Abstract
Small cell lung cancer (SCLC) is a deadly neuroendocrine malignancy, notorious for its rapid tumor growth, early metastasis, and relatively “cold” immune environment. Only standard chemotherapies and a few immune checkpoint inhibitors have been approved for SCLC treatment, revealing an urgent need for novel therapeutic approaches. Moreover, SCLC has been recently recognized as a malignancy with high intratumoral and intertumoral heterogeneity, which explains the modest response rate in some patients and the early relapse. Molecular subtypes defined by the expression of lineage-specific transcription factors (ASCL1, NEUROD1, POU2F3, and, in some studies, YAP1) or immune-related genes display different degrees of neuroendocrine differentiation, immune cell infiltration, and response to treatment. Despite the complexity of this malignancy, a few biomarkers and targets have been identified and many promising drugs are currently undergoing clinical trials. In this review, we integrate the current progress on the genomic landscape of this shapeshifting malignancy, the characteristics and treatment vulnerabilities of each subtype, and promising drugs in clinical phases.
1. Introduction
Lung cancer is the most common cancer worldwide, with a high incidence (11.6%) and mortality rate (18.4%) in both men and women [1]. Small cell lung cancer (SCLC) constitutes 15% of all lung cancer cases and is known for its aggressive and highly metastatic nature [2]. SCLC typically originates in the bronchi and is most frequently associated with smoking. The incidence rate of SCLC was 9.5 per 10,000 population in 1975 and peaked at 15.3 in 1988. Since the initiation of tobacco control programs around the world, the incidence of SCLC declined to 6.5 in 2019 [3]. Unfortunately, SCLC often goes undiagnosed until it reaches an advanced state due to its rapid tumor growth, lack of symptoms, and symptoms being mistaken for those caused by smoking and air pollution. The percentage of advanced stage IV at diagnosis has increased from 58.6% in 1988 to 70.8% in 2010 [3]. As a result, the 5-year survival rate for SCLC patients is less than 6% in approximately 70% of cases [2].
SCLC is a high-grade neuroendocrine tumor that exhibits abnormal neurotransmitter or hormone signaling [4]. This is thought to be linked to genetic variations, such as the inactivation of TP53 and RB1, as well as disruptions in signaling pathways. The neuroendocrine nature of SCLC has opened up new avenues for targeted therapy and immunotherapy. Several clinical studies are currently ongoing with some promising results.
In addition to developing new drugs for known targets, substantial efforts have been invested in understanding and categorizing different SCLC subtypes. Traditionally treated as a single disease, recent evidence suggests significant heterogeneity in the degree of neuroendocrine differentiation and the regulation of neuronal lineage-specific transcription factors within SCLC. This has led to the identification of several SCLC subtypes, including SCLC-A (ASCL1-positive), SCLC-N (NEUROD1-positive), SCLC-P (POU2F3-positive), and SCLC-Y (YAP1-positive) or SCLC-inflamed [5,6].
This review aims to provide an overview of SCLC’s genomic variations, differences among its subtypes, and a summary of current and emerging therapeutic and diagnostic strategies. The goal is to assess the most promising approaches to improving outcomes for SCLC patients.
2. Unveiling the Genomic Landscape of SCLC
In 1988, a pivotal discovery was made by Harbour and colleagues: they found that the RB1 gene was exclusively inactivated in SCLC, setting it apart from non-SCLC cell lines and normal human lungs [7]. Shortly thereafter, in 1989, Takahashi and colleagues identified abnormal TP53 mutations in lung cancer cells [8]. This paved the way for the confirmation that SCLC is characterized by the nearly universal inactivation of both TP53 and RB1. This inactivation is believed to be the catalyst for subsequent genomic and epigenetic alterations that bestow SCLC with its neuroendocrine characteristics [9].
Fast forwarding to 2010, Pleasance and colleagues achieved a remarkable milestone by conducting the first whole genome sequencing of an SCLC cell line, NCI-H209. This cell line was derived from a bone marrow metastasis of a 55-year-old male with SCLC before chemotherapy, and the sequencing revealed that tobacco smoking significantly amplifies the mutational burden in SCLC. Additionally, it unveiled various tumor signatures, including APOBEC, a cytidine deaminase involved in RNA editing [10,11].
Following Pleasance’s groundbreaking work, advanced sequencing tools have empowered researchers to create more comprehensive genomic profiles of SCLC [12]. A deeper analysis of these genomic alterations revealed that many of the mutated genes in SCLC can be grouped into four main categories: regulators of cell cycle and death, epigenetic regulators, members of the Notch signaling pathway, and regulators of cytoskeleton dynamics and cell adhesion [12].
As we continue to explore these genomic and epigenomic changes, significant efforts have been invested in identifying potential biomarkers and developing targeted therapies against them.
2.1. Loss of the RB and TP53 Families in SCLC
In SCLC, the loss of RB1 and TP53 is nearly universal, but it does not stop there. The inactivation of their homologs, like RBL1 (3–4%), RBL2 (5–7%), and TP73 (13%), has also been reported in SCLC [5]. RBL1 and RBL2, also known as p107 and p130, respectively, share crucial functions with RB1, regulating E2F transcription factors that control the cell cycle. It has been suggested that RBL1 and RBL2 may compensate for the functional loss of other RB family members, contributing to the latent development of tumors in SCLC. This idea gained support in a study where Rb1/Tp53-mutant genetically engineered mouse models (GEMMs) with the homozygous or heterozygous deletion of Rbl2 showed significantly higher tumor incidence and shorter tumor latency compared to those with wild-type Rbl2 [13]. Similarly, TP73, along with p63, comprises the p53 family of transcription factors, which controls cell cycle arrest and apoptosis by inducing the expression of related genes [14].
Although the mutational pattern of TP53 aligns with what is expected from tobacco exposure, there is no direct evidence linking tobacco exposure to RB1 loss in SCLC [15,16]. Interestingly, even though RB1 is among the significantly mutated genes in SCLC, approximately 10% of patients do not exhibit RB1 mutations [12]. The RB1 protein undergoes initial mono-phosphorylation mediated by the CDK4/6-Cyclin D complex during the G1 phase, leading to the release of the E2F transcription factors. Studies have revealed that SCLC tumors with functional RB1 protein are more responsive to CDK4/6 inhibitors like palbociclib and abemaciclib, and this sensitivity is dependent on the presence of RB1 [17].
2.2. Rare Kinase Alterations in SCLC
A genome-wide analysis of the kinome reveals that kinases have diverse biological functions and play crucial roles in oncogenesis, with kinase inhibitors accounting for a significant portion of small molecule inhibitors used in cancer treatment [18]. Unfortunately, in comparison to non-small cell lung cancer (NSCLC), SCLC exhibits fewer kinase mutations. Commonly mutated kinases in NSCLC, such as epidermal growth factor receptor (EGFR) and MAPK pathway kinases (KRAS, RAF, MEK, and ERK), are seldom mutated in SCLC [12]. However, SCLC patients who do have kinase mutations might benefit from genetic sequencing and targeted kinase inhibitors.
Two well-known receptor tyrosine kinases with abnormal expression in SCLC are c-Kit (or CD117) and fibroblast growth factor receptor 1 (FGFR1). C-Kit is a proto-oncogene that encodes a tyrosine kinase receptor and is believed to create an autocrine loop that drives cell proliferation with its ligand stem cell factor (SCF) [19]. Positive c-Kit expression has been detected in 37% of SCLC patient samples and is associated with reduced survival [20]. Activating mutations of c-Kit have also been identified, with S476I and P551A, mutations occurring in 3.3% and 5% of cases, respectively [12,21]. Imatinib mesylate (Gleevec), a small molecule inhibitor for several tyrosine kinase receptors, including c-Kit, has shown efficacy in gastrointestinal cancer and acute lymphoblastic leukemia linked to c-Kit S476I and P551A mutations. However, in a phase II clinical trial, imatinib mesylate failed to demonstrate therapeutic efficacy in SCLC patients [22], possibly due to the low incidence of these mutations. Nevertheless, alternative approaches to target c-Kit are being explored [23].
In contrast, the ectopic expression of Fgfr1 in precancerous neuroendocrine cells (preSCs) has been found to increase in vitro cell growth and tumor formation in immune-compromised mice, along with enhanced proliferation-related gene expression changes [24]. FGFR1 amplification has been reported in a subset of human SCLC tumor samples (6–30%). The study in [12] and subsequent research revealed that the constitutive activation of FGFR1 inhibits SCLC initiated from calcitonin gene-related peptide (CGRP)-positive neuroendocrine cells but promotes SCLC initiated from K14-positive tracheobronchial-basal epithelial cells. This suggests that SCLC may originate from a more diverse set of cell lineages than previously assumed, and the role of FGFR1 is context-dependent [25].
Aurora kinases A and B (AURKA and AURKB) are key serine/threonine kinases that regulate mitosis and coordinate the G2-M transition [26]. The overexpression of Aurora kinases has been reported in various cancers, including lung cancer [27]. Small molecule inhibitors of AURKA and AURKB have shown antitumor activity by inducing polyploidy in SCLC cell lines with MYC amplification and RB1 inactivation [28]. The effectiveness of these inhibitors correlated with the levels of MYC amplification and expression, making Aurora kinases promising therapeutic targets in SCLC with high MYC expression. Additionally, other kinases involved in cell cycle and DNA repair, such as ATR and WEE1, have emerged as promising targets, with several inhibitors in clinical trials [29,30].
2.3. MYC Amplification in SCLC
The amplification of MYC family genes, including MYC, MYCL, and MYCN, is a common oncogenic event in SCLC [31]. Approximately 20% of SCLC tumors and 30–50% of SCLC cell lines exhibit MYC amplification [31]. This genetic alteration is associated with a grim prognosis in SCLC patients, reducing their survival from 26 weeks to just 4 weeks [32].
SCLC cell lines with frequent MYC amplification tend to have a faster doubling time, indicating more aggressive growth [31]. The expression of Myc family members, especially Mycl, can restore the tumor growth capacity in non-tumorigenic, preneoplastic SCLC cells within weeks [33]. Conversely, inhibiting MYC amplification in SCLC cell lines hampers tumor cell growth [34].
