



Bilateral Uveal Melanoma: An Insight into Genetic Predisposition in Four New Unrelated Patients and Review of Published Cases

 ,
,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patient Cohort
2.2. Exome Sequencing and Genetic Testing
2.3. Variant Prioritization and Candidate Gene Selection
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Singh, A.D.; Shields, C.L.; Shields, J.A.; De Potter, P. Bilateral primary uveal melanoma. Bad luck or bad genes? Ophthalmology 1996, 103, 256–262. [Google Scholar] [CrossRef] [PubMed]
- Kiratli, H.; Bilgic, S. Sequential development of bilateral primary choroidal melanoma. Acta Ophthalmol. Scand. 2000, 78, 474–476. [Google Scholar] [CrossRef] [PubMed]
- Wiesinger, H.; Phipps, G.W.; Guerry, D., 3rd. Bilateral melanoma of the choroid associated with leukemia and meningioma. Arch. Ophthalmol. 1959, 62, 889–893. [Google Scholar] [CrossRef] [PubMed]
- Gonder, J.R.; Shields, J.A.; Shakin, J.L.; Albert, D.M. Bilateral ocular melanocytosis with malignant melanoma of the choroid. Br. J. Ophthalmol. 1981, 65, 843–845. [Google Scholar] [CrossRef] [PubMed]
- Migdal, C.; Macfarlane, A. Bilateral primary choroidal melanoma. Br. J. Ophthalmol. 1984, 68, 268–271. [Google Scholar] [CrossRef] [PubMed]
- Turaka, K.; Shields, C.L.; Shah, C.P.; Say, E.A.; Shields, J.A. Bilateral uveal melanoma in an arc welder. Graefes Arch. Clin. Exp. Ophthalmol. 2011, 249, 141–144. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.D.; Masoomian, B.; Shields, J.A.; Shields, C.L. BAP1 Germline Mutation Associated with Bilateral Primary Uveal Melanoma. Ocul. Oncol. Pathol. 2020, 6, 10–14. [Google Scholar] [CrossRef]
- Scott, J.F.; Vyas, R.; Galvin, J.; Gotow, E.; Fiessinger, L.; Gerstenblith, A.T.; Gerstenblith, M.R. Primary bilateral uveal melanoma: A population-based study and systematic review. Clin. Exp. Ophthalmol. 2018, 46, 502–510. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.D.; Bergman, L.; Seregard, S. Uveal melanoma: Epidemiologic aspects. Ophthalmol. Clin. N. Am. 2005, 18, 75–84. [Google Scholar] [CrossRef]
- Honavar, S.G.; Singh, A.D.; Shields, C.L.; Shields, J.A.; Eagle, R.C., Jr. Iris melanoma in a patient with neurofibromatosis. Surv. Ophthalmol. 2000, 45, 231–236. [Google Scholar] [CrossRef]
