. 2000, 15, 791–798. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Yu, Y.; Gao, Y.; Zhao, F.; Liang, Z.; Gao, J. Comparative Effectiveness of Rituximab and Common Induction Therapies for Lupus Nephritis: A Systematic Review and Network Meta-Analysis. Front. Immunol. 2022, 13, 859380. [Google Scholar] [CrossRef] [PubMed]
- Cobo-Ibáñez, T.; Loza-Santamaría, E.; Pego-Reigosa, J.M.; Marqués, A.O.; Rúa-Figueroa; Fernandez-Nebro, A.; Cáliz, R.C.; Longo, F.J.L.; Muñoz-Fernández, S. Efficacy and safety of rituximab in the treatment of non-renal systemic lupus erythematosus: A systematic review. Semin. Arthritis Rheum. 2014, 44, 175–185. [Google Scholar] [CrossRef]
- Pongtarakulpanit, N.; Pisitkun, P.; Ngamjanyaporn, P. Efficacy and safety of rituximab biosimilar in refractory lupus. Lupus Sci. Med. 2020, 7, e000442. [Google Scholar] [CrossRef]
- da Costa, R.Q.; Aguirre-Alastuey, M.E.; Isenberg, D.A.; Saracino, A.M. Assessment of Response to B-Cell Depletion Using Rituximab in Cutaneous Lupus Erythematosus. JAMA Dermatol. 2018, 154, 1432–1440. [Google Scholar] [CrossRef]
- Merrill, J.T.; Neuwelt, C.M.; Wallace, D.J.; Shanahan, J.C.; Latinis, K.M.; Oates, J.C.; Utset, T.O.; Gordon, C.; Isenberg, D.A.; Hsieh, H.-J.; et al. Efficacy and safety of rituximab in moderately-to-severely active systemic lupus erythematosus: The randomized, double-blind, phase ii/iii systemic lupus erythematosus evaluation of rituximab trial. Arthritis Rheum. 2010, 62, 222–233. [Google Scholar] [CrossRef]
- Rovin, B.H.; Furie, R.; Latinis, K.; Looney, R.J.; Fervenza, F.C.; Sanchez-Guerrero, J.; Maciuca, R.; Zhang, D.; Garg, J.P.; Brunetta, P.; et al. Efficacy and safety of rituximab in patients with active proliferative lupus nephritis: The lupus nephritis assessment with rituximab study. Arthritis Rheum. 2012, 64, 1215–1226. [Google Scholar] [CrossRef]
- Ramos, L.; Isenberg, D. Rituximab: The Lupus Journey. Curr. Treat. Options Rheumatol. 2015, 1, 30–41. [Google Scholar] [CrossRef]
- Ehrenstein, M.; Wing, M.R.E.C. The BAFFling effects of rituximab in lupus: Danger ahead? Nat. Rev. Rheumatol. 2016, 12, 367–372. [Google Scholar] [CrossRef] [PubMed]
- Fanouriakis, A.; Kostopoulou, M.; Cheema, K.; Anders, H.-J.; Aringer, M.; Bajema, I.; Boletis, J.; Frangou, E.; A Houssiau, F.; Hollis, J.; et al. 2019 Update of the Joint European League Against Rheumatism and European Renal Association–European Dialysis and Transplant Association (EULAR/ERA–EDTA) recommendations for the management of lupus nephritis. Ann. Rheum. Dis. 2020, 79, 713–723. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Shi, X.; Xue, H.; Lv, H.; Yu, L.; Wu, X.; Wang, Q.; Wu, H.; Han, F.; Xue, J. Rituximab as maintenance therapy following remission induction in relapsing or refractory systemic lupus erythematosus. Rheumatology 2023, 62, 1145–1152. [Google Scholar] [CrossRef] [PubMed]
- Adams, M.D.; Kerlavage, A.R.; Fleischmann, R.D.; A Fuldner, R.; Bult, C.J.; Lee, N.H.; Kirkness, E.F.; Weinstock, K.G.; Gocayne, J.D.; White, O. Initial assessment of human gene diversity and expression patterns based upon 83 million nucleotides of cDNA sequence. Nature 1995, 377, 3–174. [Google Scholar] [PubMed]