In vivo studies utilizing GEMMs also shed light on the potential tumorigenic role of Myc in SCLC. One study involved creating experimental Rb1fl/fl p53fl/fl MycLSL/LSL (RMP) mice by crossing Rb1fl/fl p53fl/fl (RP) mice with mice carrying Lox-Stop-Lox (LSL)-MycT58A-IRES-Luciferase and having a Cre-MycT58A recombinase [31]. RPM mice were infected intratracheally with adenoviruses containing Cre driven by the CGRP promoter, a marker expressed only in the predominant cell of origin of SCLC in the RP model [35]. RPM mice experienced significantly higher mortality compared to Rb1fl/fl Trp53fl/fl Ptenfl/fl (RPP) mice, with a median survival of 60 versus 164 days, respectively [36,37]. Heterozygous RPM mice (Rb1fl/fl Trp53fl/fl MycLSL/+) had a slightly longer median survival of 81 days than RPM mice. Although the precise mechanism remains unclear, both in vitro and in vivo studies suggest that MYC amplification accelerates tumor growth, thereby shortening the survival of SCLC patients. This indicates that MYC could be a potential therapeutic target in the MYC-amplified SCLC subset.
Furthermore, increasing evidence suggests that MYC may play a role in driving the evolution of SCLC subtypes by regulating neuroendocrine and metabolic processes [31,38,39]. This aspect will be explored in more detail in a later section.
2.4. Notch Signaling Pathway in SCLC
The Notch signaling pathway plays a critical role in cell–cell communication, regulating transcription, and cell differentiation. In normal cells, the transcription factor achaete-scute homolog 1 (ASCL1) induces neuroendocrine differentiation, leading to the expression of neuroendocrine markers and activating the transcription of DLL3 and other genes in the Notch pathway. However, in lung cancers, the role of Notch pathway genes can vary, with their main function being that of tumor suppressors in SCLC [40].
A comprehensive study of SCLC cases involving genome sequencing revealed mutations in Notch family genes in approximately 25% of the cases examined [2], and these genes are generally suppressed in the majority of neuroendocrine SCLC cases [40]. Yet, recent research suggests that Notch signaling may also have a pro-tumorigenic role in SCLC. The non-neuroendocrine subtype of SCLC, which has an active Notch pathway, tends to be more resistant to chemotherapy and can support the growth of neighboring neuroendocrine SCLC cells [41].
There are three known mechanisms for inactivating the Notch pathway in SCLC: (1) the mutational inactivation of Notch pathway genes; (2) the inhibition of Notch receptors by canonical Notch ligand Delta-like ligand 3 (DLL3) or Delta-like non-canonical Notch ligand 1 (DLK1); and (3) the degradation of Notch receptors guided by DLL3 in the endosomes [42].
As a result, Notch pathway proteins, especially DLL3, are considered potential therapeutic targets. DLL3 is exclusively overexpressed in SCLC. However, the development of the experimental drug Rovalpituzumab tesirine (Rova-T), a DLL3-targeted antibody–drug conjugate, was halted after the phase III trial due to its lack of significant benefit and toxicity [43]. This could be partly attributed to the toxicity of the anti-cancer payload pyrrolobenzodiazepine (PBD). Presently, alternative approaches using DLL3 for T cell-redirecting therapies are in clinical trials [44], and the experience gained from these therapies will help determine whether DLL3 or other Notch pathway inhibitors are promising targets in SCLC, considering the pro-tumorigenic role of the Notch pathway in the non-neuroendocrine subgroup.
2.5. Epigenetic Alterations in SCLC
SCLC presents a unique epigenetic regulation pattern for DNA methylation, histone methylation, and histone acetylation when compared to other lung cancers. This distinct pattern offers an opportunity for the development of SCLC-targeted epigenetic drugs. One of the most crucial and extensively studied oncogenes related to histone methylation is the Enhancer of Zeste Homolog 2 (EZH2). EZH2 is the enzymatic catalytic subunit of Polycomb Repressive Complex 2 (PRC2), responsible for tri-methylating H3K27 and silencing gene expression. In SCLC, due to the universal loss of RB1, EZH2 is expressed at higher levels compared to NSCLC and normal lung epithelial cells [45]. EZH2 has been found to epigenetically silence the TGF-ß type II receptor (TßRII) and suppress the TGF-ß-Smad-ASCL1 pathway, resulting in elevated ASCL1 expression and promoting SCLC progression [45]. Another study demonstrated that EZH2 also drives acquired chemoresistance in relapsed SCLC through the EZH2-SLFN11 axis [46]. These promising preclinical findings have led to phase I/II clinical trials of EZH2 inhibitor DS-3201b in combination with irinotecan and a phase I trial of another EZH2 inhibitor, PF-06821497, in patients with recurrent SCLC [47,48].
Another potential epigenetic target related to histone methylation is Lysine-specific demethylase 1A (LSD1). LSD1 inhibitors have shown SCLC-specific activity by reactivating the Notch pathway and reducing the expression of ASCL1 and neuroendocrine lineage genes [49]. Currently, the LSD inhibitor CC-90011 is undergoing two clinical trials, one in combination with standard chemotherapies and another with Nivolumab for SCLC [50,51].
CREB-binding protein (CREBBP) and E1A-associated p300 (EP300) are lysine acetyltransferases (KATs) and are two major inactivated genes in SCLC, with a mutation frequency of 15% and 13%, respectively [12]. These two proteins mediate the acetylation of H3K27, which leads to the transcription of regulated downstream genes. The majority of these hotspot inactivation mutations occur in KAT domains, resulting in decreased chromatin accessibility. These mutations in CREBBP and EP300 have a mutually exclusive pattern, indicating they may share similar functions in SCLC development. The functional loss of CREBBP and EP300 has been linked to various neuroendocrine cancers by activating oncogenes and suppressing tumor suppressor genes through epigenetic modifications [5]. One study demonstrated that Crebbp loss resulted in the reduced expression of tight junction and cell adhesion genes, including Cdh1, in SCLC mouse models [52]. The treatment of lysine deacetylase (KDAC) inhibitor was able to restore histone acetylation and the expression of CDH1 [52]. Several KDAC inhibitors are currently in phase I trials as a monotherapy or in combination therapies [53,54,55]. Given the diverse role and reversible alterations of epigenetics in cancer, targeting epigenome in SCLC is a very promising treatment strategy.
2.6. Alterations of Cytoskeletal and Cell Adhesion Genes in SCLC
While SCLC is notorious for its frequent metastasis, the precise mechanisms by which genetic and transcriptional alterations affect metastasis are not fully understood. The whole-genome sequencing of clinical SCLC samples has revealed somatic mutations in genes such as ALMS1, ASPM, COBL, COL4A2, COL22A1, FMN2, KIAA1211, PDE4DIP, ROBO1, and SLIT2. These proteins are known for their functions related to cytoskeleton formation and rearrangements associated with cell–cell and cell–matrix interactions [5]. These genetic alterations may play a role in the metastatic process of SCLC, though the exact mechanisms and their implications are still being studied. A recent paper demonstrated that CCN1/2 (Cellular communication network factor 1/2) is regulated by transcription factor YAP1 to inhibit SCLC metastasis. CCN1/2 blocked the actin polymerization and thereby inhibited the migration of SCLC cells [56]. Another group identified CUL5 (Cullin5) and SOCS3 (suppressor of cytokine signaling 3) from the pooled CRISPR/Cas9 library as candidate regulators of SCLC metastasis. The depletion of CUL5/SOCS3 stabilized integrin ß1 and promoted metastasis through focal adhesion kinase/SRC signaling pathway, which predicts the potential benefit for CUL5-deficient SCLC patients from receiving SRC inhibitor treatment [57].
3. SCLC Heterogeneity and Phenotypic Switching
In recent years, research has unveiled that SCLC should no longer be analyzed and treated as a single disease. It is a malignancy with unique subtypes, each with distinct genomic profiles, including immune-related genes, and therapy responses. Over 30 years ago, human SCLCs were initially divided into two subgroups: the neuroendocrine (NE) subgroup, originating from pulmonary neuroendocrine cells, and the non-neuroendocrine (variant) (non-NE) subgroup [58]. Subsequent studies have indicated the presence of non-NE tumor cells within single SCLC cell lines and mouse models, revealing intratumoral heterogeneity [41,59]. The plasticity of tumor cells and their ability to transform between subtypes poses a significant challenge in targeting SCLC and may contribute to poor treatment outcomes and relapse (Figure 1).
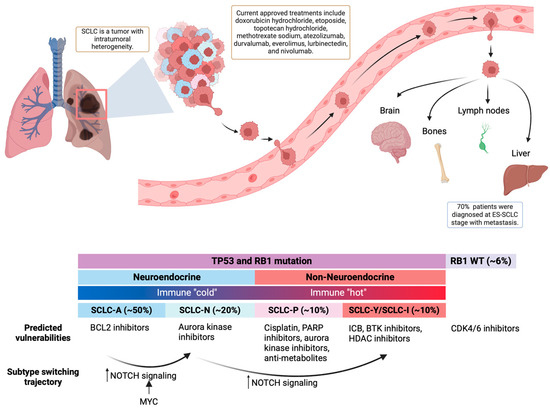
Figure 1.
Basics of SCLC. Top: SCLC is a malignancy with high plasticity and early metastasis. Bottom: SCLC can be classified into four major subtypes that differ in neuroendocrine differentiation, immune response, and treatment vulnerabilities.
3.1. Intertumoral Subtypes
Over the past decade, increased clinical cases unveiled a more complex landscape of SCLC subtypes. The computational modeling of transcriptomics data has defined SCLC subtypes using the expression of four key transcription factors: achaete-scute homolog 1 (ASCL1), Neuronal Differentiation 1 (NEUROD1), POU class 2 homeobox 3 (POU2F3), and Yes-Associated Protein 1 (YAP1) [60]. So far, no reported SCLC case lacks the expression of any of these four genes.
- Neuroendocrine SCLC has a high expression of NE markers, including Synaptophysin (SYP), Chromogranin-A (CHGA), and Neural cell adhesion molecule 1 (NCAM1 or CD56). NE SCLC can be divided into two subtypes:
- ο
- SCLC-A: This neuroendocrine subtype is characterized by high ASCL1 expression and accounts for approximately 50% of primary SCLC cases [60]. ASCL1 is an NE-lineage-specific transcription factor essential for SCLC tumorigenesis [61]. It exhibits super-enhancers associated with genes like MYCL1, NFIB, BCL2, NKX2-1, FOXA1, and FOXA2 [61]. Gene ontology analysis reveals enrichment in neuronal systems, potassium channel genes, and epithelial cell differentiation [61,62].
- ο
- SCLC-N: This neuroendocrine subtype, comprising 20% of primary SCLC cases, is characterized by high NEUROD1 expression and low ASCL1. SCLC-N often exhibits super-enhancers associated with NEUROD1 and the oncogene MYC [61]. This subtype may respond to chemotherapy but can develop resistance [31].
- Non-NE SCLC has low expression of both ASCL1 and NEUROD1 and can be divided into two subtypes:
- ο
- SCLC-P: Characterized by high POU2F3 expression, it exhibits the unique expression of other transcription factors, including SOX9 and ASCL2, and the tyrosine kinase receptor insulin-like growth factor 1 receptor (IGF1R) [63].