- Honavar, S.G.; Shields, C.L.; Singh, A.D.; Demirci, H.; Rutledge, B.K.; Shields, J.A.; Eagle, R.C., Jr. Two discrete choroidal melanomas in an eye with ocular melanocytosis. Surv. Ophthalmol. 2002, 47, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Rao, R.; Honavar, S.G.; Mulay, K. Multifocal choroidal melanoma in oculodermal melanocytosis in an Asian Indian. Indian. J. Ophthalmol. 2019, 67, 2089–2091. [Google Scholar] [CrossRef] [PubMed]
- Houlston, R.S.; Damato, B.E. Genetic predisposition to ocular melanoma. Eye 1999, 13 Pt 1, 43–46. [Google Scholar] [CrossRef] [PubMed]
- Iscovich, J.; Abdulrazik, M.; Cour, C.; Fischbein, A.; Pe’er, J.; Goldgar, D.E. Prevalence of the BRCA2 6174 del T mutation in Israeli uveal melanoma patients. Int. J. Cancer 2002, 98, 42–44. [Google Scholar] [CrossRef]
- Melzer, C.; Sharma, A.; Peters, S.; Aretz, S.; Biswas, A.; Holz, F.G.; Loeffler, K.U.; Herwig-Carl, M.C. Basal cell carcinomas developing independently from BAP1-tumor predisposition syndrome in a patient with bilateral uveal melanoma: Diagnostic challenges to identify patients with BAP1-TPDS. Genes. Chromosomes Cancer 2019, 58, 357–364. [Google Scholar] [CrossRef] [PubMed]
- Derrien, A.C.; Rodrigues, M.; Eeckhoutte, A.; Dayot, S.; Houy, A.; Mobuchon, L.; Gardrat, S.; Lequin, D.; Ballet, S.; Pierron, G.; et al. Germline MBD4 Mutations and Predisposition to Uveal Melanoma. J. Natl. Cancer Inst. 2021, 113, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Repo, P.; Jantti, J.E.; Jarvinen, R.S.; Rantala, E.S.; Tall, M.; Raivio, V.; Kivela, T.T.; Turunen, J.A. Germline loss-of-function variants in MBD4 are rare in Finnish patients with uveal melanoma. Pigment. Cell Melanoma Res. 2020, 33, 756–762. [Google Scholar] [CrossRef]
- Palles, C.; West, H.D.; Chew, E.; Galavotti, S.; Flensburg, C.; Grolleman, J.E.; Jansen, E.A.M.; Curley, H.; Chegwidden, L.; Arbe-Barnes, E.H.; et al. Germline MBD4 deficiency causes a multi-tumor predisposition syndrome. Am. J. Hum. Genet. 2022, 109, 953–960. [Google Scholar] [CrossRef]
- Nathan, V.; Palmer, J.M.; Johansson, P.A.; Hamilton, H.R.; Warrier, S.K.; Glasson, W.; McGrath, L.A.; Kahl, V.F.S.; Vasireddy, R.S.; Pickett, H.A.; et al. Loss-of-function variants in POT1 predispose to uveal melanoma. J. Med. Genet. 2021, 58, 234–236. [Google Scholar] [CrossRef]