- Mackay, F.; Woodcock, S.A.; Lawton, P.; Ambrose, C.; Baetscher, M.; Schneider, P.; Tschopp, J.; Browning, J. Mice Transgenic for Baff Develop Lymphocytic Disorders along with Autoimmune Manifestations. J. Exp. Med. 1999, 190, 1697–1710. [Google Scholar] [CrossRef] [PubMed]
- Cheema, G.S.; Roschke, V.; Hilbert, D.M.; Stohl, W. Elevated serum B lymphocyte stimulator levels in patients with systemic immune-based rheumatic diseases. Arthritis Rheum. 2001, 44, 1313–1319. [Google Scholar] [CrossRef] [PubMed]
- Stohl, W.; Hilbert, D.M. The discovery and development of belimumab: The anti-BLyS–lupus connection. Nat. Biotechnol. 2012, 30, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.J.; Stohl, W.; Furie, R.A.; Lisse, J.R.; Mckay, J.; Merrill, J.T.; Petri, M.A.; Ginzler, E.M.; Chatham, W.W.; McCune, W.J.; et al. A phase II, randomized, double-blind, placebo-controlled, dose-ranging study of belimumab in patients with active systemic lupus erythematosus. Arthritis Rheum. 2009, 61, 1168–1178. [Google Scholar] [CrossRef]
- Navarra, S.V.; Guzmán, R.M.; E Gallacher, A.; Hall, S.; A Levy, R.; E Jimenez, R.; Li, E.K.-M.; Thomas, M.; Kim, H.-Y.; León, M.G.; et al. Efficacy and safety of belimumab in patients with active systemic lupus erythematosus: A randomised, placebo-controlled, phase 3 trial. Lancet 2011, 377, 721–731. [Google Scholar] [CrossRef]
- Furie, R.; Petri, M.; Zamani, O.; Cervera, R.; Wallace, D.J.; Tegzová, D.; Sanchez-Guerrero, J.; Schwarting, A.; Merrill, J.T.; Chatham, W.W.; et al. A phase III, randomized, placebo-controlled study of belimumab, a monoclonal antibody that inhibits B lymphocyte stimulator, in patients with systemic lupus erythematosus. Arthritis Rheum. 2011, 63, 3918–3930. [Google Scholar] [CrossRef]
- Zhang, F.; Bae, S.-C.; Bass, D.; Chu, M.; Egginton, S.; Gordon, D.; A Roth, D.; Zheng, J.; Tanaka, Y. A pivotal phase III, randomised, placebo-controlled study of belimumab in patients with systemic lupus erythematosus located in China, Japan and South Korea. Ann. Rheum. Dis. 2018, 77, 355–363. [Google Scholar] [CrossRef] [PubMed]
- Ginzler, E.; Barbosa, L.S.G.; D’Cruz, D.; Furie, R.; Maksimowicz-McKinnon, K.; Oates, J.; Santiago, M.B.; Saxena, A.; Sheikh, S.; Bass, D.L.; et al. Phase III/IV, Randomized, Fifty-Two –Week Study of the Efficacy and Safety of Belimumab in Patients of Black African Ancestry With Systemic Lupus Erythematosus. Arthritis Rheumatol. 2022, 74, 112–123. [Google Scholar] [CrossRef] [PubMed]
- I Brunner, H.; Abud-Mendoza, C.; O Viola, D.; Penades, I.C.; Levy, D.; Anton, J.; E Calderon, J.; Chasnyk, V.G.; A Ferrandiz, M.; Keltsev, V.; et al. Safety and efficacy of intravenous belimumab in children with systemic lupus erythematosus: Results from a randomised, placebo-controlled trial. Ann. Rheum. Dis. 2020, 79, 1340–1348. [Google Scholar] [CrossRef]
- Stohl, W.; Schwarting, A.; Okada, M.; Scheinberg, M.; Doria, A.; Hammer, A.E.; Kleoudis, C.; Groark, J.; Bass, D.; Fox, N.L.; et al. Efficacy and Safety of Subcutaneous Belimumab in Systemic Lupus Erythematosus: A Fifty-Two–Week Randomized, Double-Blind, Placebo-Controlled Study. Arthritis Rheumatol. 2017, 69, 1016–1027. [Google Scholar] [CrossRef] [PubMed]