- ο
- The remaining SCLC tumors have low expressions of ASCL1, NEUROD1, and POU2F3. Two putative subtypes are SCLC-Y and SCLC-I.
- -
- SCLC-Y: With high YAP1 expression, it is sensitive to CDK4/6 inhibitors [64].
- -
- SCLC-Inflamed: This subtype is characterized by an inflamed gene signature (including immune checkpoints and HLAs), making it benefit from immunotherapy, but this subtype is not uniquely defined by YAP1 expression [6,65].
Multiple studies showed that patients with different SCLC subtypes have different prognoses. Based on surgery-resected patient tumor samples, better prognoses after the primary tumor surgery were observed in the low-NE group compared to the high-NE group [66,67]. More specifically, patients with the SCLC-Y subtype had the best prognosis, while patients with the SCLC-A subtype had the worst [68].
3.2. Intratumoral Heterogeneity and Evolution
The study of intratumoral heterogeneity and the evolution of SCLC has revealed a complex and dynamic landscape. In recent years, it has been observed that the majority of human and mouse SCLC tumors consist of multiple subtypes, indicating a high degree of tumor plasticity [59,62]. Tumor subpopulations within the same patients have shown dynamic changes both before and after treatment, highlighting the adaptability of SCLC.
Research by Borromeo and others demonstrated that Ascl1, but not Neurod1, is necessary for tumor formation in mouse models with Tp53/Rb1/Rbl2lox/lox mutations [61]. This suggests that ASCL1 expression might serve as a precursor to both SCLC-A and SCLC-N subtypes, with SCLC-N potentially evolving from an ASCL1-expressing state.
Further investigations have shown that MYC plays a crucial role in driving the evolution of subtypes from SCLC-A to SCLC-N and SCLC-Y [39]. MYC activates the Notch pathway, promoting the emergence of SCLC-N and SCLC-Y subtypes from a cell of origin that initially expresses ASCL1. Simultaneously, it propels the SCLC-P subtype from a cell of origin that is not a pulmonary neuroendocrine cell (PNEC), club cell, or AT2 cell [39].
Studies conducted on early-stage tumor cells from mouse models (Tp53/Rb1lox/lox or RPM mice) cultured over time have revealed a temporal subtype transition from SCLC-A-dominant to SCLC-Y-dominant. Initially, cells expressed high levels of ASCL1 and other NE markers within the first 4–7 days of culture. However, by days 11–21, they exhibited high levels of non-NE markers. This MYC-driven subtype evolution is dependent on Notch pathway activation [39].
Considering that Notch pathway loss-of-function mutations have been reported in approximately 25% of human SCLCs [12], it has been proposed that SCLC tumor cells with defective Notch signaling remain in an NE-high state, while tumor cells with intact and activated Notch are reprogrammed by MYC to a non-NE state.
Although the above studies all showed an NE to non-NE transition, so far, there is no reported non-NE to NE transition. Advanced lineage-tracing techniques during SCLC development are expected to provide a better understanding of the SCLC evolution and help in the development of subtype-targeted or plasticity-targeted therapeutic strategies.
3.3. The Immune Microenvironment in SCLC Subtypes
Immunotherapies for SCLC primarily involve immune checkpoint blockers (ICBs) targeting PD1, PD-L1, and CTLA4. However, the clinical benefits of immunotherapy in SCLC have been limited, with fewer than 20% of patients experiencing substantial improvements [69]. This can be attributed to the overall “cold” status of the immune environment in neuroendocrine SCLC. SCLC typically exhibits a low expression of class 1 major histocompatibility complex (MHC) antigens and a limited infiltration of cytotoxic immune cells compared to other tumor types [70].
As researchers have characterized SCLC subtypes, questions have arisen regarding whether the immune microenvironment varies among these subtypes and whether they respond differently to ICB. There is substantial evidence indicating increased MHC I expression and immunogenicity in non-NE subtypes [6,65,71]. SCLC subtypes display distinct immune properties. SCLC-N, for instance, exhibits the lowest expression of immune-related genes, including those involved in MHC and antigen presentation, immune checkpoints, and natural killer (NK) cells. In contrast, the SCLC-P subtype has the highest expression of immune-related genes [71]. Studies have shown that low MHC I antigen presentation in NE SCLC is associated with the epigenetic silencing of TAP1 by EZH2. The inhibition of EZH2 can reverse this process, converting NE SCLC into an antigenic non-NE phenotype [65].
As previously mentioned, SCLC-Inflamed (SCLC-I) has shown elevated immune infiltrate, including T cells, NK cells, and macrophages, as well as a high cytolytic activity score [6]. SCLC-I does not exhibit prevailing signatures of ASCL1, NEUROD1, POU2F3, and, in some cases, YAP1. Interestingly, drug response analysis has shown that SCLC-I is more resistant to cisplatin treatment. Unsurprisingly, SCLC-I has experienced greater benefits from the combination of ICBs, such as atezolizumab (an anti-PD-L1 antibody), with chemotherapy [6]. These findings highlight the importance of understanding the immune microenvironment and its variations among SCLC subtypes when developing effective therapeutic strategies.
3.4. Therapeutic Vulnerabilities in SCLC Subtypes
The distinct profiles of genomic alteration, epigenetic regulation, and the immune microenvironment observed across SCLC subtypes have raised the possibility of developing unique subtype-specific therapeutic strategies. In vitro drug response data from SCLC cell lines have provided valuable insights into potential vulnerabilities and treatment options for each subtype:
SCLC-A (ASCL1-high): This subtype appears to have a high expression of BCL2 protein and is predicted to be sensitive to BCL2 inhibitors [6]. Targeting the BCL2 protein, which plays a role in inhibiting cell death, may be a viable therapeutic approach for SCLC-A.
SCLC-N (NEUROD1-high): SCLC-N has shown increased sensitivity to aurora kinase inhibitors, but also resistance to standard cisplatin treatment [6]. Aurora kinase inhibitors can disrupt cell division and may offer a promising treatment strategy for SCLC-N.
SCLC-P (POU2F3-high): SCLC-P is most sensitive to cisplatin treatment and shows sensitivity to poly-ADP ribose polymerase (PARP) inhibitors and anti-metabolites, such as anti-folates, as well as aurora kinase inhibitors [6,31,62]. PARP inhibitors target DNA repair processes, while anti-metabolites interfere with the production of DNA and RNA in cancer cells.
SCLC-I (SCLC-Inflamed): This subtype shows resistance to cisplatin but is predicted to benefit from ICBs and Bruton’s tyrosine kinase (BTK) inhibitors [6]. Patients with MHC Ihi showed significantly more durable responses to ICBs and increased overall survival [65]. Also, due to the most mesenchymal phenotype among all subtypes, it is predicted that this subtype will also benefit from treatment strategies targeting EMT, such as HDAC inhibitors [6].
Beyond the drug response analysis, the distinct expression patterns of surface proteins in SCLC subtypes present an alternative avenue for targeted therapies by antibody—drug-conjugates (ADCs) and chimeric antigen receptor (CAR) strategies. By tailoring treatments to the unique characteristics of each SCLC subtype, we can improve therapeutic outcomes and address the challenges posed by SCLC’s heterogeneity and phenotypic switching.
4. Current Treatment
Therapeutic options for SCLC have seen little progress over the last three decades, with the major strategies centered around conventional chemotherapy and radiation therapy. The standard of care for SCLC continues to involve platinum-based alkylating agents, such as cisplatin or carboplatin, typically in combination with topoisomerase inhibitors like etoposide or irinotecan.
SCLC is classified into two stages: limited-stage (LS, stage I-III) and extensive-stage (ES, stage IV). Although surgery is not considered the main treatment option for SCLC due to the early metastasis, for patients with LS-SCLC at clinical stage I-IIA (about 5% in patients with SCLC), surgical resection is the recommended primary treatment according to the NCCN Guidelines for SCLC. For those with LS-SCLC stage IIB-IIIC, the standard practice recommends concurrent or sequential radiotherapy directed at the thorax and mediastinum, alongside platinum-based chemotherapy [2]. For extensive-stage SCLC (ES-SCLC), the initial course of treatment predominantly revolves around chemotherapy. The role and merit of thoracic radiation and prophylactic cranial irradiation (PCI) in ES-SCLC therapy remain subjects of contention and are not universally advised for all patients [72].
A significant hurdle in advancing therapeutic options for SCLC was the perception of this cancer as a “homogenous” tumor, resulting in a standard approach involving a combination of chemotherapy and radiotherapy for all SCLC patients [42]. This categorization, which has persisted for years due to the intertumoral pathological similarities, has contributed to the slow progress. Additionally, the list of drugs approved for the specific treatment of SCLC remains exceptionally short as of April 2024. Apart from common chemotherapy drugs like Doxorubicin Hydrochloride, Etoposide Phosphate, Topotecan Hydrochloride, and Methotrexate Sodium, only five drugs are approved for SCLC treatment.
Everolimus (Afinitor, Novartis) is an mTOR inhibitor approved for the treatment of adult patients with progressive, well-differentiated non-functional, neuroendocrine tumors (NETs) of gastrointestinal (GI) or lung origin with unresectable, locally advanced or metastatic disease [73]. The inhibition of mTOR blocks the translation of genes that promote tumor cell growth and survival, including angiogenesis and metabolism.
Lurbinectedin (ZEPZELCA, Pharma Mar S.A.) is a DNA alkylating agent that causes more double-strand DNA breaks and cell death in hyper-proliferating tumor cells. It has been recently approved for adult patients with metastatic SCLC upon or after platinum-based treatment. The complete mechanism of action remains elusive, but it was revealed that Lurbinectedin can also inhibit hyperactivated RNA polymerase II, resulting in reduced oncogene expression [74,75]. Lurbinectedin was granted accelerated approval after the multicenter PM1183-B-005-14 trial (Study B-005; ClinicalTrials.gov identifier NCT02454972) showed a 35% overall response rate was achieved among all patients in the trial, with a 5.3-month median response duration [74]. Despite its accelerated approval as a new monotherapy for metastatic SCLC, the combination of lurbinectedin with doxorubicin did not achieve an improved overall survival compared with the current second-line standard treatment of topotecan or cyclophosphamide, doxorubicin, and vincristine (CAV) in the multicenter, randomized, controlled, phase 3 ATLANTIS study [76]. These trials indicate that lurbinectedin as an active agent in SCLC could be further developed in both monotherapy and combinational therapy against different stages of SCLC.