- Cruz, C.; Teule, A.; Caminal, J.M.; Blanco, I.; Piulats, J.M. Uveal melanoma and BRCA1/BRCA2 genes: A relationship that needs further investigation. J. Clin. Oncol. 2011, 29, e827–e829. [Google Scholar] [CrossRef]
- Lobo, J.; Pinto, C.; Freitas, M.; Pinheiro, M.; Vizcaino, R.; Oliva, E.; Teixeira, M.R.; Jeronimo, C.; Bartosch, C. Ovarian metastasis from uveal melanoma with MLH1/PMS2 protein loss in a patient with germline MLH1 mutated Lynch syndrome: Consequence or coincidence? Virchows Arch. 2017, 470, 347–352. [Google Scholar] [CrossRef]
- Kannengiesser, C.; Avril, M.F.; Spatz, A.; Laud, K.; Lenoir, G.M.; Bressac-de-Paillerets, B. CDKN2A as a uveal and cutaneous melanoma susceptibility gene. Genes. Chromosomes Cancer 2003, 38, 265–268. [Google Scholar] [CrossRef] [PubMed]
- Marous, C.L.; Marous, M.R.; Welch, R.J.; Shields, J.A.; Shields, C.L. Choroidal Melanoma, Sector Melanocytosis, and Retinal Pigment Epithelial Microdetachments in Birt-Hogg-Dube Syndrome. Retin. Cases Brief. Rep. 2019, 13, 202–206. [Google Scholar] [CrossRef]
- Fontcuberta, I.C.; Salomao, D.R.; Quiram, P.A.; Pulido, J.S. Choroidal melanoma and lid fibrofoliculomas in Birt-Hogg-Dube syndrome. Ophthalmic Genet. 2011, 32, 143–146. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Rahman, M.H.; Sample, K.M.; Pilarski, R.; Walsh, T.; Grosel, T.; Kinnamon, D.; Boru, G.; Massengill, J.B.; Schoenfield, L.; Kelly, B.; et al. Whole Exome Sequencing Identifies Candidate Genes Associated with Hereditary Predisposition to Uveal Melanoma. Ophthalmology 2020, 127, 668–678. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Silva-Rodriguez, P.; Bande, M.; Fernandez-Diaz, D.; Lago-Baameiro, N.; Pardo, M.; Jose Blanco-Teijeiro, M.; Dominguez, F.; Loidi, L.; Pineiro, A. Role of somatic mutations and chromosomal aberrations in the prognosis of uveal melanoma in a Spanish patient cohort. Acta Ophthalmol. 2021, 99, e1077–e1089. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alfoldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. Author Correction: The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2021, 590, E53. [Google Scholar] [CrossRef] [PubMed]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018, 46, D1062–D1067. [Google Scholar] [CrossRef]