- Dooley, M.; Houssiau, F.; Aranow, C.; D’cruz, D.; Askanase, A.; Roth, D.; Zhong, Z.; Cooper, S.; Freimuth, W.; Ginzler, E. Effect of belimumab treatment on renal outcomes: Results from the phase 3 belimumab clinical trials in patients with SLE. Lupus 2013, 22, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Furie, R.; Rovin, B.H.; Houssiau, F.; Malvar, A.; Teng, Y.O.; Contreras, G.; Amoura, Z.; Yu, X.; Mok, C.-C.; Santiago, M.B.; et al. Two-Year, Randomized, Controlled Trial of Belimumab in Lupus Nephritis. N. Engl. J. Med. 2020, 383, 1117–1128. [Google Scholar] [CrossRef] [PubMed]
- Parodis, I.; Vital, E.M.; Hassan, S.-U.; Jönsen, A.; A Bengtsson, A.; Eriksson, P.; Leonard, D.; Gunnarsson, I.; Rönnblom, L.; Sjöwall, C. De novo lupus nephritis during treatment with belimumab. Rheumatology 2020, 60, 4348–4354. [Google Scholar] [CrossRef]
- Sjöwall, C.; Cöster, L. Belimumab may not prevent lupus nephritis in serologically active patients with ongoing non-renal disease activity. Scand. J. Rheumatol. 2014, 43, 428–430. [Google Scholar] [CrossRef]
- Staveri, C.; Karokis, D.; Liossis, S.-N.C. New onset of lupus nephritis in two patients with SLE shortly after initiation of treatment with belimumab. Semin. Arthritis Rheum. 2017, 46, 788–790. [Google Scholar] [CrossRef]
- Sheikh, S.Z.; A Scheinberg, M.; Wei, J.C.-C.; Tegzova, D.; Stohl, W.; de Toledo, R.A.; Mucenic, T.; Banfi, M.R.A.; Maksimowicz-McKinnon, K.; Abud-Mendoza, C.; et al. Mortality and adverse events of special interest with intravenous belimumab for adults with active, autoantibody-positive systemic lupus erythematosus (BASE): A multicentre, double-blind, randomised, placebo-controlled, phase 4 trial. Lancet Rheumatol. 2021, 3, e122–e130. [Google Scholar] [CrossRef]
- Xie, W.; Huang, H.; Zhan, S.; Zhang, Z. Risk of psychiatric disorders and all-cause mortality with belimumab therapy in patients with systemic lupus erythematosus: A meta-analysis of randomised controlled trials. Lupus Sci. Med. 2021, 8, e000534. [Google Scholar] [CrossRef] [PubMed]
- Ramaswamy, M.; Tummala, R.; Streicher, K.; da Costa, A.N.; Brohawn, P.Z. The Pathogenesis, Molecular Mechanisms, and Therapeutic Potential of the Interferon Pathway in Systemic Lupus Erythematosus and Other Autoimmune Diseases. Int. J. Mol. Sci. 2021, 22, 11286. [Google Scholar] [CrossRef] [PubMed]
- Furie, R.; Khamashta, M.; Merrill, J.T.; Werth, V.P.; Kalunian, K.; Brohawn, P.; Illei, G.G.; Drappa, J.; Wang, L.; Yoo, S.; et al. Anifrolumab, an Anti-Interferon-α Receptor Monoclonal Antibody, in Moderate-to-Severe Systemic Lupus Erythematosus. Arthritis Rheumatol. 2017, 69, 376–386. [Google Scholar] [CrossRef] [PubMed]
- A Furie, R.; Morand, E.F.; Bruce, I.N.; Manzi, S.; Kalunian, K.C.; Vital, E.M.; Ford, T.L.; Gupta, R.; Hiepe, F.; Santiago, M.; et al. Type I interferon inhibitor anifrolumab in active systemic lupus erythematosus (TULIP-1): A randomised, controlled, phase 3 trial. Lancet Rheumatol. 2019, 1, e208–e219. [Google Scholar] [CrossRef]
- Bruce, I.N.; A Furie, R.; Morand, E.F.; Manzi, S.; Tanaka, Y.; Kalunian, K.C.; Merrill, J.T.; Puzio, P.; Maho, E.; Kleoudis, C.; et al. Concordance and discordance in SLE clinical trial outcome measures: Analysis of three anifrolumab phase 2/3 trials. Ann. Rheum. Dis. 2022, 81, 962–969. [Google Scholar] [CrossRef]