Immune checkpoints regulate the immune system, and they fall into two major groups: stimulatory and inhibitory checkpoints. Inhibitory checkpoints maintain self-tolerance, making sure healthy cells are not destroyed by the activated T cells. However, cancer cells also exploit those inhibitory immune checkpoints to evade the immune response. Two inhibitory immune checkpoint receptors have been actively studied in the past few years: cytotoxic T-lymphocyte-associated antigen 4 (CTLA4; or CD152) and programmed cell death protein 1 (PD1; or CD279). Immune checkpoint inhibitors such as ipilimumab, nivolumab, pembrolizumab, durvalumab, tremelimumab, and ulocuplumab are at the forefront of immunotherapy and have achieved approvals for certain cancer types, varying from SCLC to hematologic malignancies [77].
Atezolizumab (TECENTRIQ) is an anti-PD-L1 monoclonal antibody approved with carboplatin and etoposide as a first-line treatment for ES-SCLC. By binding to programmed death-ligand 1 (PD-L1) on some tumor cells, atezolizumab inhibits the interaction between programmed death receptor 1 (PD1), inhibitory receptor on the surface of activated T cells, and PD-L1 and further prevents the immune evasion of tumor cells. Atezolizumab and carboplatin combination for ES-SCLC was approved after the IMPower133 (NCT02763579) trial, which showed significant improvement in the overall survival and progression-free rate (PFS) compared with the placebo group [69,78].
Like atezolizumab, durvalumab (IMFINZI) is another PD1/PD-L1 checkpoint monoclonal antibody inhibitor that binds PD-L1. It was approved with etoposide and either carboplatin or cisplatin as a first-line treatment for ES-SCLC based on the result in CASPIAN, a randomized, multicenter, active-controlled, open-label trial (NCT03043872). The combination treatment showed efficacious clinical outcomes in overall survival but not antibody-dependent cytotoxicity among patients. Both atezolizumab (single-mutation) and durvalumab (three mutations) are IgG1 isotypes and have engineered Fc domains to bypass the attack of PD-L1-expressed T cells and other antibody-dependent cytotoxicity [79]. The undesirable antidrug antibody (ADA) formation indicates the drug’s immunogenicity and it can affect the drug’s pharmacokinetics, pharmacodynamics, and efficacy [79]. The complete mechanisms of immunotherapy that elicited ADA formation remain elusive, but due to the different designs of atezolizumab and durvalumab, they have different ADA formation incidents. Atezolizumab has a reported treatment-emergent antidrug antibody (ADA) of 30% to 48%, while durvalumab’s is only 3.1% [80].
Besides targeting PD-L1, another strategy is to target PD1 receptors on the immune cells to block PD1/PD-L1 interaction. Nivolumab (Opdivo) is an anti-PD1 monoclonal antibody approved in the U.S. as a third-line treatment for metastatic SCLC with progression upon or after platinum-based chemotherapy and at least one other line of therapy. The accelerated approval was based on the efficacy outcome measures from the metastatic SCLC patients in CheckMate-032 (NCT01928394), a multicenter, open-label trial in patients with metastatic solid tumors. The overall response rate (ORR) was 12% (95% CI: 6.5, 19.5). Responses were durable for 6 months or longer in 77%, 12 months or longer in 62%, and 18 months or longer in 39% of the 13 responding patients [81].
5. Promising Treatment Options for SCLC
Besides the approved drugs for SCLC treatment, researchers are coming up with creative ways to target SCLC, which can be divided into three major areas: (1) targeted therapy (including drug conjugates and small molecule inhibitors), (2) immunotherapy, and (3) chemotherapy. Other innovative strategies include systemic gene therapy utilizing lipid nanoparticles that encapsulate plasmid to express tumor suppressor gene TUSC2 [82].
5.1. Targeted Therapies
Antibody–drug conjugates (ADCs) are innovative biopharmaceutical drugs that combine the advantages of immuno- and chemotherapy [Table 1]. Highly specific monoclonal antibodies against antigens presented on tumor cells are chemically linked to active antitumor agents, which significantly decreases systematic toxicity. The high selectivity and high lethality against tumor cells, while sparing healthy cells, have made ADC drugs a very powerful and promising cancer treatment option. So far, there are several ADC drugs on the market and about 100 ADCs in clinical trials for various cancer types. For SCLC, although there is no approved ADC, there are several in clinical trials.

Table 1.
Targeted therapies for SCLC currently in clinical trials.
One promising antigen is B7-H3 (CD276), a transmembrane immune checkpoint protein selectively overexpressed in cancer cells to promote immune evasion. High B7-H3 expression has been detected in 65% of SCLC patients, making it a candidate target for immunotherapy and targeted therapy [83]. Currently, there are three B7-H3-directed ADCs in SCLC clinical trials. Ifinatamab Deruxtecan (I-DXd) is in phase III trial for patients with relapsed SCLC based on its encouraging ORR of 52.4% from phase I/II subgroup analysis [84,85]. The other two B7-H3-directed ADCs are HS-20093 and ABBV-155 (Mirzotamab clezutoclax; Mirzo-C), which have a BCL-XL inhibitor payload [86].
Other antigens targeted in the ADCs include SEZ6, a cell-surface protein that is highly expressed in neuroendocrine tumors including SCLC [87] and TROP2, a glycoprotein overexpressed in epithelial tumors like SCLC [88]. DLL3, a ligand that inhibits Notch pathway activation and is selectively overexpressed in SCLC (~80%), was proposed to be a promising ADC target based on the efficacy results of the Rova-T phase I trial [89]. Unfortunately, the following trials of Rova-T failed to demonstrate efficacy, and the development of the drug was discontinued [43], which leads to the question of whether DLL3 is a valid target. Currently, there are still multiple anti-DLL3 agents in clinical trials, including ZL-1310 (anti-DLL3 ADC), BI 764532 (bispecific DLL3/CD3 T cell engager), HPN328 (anti-DLL3 T cell engager), and DLL3-directed CAR-T and CAR-NK therapies. The results of these trials will provide a clear understanding of DLL3 expression as a biomarker for SCLC treatment.
A new format of “drug conjugates” has started to exploit interactions other than antigen–antibody recognition to deliver the payload. CBX-12 is an alphalex peptide drug conjugate (PDC) that consists of a pH-sensitive alphalex peptide, a linker, and a topoisomerase inhibitor exatecan. In the tumor microenvironment, where the pH is lower than 7.0, the peptide forms an alpha helix that inserts into the cell membrane to release the linker and payload [90].
Another major advance in the targeted therapy of SCLC is the development of new small molecule inhibitors and agonists, which target various tumor activities including histone modification (HDAC, EZH1/2), cell cycle regulation (CDK2/4/6), DNA damage repair (ATR, PARP), angiogenesis (VEGFR, PDGFR), proteasome activity, and other important kinase activities (e.g., Aurora, PERK, and PP2A) [Table 1].
5.2. Immunotherapies
Despite SCLC’s reputation of being an “immune desert”, significant progress has been made with FDA-approved monoclonal antibodies targeting PD1 and PD-L1. These agents, often used in combination with chemotherapy, have shown positive outcomes, sparking further interest and development in this area (Figure 2).
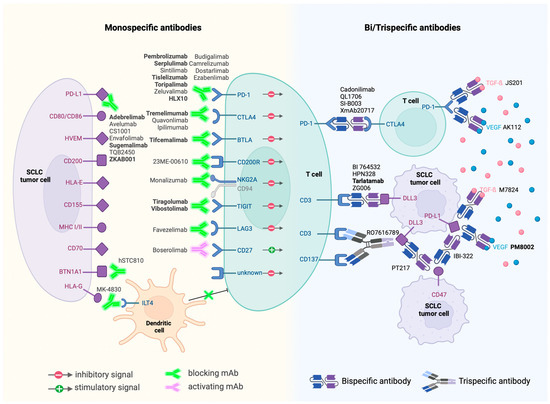
Figure 2.
Immunotherapies currently in clinical trials and their targets. Antibodies in bold are currently in phase III trials.
Monoclonal Antibodies: A significant aspect of current research focuses on monoclonal antibodies that modulate the immune checkpoints or influence the immune response directly. As of April 2024, around fifty monoclonal antibodies are under clinical evaluation. The exploration extends beyond PD1 and PD-L1 targets to include cytotoxic T-lymphocyte antigen 4 (CTLA-4), butyrophilin 1A1 (BTN1A1), T cell immunoreceptor with Ig and ITIM domains (TIGIT), T cell immunoglobulin domain and mucin domain 3 (TIM3), CD200 receptor 1 (CD200R1), B- and T-lymphocyte attenuator (BTLA), CD94/NK group 2 member A (NKG2A), immunoglobulin-like transcript 4 (ILT4) receptor, CD27, and lymphocyte-activation gene 3 (LAG3). This broadening of targets underscores the evolving understanding of the immune landscape in SCLC.
Next-Generation Antibodies: The advent of bispecific monoclonal antibodies marks a notable advancement. These antibodies, capable of targeting two antigens simultaneously, exhibit increased target-binding efficiency and potential for enhanced antitumor activity. Examples include the bispecific checkpoint inhibitors PSB 205 (targeting PD1 and CTLA-4) and XmAb22841 (targeting CTLA-4 and LAG3).
BiTEs and Beyond: Among bispecific antibodies, BiTEs (Bispecific T cell Engagers) like AMG-757, targeting the inhibitory Notch pathway ligand DLL3, have shown promise. Early studies indicate that AMG-757 can effectively redirect T cells to eliminate DLL3-positive cancer cells [44,91].
Moreover, bispecific antibodies are exploring combinations beyond ICB, venturing into areas such as anti-angiogenesis. One example is PM8002, a bispecific antibody that combines PD-L1 inhibition with VEGF blockade. The early phase II results of PM8002 and paclitaxel combination treatment showed an impressive overall response rate of 72.7% in immunotherapy-naïve patients [92].
Other innovative immunotherapy agents include cytokines and agonists to stimulate immune response (IL7, IL12, IL15, and CD137 agonist), CAR-T (DLL3-directed and GD2-directed), CAR-NK (DLL3-directed), and small molecule inhibitor and antisense oligonucleotides (ASOs) to relieve immunosuppression. RRx-001 is a small molecule Myc inhibitor currently in phase III trial. The proposed mechanism of action is that RRx-001 downregulates the expression of immune checkpoints CD47 and PD-L1 to sensitize macrophages and T cells through c-Myc inhibition [93]. One clinical ASO candidate is AZD8701, which degrades Forkhead-box P3 transcription factor (FOXP3) mRNA that promotes the regulatory T cell’s immunosuppression activity [94]. This first-in-class strategy to regulate immune response through ASO provides a novel approach to fight against SCLC.