- Stenson, P.D.; Mort, M.; Ball, E.V.; Chapman, M.; Evans, K.; Azevedo, L.; Hayden, M.; Heywood, S.; Millar, D.S.; Phillips, A.D.; et al. The Human Gene Mutation Database (HGMD((R))): Optimizing its use in a clinical diagnostic or research setting. Hum. Genet. 2020, 139, 1197–1207. [Google Scholar] [CrossRef]
- Rahman, N. Realizing the promise of cancer predisposition genes. Nature 2014, 505, 302–308. [Google Scholar] [CrossRef]
- Ruttan, C.C.; Glickman, B.W. Coding variants in human double-strand break DNA repair genes. Mutat. Res. 2002, 509, 175–200. [Google Scholar] [CrossRef] [PubMed]
- Kohler, S.; Gargano, M.; Matentzoglu, N.; Carmody, L.C.; Lewis-Smith, D.; Vasilevsky, N.A.; Danis, D.; Balagura, G.; Baynam, G.; Brower, A.M.; et al. The Human Phenotype Ontology in 2021. Nucleic Acids Res. 2021, 49, D1207–D1217. [Google Scholar] [CrossRef] [PubMed]
- Hamosh, A.; Amberger, J.S.; Bocchini, C.; Scott, A.F.; Rasmussen, S.A. Online Mendelian Inheritance in Man (OMIM(R)): Victor McKusick’s magnum opus. Am. J. Med. Genet. A 2021, 185, 3259–3265. [Google Scholar] [CrossRef] [PubMed]
- Lindskog, C. The potential clinical impact of the tissue-based map of the human proteome. Expert. Rev. Proteomics 2015, 12, 213–215. [Google Scholar] [CrossRef] [PubMed]
- UniProt, C. UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [Google Scholar] [CrossRef]
- Kivelä, T.; Simpson, E.; Grossniklaus, H.; Jager, M.; Singh, A.; Caminal, J.; Pavlick, A.; Kujala, E.; Finger, P. Uveal melanoma. In AJCC Cancer Staging Manual, 8th ed.; Springer: New York, NY, USA, 2016; pp. 805–817. [Google Scholar]
- Aoude, L.G.; Pritchard, A.L.; Robles-Espinoza, C.D.; Wadt, K.; Harland, M.; Choi, J.; Gartside, M.; Quesada, V.; Johansson, P.; Palmer, J.M.; et al. Nonsense mutations in the shelterin complex genes ACD and TERF2IP in familial melanoma. J. Natl. Cancer Inst. 2015, 107, dju408. [Google Scholar] [CrossRef]
- Aoude, L.G.; Wadt, K.A.; Pritchard, A.L.; Hayward, N.K. Genetics of familial melanoma: 20 years after CDKN2A. Pigment. Cell Melanoma Res. 2015, 28, 148–160. [Google Scholar] [CrossRef]