- Morand, E.F.; Furie, R.; Tanaka, Y.; Bruce, I.N.; Askanase, A.D.; Richez, C.; Bae, S.-C.; Brohawn, P.Z.; Pineda, L.; Berglind, A.; et al. Trial of Anifrolumab in Active Systemic Lupus Erythematosus. N. Engl. J. Med. 2020, 382, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Mössner, E.; Brünker, P.; Moser, S.; Püntener, U.; Schmidt, C.; Herter, S.; Grau, R.; Gerdes, C.; Nopora, A.; van Puijenbroek, E.; et al. Increasing the efficacy of CD20 antibody therapy through the engineering of a new type II anti-CD20 antibody with enhanced direct and immune effector cell–mediated B-cell cytotoxicity. Blood 2010, 115, 4393–4402. [Google Scholar] [CrossRef] [PubMed]
- A Furie, R.; Aroca, G.; Cascino, M.D.; Garg, J.P.; Rovin, B.H.; Alvarez, A.; Fragoso-Loyo, H.; Zuta-Santillan, E.; Schindler, T.; Brunetta, P.; et al. B-cell depletion with obinutuzumab for the treatment of proliferative lupus nephritis: A randomised, double-blind, placebo-controlled trial. Ann. Rheum. Dis. 2022, 81, 100–107. [Google Scholar] [CrossRef]
- Robles-Carrillo, L.; Meyer, T.; Hatfield, M.; Desai, H.; Dávila, M.; Langer, F.; Amaya, M.; Garber, E.; Francis, J.L.; Hsu, Y.-M.; et al. Anti-CD40L Immune Complexes Potently Activate Platelets In Vitro and Cause Thrombosis in FCGR2A Transgenic Mice. J. Immunol. 2010, 185, 1577–1583. [Google Scholar] [CrossRef]
- Tocoian, A.; Buchan, P.; Kirby, H.; Soranson, J.; Zamacona, M.; Walley, R.; Mitchell, N.; Esfandiari, E.; Wagner, F.; Oliver, R. First-in-human trial of the safety, pharmacokinetics and immunogenicity of a PEGylated anti-CD40L antibody fragment (CDP7657) in healthy individuals and patients with systemic lupus erythematosus. Lupus 2015, 24, 1045–1056. [Google Scholar] [CrossRef]
- Chamberlain, C.; Colman, P.J.; Ranger, A.M.; Burkly, L.C.; I Johnston, G.; Otoul, C.; Stach, C.; Zamacona, M.; Dörner, T.; Urowitz, M.; et al. Repeated administration of dapirolizumab pegol in a randomised phase I study is well tolerated and accompanied by improvements in several composite measures of systemic lupus erythematosus disease activity and changes in whole blood transcriptomic profiles. Ann. Rheum. Dis. 2017, 76, 1837–1844. [Google Scholar] [CrossRef] [PubMed]
- A Furie, R.; Bruce, I.N.; Dörner, T.; Leon, M.G.; Leszczyński, P.; Urowitz, M.; Haier, B.; Jimenez, T.; Brittain, C.; Liu, J.; et al. Phase 2, randomized, placebo-controlled trial of dapirolizumab pegol in patients with moderate-to-severe active systemic lupus erythematosus. Rheumatology 2021, 60, 5397–5407. [Google Scholar] [CrossRef] [PubMed]
- Hoy, S.M. Deucravacitinib: First Approval. Drugs 2022, 82, 1671–1679. [Google Scholar] [CrossRef] [PubMed]
- Morand, E.; Pike, M.; Merrill, J.T.; van Vollenhoven, R.; Werth, V.P.; Hobar, C.; Delev, N.; Shah, V.; Sharkey, B.; Wegman, T.; et al. Deucravacitinib, a Tyrosine Kinase 2 Inhibitor, in Systemic Lupus Erythematosus: A Phase II, Randomized, Double-Blind, Placebo-Controlled Trial. Arthritis Rheumatol. 2023, 75, 242–252. [Google Scholar] [CrossRef]
- Winthrop, K.L. The emerging safety profile of JAK inhibitors in rheumatic disease. Nat. Rev. Rheumatol. 2017, 13, 234–243. [Google Scholar] [CrossRef]
- Dzionek, A.; Fuchs, A.; Schmidt, P.; Cremer, S.; Zysk, M.; Miltenyi, S.; Buck, D.W.; Schmitz, J. BDCA-2, BDCA-3, and BDCA-4: Three Markers for Distinct Subsets of Dendritic Cells in Human Peripheral Blood. J. Immunol. 2000, 165, 6037–6046. [Google Scholar] [CrossRef]