6. Advance in Early Lung Cancer Diagnosis and Assessment of Therapy Response
Despite the advancement in understanding the biological pathways and identifying therapeutic targets, there is still a lack of non-invasive and sensitive diagnosis and screening tools for lung cancer. One contributing factor to poor prognoses in SCLC is the late diagnosis, making the conventional treatments less effective. Another factor is that the assessment of therapy is often limited by invasive biopsy procedures and complex tumor dynamics. Tumors can have delayed shrinkage and transiently enlarge due to inflammation, especially after immunotherapies, which makes it hard to interpret the treatment response simply from the serial imaging [95]. In the past decade, the technological advances in measuring and analyzing circulating tumor DNA (ctDNA) or other markers (exosomes) in blood have shown the potential of “liquid biopsy” for early detection and therapy assessment in both leukemia and solid malignancies, including lung cancer [96,97]. Compared with traditional tissue biopsy, liquid biopsy is less limited by tumor accessibility, sampling frequency, and complicated tumor dynamics.
Although there is currently no ctDNA-based diagnostic tool for SCLC on the market, the FDA approved Guardant360 CDx as a companion diagnostic for NSCLC patients with epidermal growth factor receptor (EGFR) alterations who may benefit from treatment with osimertinib (Tagrisso) in August 2020 [98]. The approval of Guardant360 CDx paved the way for repurposing the biomarkers into diagnostic and prognostic tools in all solid neoplasms, especially in lung tumors.
Besides genomic analysis such as Guardant360 CDx, epigenetic-based ctDNA testing is potentially also a powerful screening tool. The hypermethylation of Septin9 in ctDNA is observed in various cancers, including colorectal cancer and lung cancer [99]. mSEPT9 test was approved by the FDA as a commercial test for colorectal cancer (CRC)-screening tests after clinical trials. LUNAR-2, which combines the genomic and epigenomic ctDNA analyses, is currently under clinical trials to study the risk stratification in lung cancer screening [100]. SUMMIT, another early detection blood test clinical trial, is ongoing in the UK to validate the blood test in individuals with high-risk lung cancer [101].
Still, despite the advantages of the liquid biopsy, the development of biological techniques, and the promising results in clinical trials, there is uncertainty about the ctDNA-based blood test for lung cancer, including its accuracy and effectiveness [102]. With careful biomarker design, rational clinical implementation, and result interpretation considering unique lung tumor characteristics, ctDNA-based assays are likely to have an impact on lung cancer care.
7. Remaining Challenges and Future Directions
Although most patients respond very well to primary treatments like chemotherapy and radiation, relapse and developed drug resistance are still major clinical challenges. The heterogenous nature of SCLC contributes to this phenomenon as different subtypes display distinct resistance to treatment. The less-sensitive cells that survive from initial treatment are the origins of the relapsed tumor. To date, many efforts have focused on exploring drug resistance in SCLC. Besides the previously mentioned EZH2-SLFN11 axis [46], another driver of chemoresistance identified is MYCN and the potential of inhibiting USP7 to restore chemosensitivity [103]. Despite these preliminary results, drug resistance in SCLC remains a big obstacle. Combinational regimens might be one strategy to increase efficacy while reducing the likelihood of developing resistance. Currently, there are many combinational treatments and a few targeted therapies for refractory or relapsed SCLC in clinical trials [Table 1], including one EZH2 inhibitor, PF-06821497. The results of these trials will provide valuable insights into developing strategies for fighting chemoresistance.
Treatment toxicity should also be carefully evaluated and monitored in clinical practice and in trials. One example is the failure of Rova-T with the toxic payload PBD. Unlike other ADCs utilizing topoisomerase inhibitor or tubulin inhibitor monomethyl auristatin E (MMAE), PBD is more potent yet toxic. The linker of Rova-T is also predicted to have early cleavage, resulting in PBD systematic exposure [104].
- It is essential to recognize that SCLC management is evolving, with ongoing research and clinical trials exploring novel and potentially more effective therapies for this aggressive cancer. Recent insights into the heterogeneous nature of SCLC, as well as advancements in understanding its plasticity, offer the potential for tailored and targeted treatment approaches. These may encompass subtype-specific therapies, immunotherapies, and innovative treatments based on epigenetics and other cutting-edge approaches. Here, we propose a few future directions for studying and targeting SCLC:
- It is exciting to observe the declining incidence rate of SCLC with the help of global tobacco control programs. It is crucial to continue public education emphasizing smoking as the primary cause of SCLC and advocating for reduced tobacco consumption.
- The identification of predictive biomarkers will be crucial for treating SCLC. Although different subtype’s therapeutic vulnerabilities have been predicted with drug library screening [62], the exact difference among subtypes should be more closely investigated. The inclusion of subtype-specific markers (ASCL1, NEUROD1, POU2F3, and maybe YAP1) for immunohistochemistry staining besides neuroendocrine markers, such as SYP and NCAM1, will benefit the physicians in diagnosing patients with specific SCLC subtypes and predicting the potential treatment response. Stratifying patients based on molecular subtypes should also be incorporated into clinical trial design. The failure of certain targets in the clinical trials might be due to not targeting the proper patient subpopulation. Tumor shapeshifting after treatment, especially chemotherapy, should also be considered when designing clinical trials. One example is SLFN11, which is utilized as a predictive marker for PARP1/2-targeted therapies [Table 1].
- Another big direction will be to improve the immunotherapy response. Since non-neuroendocrine subtypes (especially triple-negative for ASCL1, NEUROD1, and POU2F3) showed more immune infiltration, identifying the genes switching neuroendocrine SCLC to non-neuroendocrine SCLC will be critically important to achieve durable immune therapy response by directing immune “cold” NE to immune “hot” non-NE SCLC. One major player for the switch is the activation of the MYC-Notch signaling pathway, which has been shown to drive the SCLC-A subtype to SCLC-N and eventually to SCLC-Y [39]. Treatments targeting this pathway and other mechanisms underlying the NE-to-non-NE switch should be investigated.
- Considering the general immune “cold” phenotype in the classic neuroendocrine SCLC, immunotherapy, especially monospecific immune checkpoint inhibitors alone, might not be the best strategy, as shown by the moderate clinical ORRs, but targeting overexpressed antigens with proper payload and antigen-directed T cell engagers might have better efficacy. Another approach is to explore the combinational treatment of ICBs with non-NE-induction treatment.
- The approval of atezolizumab and durvalumab as a first-line treatment with platinum-based chemotherapy ignites the exploration of combined regimens. It provides the opportunities to target tumors while potentially bypassing the resistance; however, it also brings challenges: finding the best combination in this heterogenous and shapeshifting malignancy and determining the best dosage schedule when designing clinical trials.
- Due to the plasticity and heterogeneity of SCLC, models like patient-derived xenograft will be a powerful tool to monitor the subtype transition before, during, and after the treatment, to develop a more personalized treatment plan. Also, validating the preliminary results obtained from murine models and human SCLC cell lines in these patient-derived xenograft models will increase the probability of successful laboratory-to-clinic translation.
Author Contributions
Y.G. conceived and wrote the original draft of the manuscript and designed the figures. C.A.B. supported, reviewed, and edited the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by grants to C.A.B. from the American Lung Association (LCD-1034555).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA A Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- The American Cancer Society Medical and Editorial Content Team What Is Lung Cancer?|Types of Lung Cancer. Available online: https://www.cancer.org/cancer/lung-cancer/about/what-is.html (accessed on 4 May 2021).
- Cittolin-Santos, G.F.; Knapp, B.; Ganesh, B.; Gao, F.; Waqar, S.; Stinchcombe, T.E.; Govindan, R.; Morgensztern, D. The Changing Landscape of Small Cell Lung Cancer. Cancer 2024. [Google Scholar] [CrossRef] [PubMed]
- Fisseler-Eckhoff, A.; Demes, M. Neuroendocrine Tumors of the Lung. Cancers 2012, 4, 777–798. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.-B.; Dunn, C.T.; Park, K.-S. Recent Progress in Mapping the Emerging Landscape of the Small Cell Lung Cancer Genome. Exp. Mol. Med. 2019, 51, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Gay, C.M.; Stewart, C.A.; Park, E.M.; Diao, L.; Groves, S.M.; Heeke, S.; Nabet, B.Y.; Fujimoto, J.; Solis, L.M.; Lu, W.; et al. Patterns of Transcription Factor Programs and Immune Pathway Activation Define Four Major Subtypes of SCLC with Distinct Therapeutic Vulnerabilities. Cancer Cell 2021, 39, 346–360.e7. [Google Scholar] [CrossRef] [PubMed]
- Harbour, J.W.; Lai, S.L.; Whang-Peng, J.; Gazdar, A.F.; Minna, J.D.; Kaye, F.J. Abnormalities in Structure and Expression of the Human Retinoblastoma Gene in SCLC. Science 1988, 241, 353–357. [Google Scholar] [CrossRef]