- Shammas, H.F.; Watzke, R.C. Bilateral choroidal melanomas. Case report and incidence. Arch. Ophthalmol. 1977, 95, 617–623. [Google Scholar] [CrossRef]
- Turunen, J.A.; Markkinen, S.; Wilska, R.; Saarinen, S.; Raivio, V.; Tall, M.; Lehesjoki, A.E.; Kivela, T.T. BAP1 Germline Mutations in Finnish Patients with Uveal Melanoma. Ophthalmology 2016, 123, 1112–1117. [Google Scholar] [CrossRef]
- Aoude, L.G.; Gartside, M.; Johansson, P.; Palmer, J.M.; Symmons, J.; Martin, N.G.; Montgomery, G.W.; Hayward, N.K. Prevalence of Germline BAP1, CDKN2A, and CDK4 Mutations in an Australian Population-Based Sample of Cutaneous Melanoma Cases. Twin Res. Hum. Genet. 2015, 18, 126–133. [Google Scholar] [CrossRef]
- De Lange, T. Shelterin: The protein complex that shapes and safeguards human telomeres. Genes. Dev. 2005, 19, 2100–2110. [Google Scholar] [CrossRef]
- Shi, J.; Yang, X.R.; Ballew, B.; Rotunno, M.; Calista, D.; Fargnoli, M.C.; Ghiorzo, P.; Bressac-de Paillerets, B.; Nagore, E.; Avril, M.F.; et al. Rare missense variants in POT1 predispose to familial cutaneous malignant melanoma. Nat. Genet. 2014, 46, 482–486. [Google Scholar] [CrossRef]
- Kabir, S.; Hockemeyer, D.; de Lange, T. TALEN gene knockouts reveal no requirement for the conserved human shelterin protein Rap1 in telomere protection and length regulation. Cell Rep. 2014, 9, 1273–1280. [Google Scholar] [CrossRef]
- Deregowska, A.; Wnuk, M. RAP1/TERF2IP-A Multifunctional Player in Cancer Development. Cancers 2021, 13, 5970. [Google Scholar] [CrossRef]
- Oltvai, Z.N.; Milliman, C.L.; Korsmeyer, S.J. Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programmed cell death. Cell 1993, 74, 609–619. [Google Scholar] [CrossRef] [PubMed]
- Wei, M.C.; Zong, W.X.; Cheng, E.H.; Lindsten, T.; Panoutsakopoulou, V.; Ross, A.J.; Roth, K.A.; MacGregor, G.R.; Thompson, C.B.; Korsmeyer, S.J. Proapoptotic BAX and BAK: A requisite gateway to mitochondrial dysfunction and death. Science 2001, 292, 727–730. [Google Scholar] [CrossRef] [PubMed]
- Barlow, J.W.; Mous, M.; Wiley, J.C.; Varley, J.M.; Lozano, G.; Strong, L.C.; Malkin, D. Germ line BAX alterations are infrequent in Li-Fraumeni syndrome. Cancer Epidemiol. Biomarkers Prev. 2004, 13, 1403–1406. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.G.; Lisboa, B.C.; Achatz, M.I.; Carraro, D.M.; da Cunha, I.W.; Pearson, P.L.; Krepischi, A.C.; Rosenberg, C. Germline BAX deletion in a patient with melanoma and gastrointestinal stromal tumor. Am. J. Gastroenterol. 2013, 108, 1372–1375. [Google Scholar] [CrossRef] [PubMed]