- Blomberg, S.; Eloranta, M.-L.; Magnusson, M.; Alm, G.V.; Rönnblom, L. Expression of the markers BDCA-2 and BDCA-4 and production of interferon-α by plasmacytoid dendritic cells in systemic lupus erythematosus: BDCA-2/4 Expression and IFNα Production in SLE. Arthritis Rheum. 2003, 48, 2524–2532. [Google Scholar] [CrossRef]
- Furie, R.A.; van Vollenhoven, R.F.; Kalunian, K.; Navarra, S.; Romero-Diaz, J.; Werth, V.P.; Huang, X.; Clark, G.; Carroll, H.; Meyers, A.; et al. Trial of Anti-BDCA2 Antibody Litifilimab for Systemic Lupus Erythematosus. N. Engl. J. Med. 2022, 387, 894–904. [Google Scholar] [CrossRef]
- Kaegi, C.; Steiner, U.C.; Wuest, B.; Crowley, C.; Boyman, O. Systematic Review of Safety and Efficacy of Atacicept in Treating Immune-Mediated Disorders. Front. Immunol. 2020, 11, 433. [Google Scholar] [CrossRef]
- Carter, R.H.; Zhao, H.; Liu, X.; Pelletier, M.; Chatham, W.; Kimberly, R.; Zhou, T. Expression and occupancy of BAFF-R on B cells in systemic lupus erythematosus. Arthritis Rheum. 2005, 52, 3943–3954. [Google Scholar] [CrossRef]
- Isenberg, D.; Gordon, C.; Licu, D.; Copt, S.; Rossi, C.P.; Wofsy, D. Efficacy and safety of atacicept for prevention of flares in patients with moderate-to-severe systemic lupus erythematosus (SLE): 52-week data (APRIL-SLE randomised trial). Ann. Rheum. Dis. 2014, 74, 2006–2015. [Google Scholar] [CrossRef]
- Merrill, J.T.; Wallace, D.J.; Wax, S.; Kao, A.; Fraser, P.A.; Chang, P.; Isenberg, D.; on behalf of the ADDRESS II Investigators. Efficacy and Safety of Atacicept in Patients with Systemic Lupus Erythematosus. Arthritis Rheumatol. 2017, 70, 266–276. [Google Scholar] [CrossRef] [PubMed]
- Ginzler, E.M.; Wax, S.; Rajeswaran, A.; Copt, S.; Hillson, J.; Ramos, E.; Singer, N.G. Atacicept in combination with MMF and corticosteroids in lupus nephritis: Results of a prematurely terminated trial. Arthritis Res. Ther. 2012, 14, R33. [Google Scholar] [CrossRef] [PubMed]
- Rovin, B.H.; Teng, Y.K.O.; Ginzler, E.M.; Arriens, C.; Caster, D.J.; Romero-Diaz, J.; Gibson, K.; Kaplan, J.; Lisk, L.; Navarra, S.; et al. Efficacy and safety of voclosporin versus placebo for lupus nephritis (AURORA 1): A double-blind, randomised, multicentre, placebo-controlled, phase 3 trial. Lancet 2021, 397, 2070–2080. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.J.; A Isenberg, D.; Morand, E.F.; Vazquez–Mateo, C.; Kao, A.H.; Aydemir, A.; Pudota, K.; Ona, V.; Aranow, C.; Merrill, J.T. Safety and clinical activity of atacicept in the long-term extension of the phase 2b ADDRESS II study in systemic lupus erythematosus. Rheumatology 2021, 60, 5379–5389. [Google Scholar] [CrossRef]
- Shi, F.; Xue, R.; Zhou, X.; Shen, P.; Wang, S.; Yang, Y. Telitacicept as a BLyS/APRIL dual inhibitor for autoimmune disease. Immunopharmacol. Immunotoxicol. 2021, 43, 666–673. [Google Scholar] [CrossRef]
- Dhillon, S. Telitacicept: First Approval. Drugs 2021, 81, 1671–1675. [Google Scholar] [CrossRef]
- A Human Recombinant Fusion Protein Targeting B Lymphocyte Stimulator (BlyS) and a Proliferation-Inducing Ligand (APRIL), Telitacicept (RC18), in Systemic Lupus Erythematosus (SLE): Results of a Phase 2b Study. ACR Meeting Abstracts n.d. Available online: https://acrabstracts.org/abstract/a-human-recombinant-fusion-protein-targeting-b-lymphocyte-stimulator-blys-and-a-proliferation-inducing-ligand-april-telitacicept-rc18-in-systemic-lupus-erythematosus-sle-results-of-a-phase/ (accessed on 25 March 2023).