- Takahashi, T.; Nau, M.M.; Chiba, I.; Birrer, M.J.; Rosenberg, R.K.; Vinocour, M.; Levitt, M.; Pass, H.; Gazdar, A.F.; Minna, J.D. P53: A Frequent Target for Genetic Abnormalities in Lung Cancer. Science 1989, 246, 491–494. [Google Scholar] [CrossRef] [PubMed]
- Semenova, E.A.; Nagel, R.; Berns, A. Origins, Genetic Landscape, and Emerging Therapies of Small Cell Lung Cancer. Genes Dev. 2015, 29, 1447–1462. [Google Scholar] [CrossRef] [PubMed]
- Pleasance, E.D.; Stephens, P.J.; O’Meara, S.; McBride, D.J.; Meynert, A.; Jones, D.; Lin, M.-L.; Beare, D.; Lau, K.W.; Greenman, C.; et al. A Small Cell Lung Cancer Genome with Complex Signatures of Tobacco Exposure. Nature 2010, 463, 184–190. [Google Scholar] [CrossRef]
- Roberts, S.A.; Lawrence, M.S.; Klimczak, L.J.; Grimm, S.A.; Fargo, D.; Stojanov, P.; Kiezun, A.; Kryukov, G.V.; Carter, S.L.; Saksena, G.; et al. An APOBEC Cytidine Deaminase Mutagenesis Pattern Is Widespread in Human Cancers. Nat. Genet. 2013, 45, 970–976. [Google Scholar] [CrossRef]
- George, J.; Lim, J.S.; Jang, S.J.; Cun, Y.; Ozretić, L.; Kong, G.; Leenders, F.; Lu, X.; Fernández-Cuesta, L.; Bosco, G.; et al. Comprehensive Genomic Profiles of Small Cell Lung Cancer. Nature 2015, 524, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Schaffer, B.E.; Park, K.-S.; Yiu, G.; Conklin, J.F.; Lin, C.; Burkhart, D.L.; Karnezis, A.N.; Sweet-Cordero, E.A.; Sage, J. Loss of P130 Accelerates Tumor Development in a Mouse Model for Human Small Cell Lung Carcinoma. Cancer Res. 2010, 70, 3877–3883. [Google Scholar] [CrossRef] [PubMed]
- Costanzo, A.; Merlo, P.; Pediconi, N.; Fulco, M.; Sartorelli, V.; Cole, P.A.; Fontemaggi, G.; Fanciulli, M.; Schiltz, L.; Blandino, G.; et al. DNA Damage-Dependent Acetylation of P73 Dictates the Selective Activation of Apoptotic Target Genes. Mol. Cell 2002, 9, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Gowers, K.; Yoshida, K.; Lee-Six, H.; Chandrasekharan, D.; Maughan, E.; Millar, F.; Clarke, S.; Pennycuick, A.; Thakrar, R.; Carroll, B.; et al. Tobacco Exposure and Somatic Mutations in Normal Human Bronchial Epithelium. Am. J. Respir. Crit. Care Med. 2020, 201, A4090. [Google Scholar]
- Gibbons, D.L.; Byers, L.A.; Kurie, J.M. Smoking, P53 Mutation, and Lung Cancer. Mol. Cancer Res. 2014, 12, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Wildey, G.; Shay, A.M.; McColl, K.S.; Yoon, S.; Shatat, M.A.; Perwez, A.; Spainhower, K.B.; Kresak, A.M.; Lipka, M.; Yang, M.; et al. Retinoblastoma Expression and Targeting by CDK4/6 Inhibitors in Small Cell Lung Cancer. Mol. Cancer Ther. 2023, 22, 264–273. [Google Scholar] [CrossRef] [PubMed]
- Workman, P. Drugging the Cancer Kinome: Progress and Challenges in Developing Personalized Molecular Cancer Therapeutics. Cold Spring Harb. Symp. Quant. Biol. 2005, 70, 499–515. [Google Scholar] [CrossRef] [PubMed]
- Micke, P.; Basrai, M.; Faldum, A.; Bittinger, F.; Rönnstrand, L.; Blaukat, A.; Beeh, K.M.; Oesch, F.; Fischer, B.; Buhl, R.; et al. Characterization of C-Kit Expression in Small Cell Lung Cancer: Prognostic and Therapeutic Implications. Clin. Cancer Res. 2003, 9, 188–194. [Google Scholar]
- Voortman, J.; Lee, J.-H.; Killian, J.K.; Suuriniemi, M.; Wang, Y.; Lucchi, M.; Smith, W.I.; Meltzer, P.; Wang, Y.; Giaccone, G. Array Comparative Genomic Hybridization-Based Characterization of Genetic Alterations in Pulmonary Neuroendocrine Tumors. Proc. Natl. Acad. Sci. USA 2010, 107, 13040–13045. [Google Scholar] [CrossRef]
- Boldrini, L.; Ursino, S.; Gisfredi, S.; Faviana, P.; Donati, V.; Camacci, T.; Lucchi, M.; Mussi, A.; Basolo, F.; Pingitore, R.; et al. Expression and Mutational Status of C-Kit in Small Cell Lung Cancer: Prognostic Relevance. Clin. Cancer Res. 2004, 10, 4101–4108. [Google Scholar] [CrossRef]
- Johnson, B.E.; Fischer, T.; Fischer, B.; Dunlop, D.; Rischin, D.; Silberman, S.; Kowalski, M.O.; Sayles, D.; Dimitrijevic, S.; Fletcher, C.; et al. Phase II Study of Imatinib in Patients with Small Cell Lung Cancer. Clin. Cancer Res. 2003, 9, 5880–5887. [Google Scholar] [PubMed]
- Kim, K.-H.; Kim, J.-O.; Park, J.-Y.; Seo, M.-D.; Park, S.G. Antibody-Drug Conjugate Targeting c-Kit for the Treatment of Small Cell Lung Cancer. Int. J. Mol. Sci. 2022, 23, 2264. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.-B.; Kim, Y.; Kim, D.-W.; Park, K.-S. Oncogenic Role of FGFR1 and Vulnerability of RBL2-FGFR1 Axis in Small Cell Lung Cancer Development. bioRxiv 2019, 796607. [Google Scholar] [CrossRef]
- Ferone, G.; Song, J.-Y.; Krijgsman, O.; van der Vliet, J.; Cozijnsen, M.; Semenova, E.A.; Adams, D.J.; Peeper, D.; Berns, A. FGFR1 Oncogenic Activation Reveals an Alternative Cell of Origin of SCLC in Rb1/P53 Mice. Cell Rep. 2020, 30, 3837–3850.e3. [Google Scholar] [CrossRef] [PubMed]
- Carmena, M.; Earnshaw, W.C. The Cellular Geography of Aurora Kinases. Nat. Rev. Mol. Cell Biol. 2003, 4, 842–854. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.L.; Bowers, N.L.; Betticher, D.C.; Gautschi, O.; Ratschiller, D.; Hoban, P.R.; Booton, R.; Santibáñez-Koref, M.F.; Heighway, J. Overexpression of Aurora B Kinase (AURKB) in Primary Non-Small Cell Lung Carcinoma Is Frequent, Generally Driven from One Allele, and Correlates with the Level of Genetic Instability. Br. J. Cancer 2005, 93, 719–729. [Google Scholar] [CrossRef] [PubMed]
- Helfrich, B.A.; Kim, J.; Gao, D.; Chan, D.C.; Zhang, Z.; Tan, A.-C.; Bunn, P.A. Barasertib (AZD1152), a Small Molecule Aurora B Inhibitor, Inhibits the Growth of SCLC Cell Lines In Vitro and In Vivo. Mol. Cancer Ther. 2016, 15, 2314–2322. [Google Scholar] [CrossRef] [PubMed]
- Testing the Addition of an Anti-Cancer Drug, BAY 1895344, to Usual Chemotherapy for Advanced Stage Solid Tumors, with a Specific Focus on Patients with Small Cell Lung Cancer, Poorly Differentiated Neuroendocrine Cancer, and Pancreatic Cancer—Full Text View—ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/NCT04514497 (accessed on 26 July 2023).
- Thomas, A.; Takahashi, N.; Rajapakse, V.N.; Zhang, X.; Sun, Y.; Ceribelli, M.; Wilson, K.M.; Zhang, Y.; Beck, E.; Sciuto, L.; et al. Therapeutic Targeting of ATR Yields Durable Regressions in Small Cell Lung Cancers with High Replication Stress. Cancer Cell 2021, 39, 566–579.e7. [Google Scholar] [CrossRef] [PubMed]
- Mollaoglu, G.; Guthrie, M.R.; Böhm, S.; Brägelmann, J.; Can, I.; Ballieu, P.M.; Marx, A.; George, J.; Heinen, C.; Chalishazar, M.D.; et al. MYC Drives Progression of Small Cell Lung Cancer to a Variant Neuroendocrine Subtype with Vulnerability to Aurora Kinase Inhibition. Cancer Cell 2017, 31, 270–285. [Google Scholar] [CrossRef]
- Rita de Cássia, S.A.; Meurer, R.T.; Roehe, A.V. MYC Amplification Is Associated with Poor Survival in Small Cell Lung Cancer: A Chromogenic in Situ Hybridization Study. J. Cancer Res. Clin. Oncol. 2014, 140, 2021–2025. [Google Scholar] [CrossRef]
- Kim, D.-W.; Wu, N.; Kim, Y.-C.; Cheng, P.F.; Basom, R.; Kim, D.; Dunn, C.T.; Lee, A.Y.; Kim, K.; Lee, C.S.; et al. Genetic Requirement for Mycl and Efficacy of RNA Pol I Inhibition in Mouse Models of Small Cell Lung Cancer. Genes Dev. 2016, 30, 1289–1299. [Google Scholar] [CrossRef] [PubMed]
- Fiorentino, F.P.; Tokgün, E.; Solé-Sánchez, S.; Giampaolo, S.; Tokgün, O.; Jauset, T.; Kohno, T.; Perucho, M.; Soucek, L.; Yokota, J. Growth Suppression by MYC Inhibition in Small Cell Lung Cancer Cells with TP53 and RB1 Inactivation. Oncotarget 2016, 7, 31014–31028. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, K.D.; Proost, N.; Brouns, I.; Adriaensen, D.; Song, J.-Y.; Berns, A. Cell of Origin of Small Cell Lung Cancer: Inactivation of Trp53 and Rb1 in Distinct Cell Types of Adult Mouse Lung. Cancer Cell 2011, 19, 754–764. [Google Scholar] [CrossRef] [PubMed]
- Cui, M.; Augert, A.; Rongione, M.; Conkrite, K.; Parazzoli, S.; Nikitin, A.Y.; Ingolia, N.; MacPherson, D. PTEN Is a Potent Suppressor of Small Cell Lung Cancer. Mol. Cancer Res. 2014, 12, 654–659. [Google Scholar] [CrossRef] [PubMed]
- McFadden, D.G.; Papagiannakopoulos, T.; Taylor-Weiner, A.; Stewart, C.; Carter, S.L.; Cibulskis, K.; Bhutkar, A.; McKenna, A.; Dooley, A.; Vernon, A.; et al. Genetic and Clonal Dissection of Murine Small Cell Lung Carcinoma Progression by Genome Sequencing. Cell 2014, 156, 1298–1311. [Google Scholar] [CrossRef] [PubMed]
- Chalishazar, M.D.; Wait, S.J.; Huang, F.; Ireland, A.S.; Mukhopadhyay, A.; Lee, Y.; Schuman, S.S.; Guthrie, M.R.; Berrett, K.C.; Vahrenkamp, J.M.; et al. MYC-Driven Small Cell Lung Cancer Is Metabolically Distinct and Vulnerable to Arginine Depletion. Clin. Cancer Res. 2019, 25, 5107–5121. [Google Scholar] [CrossRef] [PubMed]
- Ireland, A.S.; Micinski, A.M.; Kastner, D.W.; Guo, B.; Wait, S.J.; Spainhower, K.B.; Conley, C.C.; Chen, O.S.; Guthrie, M.R.; Soltero, D.; et al. MYC Drives Temporal Evolution of Small Cell Lung Cancer Subtypes by Reprogramming Neuroendocrine Fate. Cancer Cell 2020, 38, 60–78.e12. [Google Scholar] [CrossRef] [PubMed]
- Meder, L.; König, K.; Ozretić, L.; Schultheis, A.M.; Ueckeroth, F.; Ade, C.P.; Albus, K.; Boehm, D.; Rommerscheidt-Fuss, U.; Florin, A.; et al. Notch, ASCL1, P53 and RB Alterations Define an Alternative Pathway Driving Neuroendocrine and Small Cell Lung Carcinomas. Int. J. Cancer 2016, 138, 927–938. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.S.; Ibaseta, A.; Fischer, M.M.; Cancilla, B.; O’Young, G.; Cristea, S.; Luca, V.C.; Yang, D.; Jahchan, N.S.; Hamard, C.; et al. Intratumoural Heterogeneity Generated by Notch Signalling Promotes Small Cell Lung Cancer. Nature 2017, 545, 360–364. [Google Scholar] [CrossRef]
- Gazdar, A.F.; Bunn, P.A.; Minna, J.D. Small Cell Lung Cancer: What We Know, What We Need to Know and the Path Forward. Nat. Rev. Cancer 2017, 17, 725–737. [Google Scholar] [CrossRef]
- AbbVie. AbbVie Discontinues Rovalpituzumab Tesirine (Rova-T) Research and Development Program. Available online: https://www.prnewswire.com/news-releases/abbvie-discontinues-rovalpituzumab-tesirine-rova-t-research-and-development-program-300909121.html (accessed on 4 May 2021).