- Ionov, Y.; Yamamoto, H.; Krajewski, S.; Reed, J.C.; Perucho, M. Mutational inactivation of the proapoptotic gene BAX confers selective advantage during tumor clonal evolution. Proc. Natl. Acad. Sci. USA 2000, 97, 10872–10877. [Google Scholar] [CrossRef]

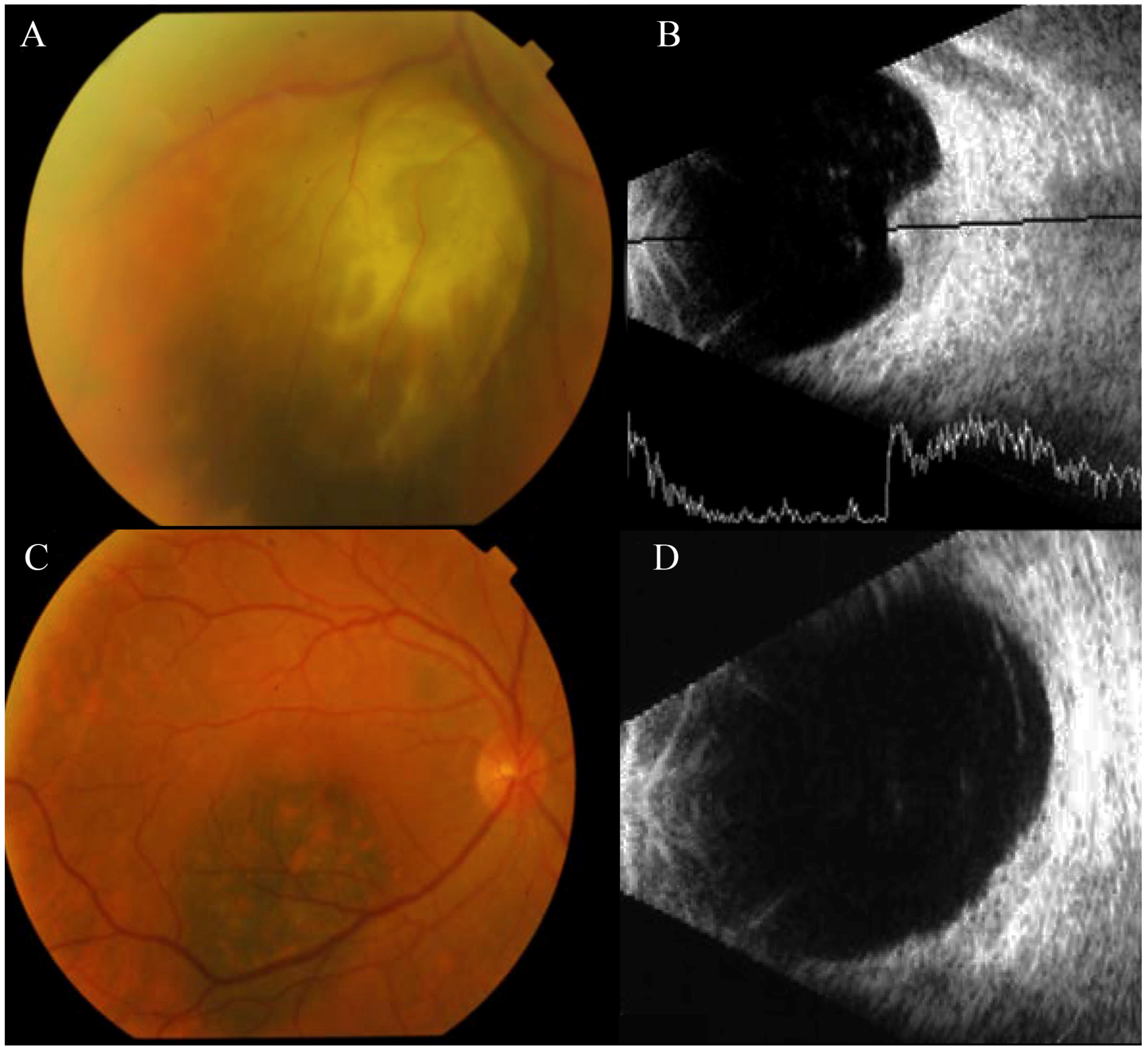

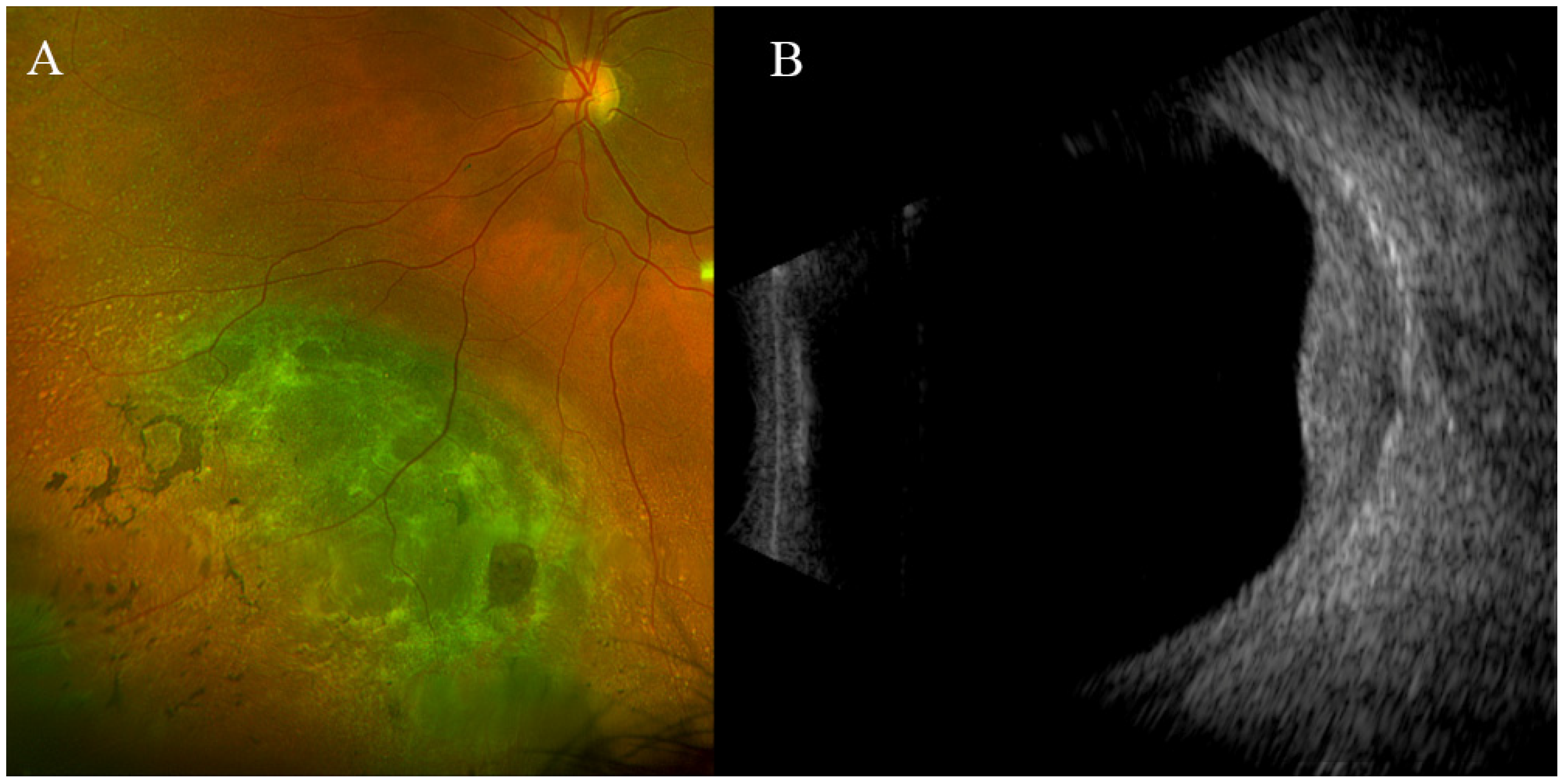

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient | Gene ab | Variant Description | Protein Effect | gnomAD Freq. | ACMG Classification * | ACMG Criteria |
|---|---|---|---|---|---|---|
| BUM3 | BAX | c.243del p.(Ala82ProfsTer51) | premature stop codon | 0 | LP | PVS1, PM2 |
| BUM4 | TERF2IP | c.1090C>T p.(R364Ter) | premature stop codon | 0.0000199 | VUS | PVS1_strong, PM2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Silva-Rodríguez, P.; Bande, M.; Pardo, M.; Domínguez, F.; Loidi, L.; Blanco-Teijeiro, M.J. Bilateral Uveal Melanoma: An Insight into Genetic Predisposition in Four New Unrelated Patients and Review of Published Cases. J. Clin. Med. 2024, 13, 3035. https://doi.org/10.3390/jcm13113035
Silva-Rodríguez P, Bande M, Pardo M, Domínguez F, Loidi L, Blanco-Teijeiro MJ. Bilateral Uveal Melanoma: An Insight into Genetic Predisposition in Four New Unrelated Patients and Review of Published Cases. Journal of Clinical Medicine. 2024; 13(11):3035. https://doi.org/10.3390/jcm13113035
Chicago/Turabian StyleSilva-Rodríguez, Paula, Manuel Bande, María Pardo, Fernando Domínguez, Lourdes Loidi, and María José Blanco-Teijeiro. 2024. "Bilateral Uveal Melanoma: An Insight into Genetic Predisposition in Four New Unrelated Patients and Review of Published Cases" Journal of Clinical Medicine 13, no. 11: 3035. https://doi.org/10.3390/jcm13113035
APA StyleSilva-Rodríguez, P., Bande, M., Pardo, M., Domínguez, F., Loidi, L., & Blanco-Teijeiro, M. J. (2024). Bilateral Uveal Melanoma: An Insight into Genetic Predisposition in Four New Unrelated Patients and Review of Published Cases. Journal of Clinical Medicine, 13(11), 3035. https://doi.org/10.3390/jcm13113035