- Telitacicept, a Human Recombinant Fusion Protein Targeting B Lymphocyte Stimulator (BlyS) and a Proliferation-Inducing Ligand (APRIL), in Systemic Lupus Erythematosus (SLE): Results of a Phase 3 Study. ACR Meeting Abstracts n.d. Available online: https://acrabstracts.org/abstract/telitacicept-a-human-recombinant-fusion-protein-targeting-b-lymphocyte-stimulator-blys-and-a-proliferation-inducing-ligand-april-in-systemic-lupus-erythematosus-sle-results-of-a-phase-3-study/ (accessed on 25 March 2023).
- Chen, R.; Fu, R.; Lin, Z.; Huang, C.; Huang, W. The efficacy and safety of telitacicept for the treatment of systemic lupus erythematosus: A real life observational study. Lupus 2023, 32, 94–100. [Google Scholar] [CrossRef]
- Merrill, J.T.; Van Vollenhoven, R.F.; Buyon, J.; A Furie, R.; Stohl, W.; Morgan-Cox, M.; Dickson, C.; Anderson, P.W.; Lee, C.; Berclaz, P.-Y.; et al. Efficacy and safety of subcutaneous tabalumab, a monoclonal antibody to B-cell activating factor, in patients with systemic lupus erythematosus: Results from ILLUMINATE-2, a 52-week, phase III, multicentre, randomised, double-blind, placebo-controlled study. Ann. Rheum. Dis. 2016, 75, 332–340. [Google Scholar] [CrossRef]
- A Isenberg, D.; Petri, M.; Kalunian, K.C.; Tanaka, Y.; Urowitz, M.B.; Hoffman, R.W.; Morgan-Cox, M.; Iikuni, N.; Silk, M.; Wallace, D.J. Efficacy and safety of subcutaneous tabalumab in patients with systemic lupus erythematosus: Results from ILLUMINATE-1, a 52-week, phase III, multicentre, randomised, double-blind, placebo-controlled study. Ann. Rheum. Dis. 2016, 75, 323–331. [Google Scholar] [CrossRef]
- A Furie, R.; Leon, G.; Thomas, M.; A Petri, M.; Chu, A.D.; Hislop, C.; Martin, R.S.; A Scheinberg, M. A phase 2, randomised, placebo-controlled clinical trial of blisibimod, an inhibitor of B cell activating factor, in patients with moderate-to-severe systemic lupus erythematosus, the PEARL-SC study. Ann. Rheum. Dis. 2015, 74, 1667–1675. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.J.; Kalunian, K.; A Petri, M.; Strand, V.; A Houssiau, F.; Pike, M.; Kilgallen, B.; Bongardt, S.; Barry, A.; Kelley, L.; et al. Efficacy and safety of epratuzumab in patients with moderate/severe active systemic lupus erythematosus: Results from EMBLEM, a phase IIb, randomised, double-blind, placebo-controlled, multicentre study. Ann. Rheum. Dis. 2014, 73, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Merrill, J.T.; Shanahan, W.R.; Scheinberg, M.; Kalunian, K.C.; Wofsy, D.; Martin, R.S. Phase III trial results with blisibimod, a selective inhibitor of B-cell activating factor, in subjects with systemic lupus erythematosus (SLE): Results from a randomised, double-blind, placebo-controlled trial. Ann. Rheum. Dis. 2018, 77, 883–889. [Google Scholar] [CrossRef] [PubMed]