- Owen, D.H.; Giffin, M.J.; Bailis, J.M.; Smit, M.-A.D.; Carbone, D.P.; He, K. DLL3: An Emerging Target in Small Cell Lung Cancer. J. Hematol. Oncol. 2019, 12, 61. [Google Scholar] [CrossRef] [PubMed]
- Murai, F.; Koinuma, D.; Shinozaki-Ushiku, A.; Fukayama, M.; Miyaozono, K.; Ehata, S. EZH2 Promotes Progression of Small Cell Lung Cancer by Suppressing the TGF-β-Smad-ASCL1 Pathway. Cell Discov. 2015, 1, 15026. [Google Scholar] [CrossRef] [PubMed]
- Gardner, E.E.; Lok, B.H.; Schneeberger, V.E.; Desmeules, P.; Miles, L.A.; Arnold, P.K.; Ni, A.; Khodos, I.; de Stanchina, E.; Nguyen, T.; et al. Chemosensitive Relapse in Small Cell Lung Cancer Proceeds through an EZH2-SLFN11 Axis. Cancer Cell 2017, 31, 286–299. [Google Scholar] [CrossRef] [PubMed]
- DS-3201b and Irinotecan for Patients with Recurrent Small Cell Lung Cancer—Full Text View—ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03879798 (accessed on 27 July 2023).
- PF-06821497 Treatment of Relapsed/Refractory SCLC, Castration Resistant Prostate Cancer, and Follicular Lymphoma—Full Text View—ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03460977 (accessed on 27 July 2023).
- Augert, A.; Eastwood, E.; Ibrahim, A.H.; Wu, N.; Grunblatt, E.; Basom, R.; Liggitt, D.; Eaton, K.D.; Martins, R.; Poirier, J.T.; et al. Targeting Notch Activation in Small Cell Lung Cancer through LSD1 Inhibition. Sci. Signal. 2019, 12, eaau2922. [Google Scholar] [CrossRef] [PubMed]
- A Safety and Efficacy Study of CC-90011 in Combination with Nivolumab in Subjects with Advanced Cancers—Full Text View—ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/NCT04350463 (accessed on 27 July 2023).
- A Safety, Tolerability and Preliminary Efficacy Evaluation of CC-90011 Given in Combination with Cisplatin and Etoposide in Subjects with First Line, Extensive Stage Small Cell Lung Cancer—Full Text View—ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03850067 (accessed on 27 July 2023).
- Jia, D.; Augert, A.; Kim, D.-W.; Eastwood, E.; Wu, N.; Ibrahim, A.H.; Kim, K.-B.; Dunn, C.T.; Pillai, S.P.S.; Gazdar, A.F.; et al. Crebbp Loss Drives Small Cell Lung Cancer and Increases Sensitivity to HDAC Inhibition. Cancer Discov. 2018, 8, 1422–1437. [Google Scholar] [CrossRef] [PubMed]
- Abexinostat in Combination with Pembrolizumab in Patients with Advanced Solid Tumor Malignancies—No Study Results Posted—ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/results/NCT03590054 (accessed on 27 July 2023).
- Study of the Safety, Pharmacokinetics and Efficacy of EDO-S101, in Patients with Advanced Solid Tumors—Full Text View—ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03345485 (accessed on 27 July 2023).
- Testing the Addition of an Anti-Cancer Drug, Entinostat, to the Usual Chemotherapy and Immunotherapy Treatment (Atezolizumab, Carboplatin and Etoposide) for Previously Untreated Aggressive Lung Cancer That Has Spread—Full Text View—ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/NCT04631029 (accessed on 27 July 2023).
- Wu, Z.; Su, J.; Li, F.; Chen, T.; Mayner, J.; Engler, A.; Ma, S.; Li, Q.; Guan, K.-L. YAP Silencing by RB1 Mutation Is Essential for Small Cell Lung Cancer Metastasis. Nat. Commun. 2023, 14, 5916. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Gong, L.; Su, D.; Jin, Y.; Guo, C.; Yue, M.; Yao, S.; Qin, Z.; Ye, Y.; Tang, Y.; et al. Cullin5 Deficiency Promotes Small Cell Lung Cancer Metastasis by Stabilizing Integrin β1. J. Clin. Investig. 2019, 129, 972–987. [Google Scholar] [CrossRef] [PubMed]
- Gazdar, A.F.; Carney, D.N.; Nau, M.M.; Minna, J.D. Characterization of Variant Subclasses of Cell Lines Derived from Small Cell Lung Cancer Having Distinctive Biochemical, Morphological, and Growth Properties. Cancer Res. 1985, 45, 2924–2930. [Google Scholar] [PubMed]
- Calbo, J.; van Montfort, E.; Proost, N.; van Drunen, E.; Beverloo, H.B.; Meuwissen, R.; Berns, A. A Functional Role for Tumor Cell Heterogeneity in a Mouse Model of Small Cell Lung Cancer. Cancer Cell 2011, 19, 244–256. [Google Scholar] [CrossRef] [PubMed]
- Rudin, C.M.; Poirier, J.T.; Byers, L.A.; Dive, C.; Dowlati, A.; George, J.; Heymach, J.V.; Johnson, J.E.; Lehman, J.M.; MacPherson, D.; et al. Molecular Subtypes of Small Cell Lung Cancer: A Synthesis of Human and Mouse Model Data. Nat. Rev. Cancer 2019, 19, 289–297. [Google Scholar] [CrossRef]
- Borromeo, M.D.; Savage, T.K.; Kollipara, R.K.; He, M.; Augustyn, A.; Osborne, J.K.; Girard, L.; Minna, J.D.; Gazdar, A.F.; Cobb, M.H.; et al. ASCL1 and NEUROD1 Reveal Heterogeneity in Pulmonary Neuroendocrine Tumors and Regulate Distinct Genetic Programs. Cell Rep. 2016, 16, 1259–1272. [Google Scholar] [CrossRef] [PubMed]
- Wooten, D.J.; Groves, S.M.; Tyson, D.R.; Liu, Q.; Lim, J.S.; Albert, R.; Lopez, C.F.; Sage, J.; Quaranta, V. Systems-Level Network Modeling of Small Cell Lung Cancer Subtypes Identifies Master Regulators and Destabilizers. PLoS Comput. Biol. 2019, 15, e1007343. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.-H.; Klingbeil, O.; He, X.-Y.; Wu, X.S.; Arun, G.; Lu, B.; Somerville, T.D.D.; Milazzo, J.P.; Wilkinson, J.E.; Demerdash, O.E.; et al. POU2F3 Is a Master Regulator of a Tuft Cell-like Variant of Small Cell Lung Cancer. Genes Dev. 2018, 32, 915–928. [Google Scholar] [CrossRef] [PubMed]
- McColl, K.; Wildey, G.; Sakre, N.; Lipka, M.B.; Behtaj, M.; Kresak, A.; Chen, Y.; Yang, M.; Velcheti, V.; Fu, P.; et al. Reciprocal Expression of INSM1 and YAP1 Defines Subgroups in Small Cell Lung Cancer. Oncotarget 2017, 8, 73745–73756. [Google Scholar] [CrossRef] [PubMed]
- Mahadevan, N.R.; Knelson, E.H.; Wolff, J.O.; Vajdi, A.; Saigí, M.; Campisi, M.; Hong, D.; Thai, T.C.; Piel, B.; Han, S.; et al. Intrinsic Immunogenicity of Small Cell Lung Carcinoma Revealed by Its Cellular Plasticity. Cancer Discov. 2021, 11, 1952–1969. [Google Scholar] [CrossRef] [PubMed]
- Hamanaka, W.; Motoi, N.; Ishikawa, S.; Ushijima, M.; Inamura, K.; Hatano, S.; Uehara, H.; Okumura, S.; Nakagawa, K.; Nishio, M.; et al. A Subset of Small Cell Lung Cancer with Low Neuroendocrine Expression and Good Prognosis: A Comparison Study of Surgical and Inoperable Cases with Biopsy. Hum. Pathol. 2014, 45, 1045–1056. [Google Scholar] [CrossRef]
- Megyesfalvi, Z.; Barany, N.; Lantos, A.; Valko, Z.; Pipek, O.; Lang, C.; Schwendenwein, A.; Oberndorfer, F.; Paku, S.; Ferencz, B.; et al. Expression Patterns and Prognostic Relevance of Subtype-Specific Transcription Factors in Surgically Resected Small Cell Lung Cancer: An International Multicenter Study. J. Pathol. 2022, 257, 674–686. [Google Scholar] [CrossRef] [PubMed]
- Qi, J.; Zhang, J.; Liu, N.; Zhao, L.; Xu, B. Prognostic Implications of Molecular Subtypes in Primary Small Cell Lung Cancer and Their Correlation with Cancer Immunity. Front. Oncol. 2022, 12, 779276. [Google Scholar] [CrossRef] [PubMed]
- Horn, L.; Mansfield, A.S.; Szczęsna, A.; Havel, L.; Krzakowski, M.; Hochmair, M.J.; Huemer, F.; Losonczy, G.; Johnson, M.L.; Nishio, M.; et al. First-Line Atezolizumab plus Chemotherapy in Extensive-Stage Small Cell Lung Cancer. N. Engl. J. Med. 2018, 379, 2220–2229. [Google Scholar] [CrossRef]
- Doyle, A.; Martin, W.J.; Funa, K.; Gazdar, A.; Carney, D.; Martin, S.E.; Linnoila, I.; Cuttitta, F.; Mulshine, J.; Bunn, P. Markedly Decreased Expression of Class I Histocompatibility Antigens, Protein, and mRNA in Human Small Cell Lung Cancer. J. Exp. Med. 1985, 161, 1135–1151. [Google Scholar] [CrossRef]
- Best, S.A.; Hess, J.B.; Souza-Fonseca-Guimaraes, F.; Cursons, J.; Kersbergen, A.; Dong, X.; Rautela, J.; Hyslop, S.R.; Ritchie, M.E.; Davis, M.J.; et al. Harnessing Natural Killer Immunity in Metastatic SCLC. J. Thorac. Oncol. 2020, 15, 1507–1521. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Zhang, Z.; Wang, Q. Emerging Therapies for Small Cell Lung Cancer. J. Hematol. Oncol. 2019, 12, 47. [Google Scholar] [CrossRef] [PubMed]
- AFINITOR® (Everolimus) Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2010/022334s6lbl.pdf (accessed on 5 May 2021).