- Clowse, M.E.B.; Wallace, D.J.; Furie, R.A.; Petri, M.A.; Pike, M.C.; Leszczyński, P.; Neuwelt, C.M.; Hobbs, K.; Keiserman, M.; Duca, L.; et al. Efficacy and Safety of Epratuzumab in Moderately to Severely Active Systemic Lupus Erythematosus: Results from Two Phase III Randomized, Double-Blind, Placebo-Controlled Trials. Arthritis Rheumatol. 2017, 69, 362–375. [Google Scholar] [CrossRef]
- Kraaij, T.; Kamerling, S.W.; de Rooij, E.N.; van Daele, P.L.; Bredewold, O.W.; Bakker, J.A.; Bajema, I.M.; Scherer, H.U.; Toes, R.E.; Huizinga, T.J.; et al. The NET-effect of combining rituximab with belimumab in severe systemic lupus erythematosus. J. Autoimmun. 2018, 91, 45–54. [Google Scholar] [CrossRef]
- Atisha-Fregoso, Y.; Malkiel, S.; Harris, K.M.; Byron, M.; Ding, L.; Kanaparthi, S.; Barry, W.T.; Gao, W.; Ryker, K.; Tosta, P.; et al. Phase II Randomized Trial of Rituximab Plus Cyclophosphamide Followed by Belimumab for the Treatment of Lupus Nephritis. Arthritis Rheumatol. 2021, 73, 121–131. [Google Scholar] [CrossRef]
- Shipa, M.M.; Embleton-Thirsk, A.; Parvaz, M.M.; Santos, L.R.; Muller, M.P.; Chowdhury, M.K.; Isenberg, D.A.; Doré, B.C.J.; Gordon, M.C.; Ehrenstein, M.M.R.; et al. Effectiveness of Belimumab After Rituximab in Systemic Lupus Erythematosus: A Randomized Controlled Trial. Ann. Intern. Med. 2021, 174, 1647–1657. [Google Scholar] [CrossRef]
- Aranow, C. Efficacy and Safety of Subcutaneous Belimumab (BEL) and Rituximab (RTX) Sequential Therapy in Patients with Systemic Lupus Erythematosus: The Phase 3, Randomized, Placebo-Controlled BLISS-BELIEVE Study. ACR Meeting Abstracts n.d. Available online: https://acrabstracts.org/abstract/efficacy-and-safety-of-subcutaneous-belimumab-bel-and-rituximab-rtx-sequential-therapy-in-patients-with-systemic-lupus-erythematosus-the-phase-3-randomized-placebo-controlled-bliss-believe-stud/ (accessed on 20 March 2023).
- Lim, S.M.; Pyo, K.-H.; A Soo, R.; Cho, B.C. The promise of bispecific antibodies: Clinical applications and challenges. Cancer Treat. Rev. 2021, 99, 102240. [Google Scholar] [CrossRef]
- Kang, C. Mosunetuzumab: First Approval. Drugs 2022, 82, 1229–1234. [Google Scholar] [CrossRef]
- Rodriguez, P.C.; Torres-Moya, R.; Reyes, G.; Molinero, C.; Prada, D.; Lopez, A.M.; Hernandez, I.M.; Hernandez, M.V.; Martinez, J.P.; Hernandez, X.; et al. A clinical exploratory study with itolizumab, an anti-CD6 monoclonal antibody, in patients with rheumatoid arthritis. Results Immunol. 2012, 2, 204–211. [Google Scholar] [CrossRef]
- Morandi, F.; Horenstein, A.L.; Costa, F.; Giuliani, N.; Pistoia, V.; Malavasi, F. CD38: A Target for Immunotherapeutic Approaches in Multiple Myeloma. Front. Immunol. 2018, 9, 2722. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Peng, Y.; Li, H.; Zhang, X. From monogenic lupus to TLR7/MyD88-targeted therapy. Innovation 2022, 3, 100299. [Google Scholar] [CrossRef] [PubMed]
- Gordon, C.; Bassi, R.; Chang, P.; Kao, A.; Jayne, D.; Wofsy, D.; Fleuranceau-Morel, P. Integrated safety profile of atacicept: An analysis of pooled data from the atacicept clinical trial programme. Rheumatol. Adv. Pr. 2019, 3, rkz021. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).