- Santamaría Nuñez, G.; Robles, C.M.G.; Giraudon, C.; Martínez-Leal, J.F.; Compe, E.; Coin, F.; Aviles, P.; Galmarini, C.M.; Egly, J.-M. Lurbinectedin Specifically Triggers the Degradation of Phosphorylated RNA Polymerase II and the Formation of DNA Breaks in Cancer Cells. Mol. Cancer Ther. 2016, 15, 2399–2412. [Google Scholar] [CrossRef] [PubMed]
- Leal, J.F.M.; Martínez-Díez, M.; García-Hernández, V.; Moneo, V.; Domingo, A.; Bueren-Calabuig, J.A.; Negri, A.; Gago, F.; Guillén-Navarro, M.J.; Avilés, P.; et al. PM01183, a New DNA Minor Groove Covalent Binder with Potent in Vitro and in Vivo Anti-Tumour Activity. Br. J. Pharmacol. 2010, 161, 1099–1110. [Google Scholar] [CrossRef] [PubMed]
- ZEPZELCA (Lurbinectedin) Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/213702s000lbl.pdf (accessed on 5 May 2021).
- Akinleye, A.; Rasool, Z. Immune Checkpoint Inhibitors of PD-L1 as Cancer Therapeutics. J. Hematol. Oncol. 2019, 12, 92. [Google Scholar] [CrossRef]
- Mansfield, A.S.; Każarnowicz, A.; Karaseva, N.; Sánchez, A.; De Boer, R.; Andric, Z.; Reck, M.; Atagi, S.; Lee, J.-S.; Garassino, M.; et al. Safety and Patient-Reported Outcomes of Atezolizumab, Carboplatin, and Etoposide in Extensive-Stage Small Cell Lung Cancer (IMpower133): A Randomized Phase I/III Trial. Ann. Oncol. 2020, 31, 310–317. [Google Scholar] [CrossRef] [PubMed]
- Vaisman-Mentesh, A.; Gutierrez-Gonzalez, M.; DeKosky, B.J.; Wine, Y. The Molecular Mechanisms That Underlie the Immune Biology of Anti-Drug Antibody Formation Following Treatment with Monoclonal Antibodies. Front. Immunol. 2020, 11, 1951. [Google Scholar] [CrossRef] [PubMed]
- Schofield, D.J.; Percival-Alwyn, J.; Rytelewski, M.; Hood, J.; Rothstein, R.; Wetzel, L.; McGlinchey, K.; Adjei, G.; Watkins, A.; Machiesky, L.; et al. Activity of Murine Surrogate Antibodies for Durvalumab and Tremelimumab Lacking Effector Function and the Ability to Deplete Regulatory T Cells in Mouse Models of Cancer. mAbs 2021, 13, 1857100. [Google Scholar] [CrossRef] [PubMed]
- OPDIVO (Nivolumab) Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/125554s112lbl.pdf (accessed on 5 May 2021).
- Genprex, Inc. A Phase 1/2 Clinical Trial of Quaratusugene Ozeplasmid and Atezolizumab Maintenance Therapy in Patients with Extensive Stage Small Cell Lung Cancer (ES-SCLC); clinicaltrials.gov; Genprex, Inc.: Austin, TX, USA, 2023. Available online: https://clinicaltrials.gov/study/NCT05703971 (accessed on 31 January 2024).
- Carvajal-Hausdorf, D.; Altan, M.; Velcheti, V.; Gettinger, S.N.; Herbst, R.S.; Rimm, D.L.; Schalper, K.A. Expression and Clinical Significance of PD-L1, B7-H3, B7-H4 and TILs in Human Small Cell Lung Cancer (SCLC). J. Immunother. Cancer 2019, 7, 65. [Google Scholar] [CrossRef]
- Daiichi Sankyo, Inc. A Phase 3, Multicenter, Randomized, Open-Label Study of Ifinatamab Deruxtecan (I-DXd), a B7-H3 Antibody Drug Conjugate (ADC), Versus Treatment of Physician’s Choice (TPC) in Subjects with Relapsed Small Cell Lung Cancer (SCLC) (IDeate-2); clinicaltrials.gov; Daiichi Sankyo, Inc.: Chūō City, Japan, 2024. Available online: https://clinicaltrials.gov/study/NCT06203210 (accessed on 31 December 2023).
- Johnson, M.; Awad, M.; Koyama, T.; Gutierrez, M.; Falchook, G.S.; Piha-Paul, S.A.; Doi, T.; Satoh, T.; Okamoto, N.; Singh, J.; et al. OA05.05 Ifinatamab Deruxtecan (I-DXd; DS-7300) in Patients with Refractory SCLC: A Subgroup Analysis of a Phase 1/2 Study. J. Thorac. Oncol. 2023, 18, S54–S55. [Google Scholar] [CrossRef]
- AbbVie. A Phase 1 First-in-Human Study with ABBV-155 Alone and in Combination with Taxane Therapy in Adults with Relapsed and/or Refractory Solid Tumors; clinicaltrials.gov; AbbVie: North Chicago, IL, USA, 2021. Available online: https://clinicaltrials.gov/ct2/show/NCT03595059 (accessed on 5 May 2021).
- Wiedemeyer, W.R.; Gavrilyuk, J.; Schammel, A.; Zhao, X.; Sarvaiya, H.; Pysz, M.; Gu, C.; You, M.; Isse, K.; Sullivan, T.; et al. ABBV-011, A Novel, Calicheamicin-Based Antibody–Drug Conjugate, Targets SEZ6 to Eradicate Small Cell Lung Cancer Tumors. Mol. Cancer Ther. 2022, 21, 986–998. [Google Scholar] [CrossRef]
- Gray, J.E.; Heist, R.S.; Starodub, A.N.; Camidge, D.R.; Kio, E.A.; Masters, G.A.; Purcell, W.T.; Guarino, M.J.; Misleh, J.; Schneider, C.J.; et al. Therapy of Small Cell Lung Cancer (SCLC) with a Topoisomerase-I-Inhibiting Antibody-Drug Conjugate (ADC) Targeting Trop-2, Sacituzumab Govitecan. Clin. Cancer Res. 2017, 23, 5711–5719. [Google Scholar] [CrossRef] [PubMed]
- Rudin, C.M.; Pietanza, M.C.; Bauer, T.M.; Ready, N.; Morgensztern, D.; Glisson, B.S.; Byers, L.A.; Johnson, M.L.; Burris, H.A.; Robert, F.; et al. Rovalpituzumab Tesirine, a DLL3-Targeted Antibody-Drug Conjugate, in Recurrent Small Cell Lung Cancer: A First-in-Human, First-in-Class, Open-Label, Phase 1 Study. Lancet Oncol. 2017, 18, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez Rivera, I.I.; Hafez, N.; Tolcher, A.W.; LoRusso, P.; Wilks, S.; Tripathy, D.; Gara, M.; Pearson, P.; DeCillis, A.P.; Meric-Bernstam, F. CBX-12-101: A First-in-Human Study of CBX-12, an Alphalex Peptide Drug Conjugate (PDC) in Patients (Pts) with Advanced or Metastatic Solid Tumors. JCO 2023, 41, 3087. [Google Scholar] [CrossRef]
- Gif, M.J.; Thomas, M.; Murawsky, C.M.; Werner, J.; Liu, S.; Lee, F.; Homann, O.; Friedrich, M.; Pearson, J.T.; Raum, T.; et al. AMG 757, a Half-Life Extended, DLL3-Targeted Bispecific T-Cell Engager, Shows High Potency and Sensitivity in Preclinical Models of Small Cell Lung Cancer. Clin. Cancer Res. 2021, 27, 1526–1537. [Google Scholar]
- Guo, Y.; Guo, J.; Cheng, Y.; Wang, Z.; Li, Y.; Lv, D.; Yin, Y.; Li, G.; Wu, L.; Huang, Y.; et al. Phase Ib/IIa Safety and Efficacy of PM8002, a Bispecific Antibody Targeting PD-L1 and VEGF-A, as a Monotherapy in Patients with Advanced Solid Tumors. JCO 2023, 41, 2536. [Google Scholar] [CrossRef]
- Tomita, Y.; Oronsky, B.; Abrouk, N.; Cabrales, P.; Reid, T.R.; Lee, M.-J.; Yuno, A.; Baker, J.; Lee, S.; Trepel, J.B. In Small Cell Lung Cancer Patients Treated with RRx-001, a Downregulator of CD47, Decreased Expression of PD-L1 on Circulating Tumor Cells Significantly Correlates with Clinical Benefit. Transl. Lung Cancer Res. 2021, 10, 274–278. [Google Scholar] [CrossRef]
- Sinclair, C.; Revenko, A.S.; Johnson, R.B.; Peter, A.; Taylor, M.A.; Hettrick, L.A.; Klein, S.; Solanki, A.; Chapman, M.; Yates, J.; et al. Abstract 2713: Discovery and Characterization of AZD8701, a High Affinity Antisense Oligonucleotide Targeting FOXP3 to Relieve Immunosuppression in Cancer. Cancer Res. 2019, 79, 2713. [Google Scholar] [CrossRef]
- Wolchok, J.D.; Hoos, A.; O’Day, S.; Weber, J.S.; Hamid, O.; Lebbé, C.; Maio, M.; Binder, M.; Bohnsack, O.; Nichol, G.; et al. Guidelines for the Evaluation of Immune Therapy Activity in Solid Tumors: Immune-Related Response Criteria. Clin. Cancer Res. 2009, 15, 7412–7420. [Google Scholar] [CrossRef]
- Chaudhuri, A.A.; Chabon, J.J.; Lovejoy, A.F.; Newman, A.M.; Stehr, H.; Azad, T.D.; Khodadoust, M.S.; Esfahani, M.S.; Liu, C.L.; Zhou, L.; et al. Early Detection of Molecular Residual Disease in Localized Lung Cancer by Circulating Tumor DNA Profiling. Cancer Discov. 2017, 7, 1394–1403. [Google Scholar] [CrossRef]
- Swisher, E.M.; Wollan, M.; Mahtani, S.M.; Willner, J.B.; Garcia, R.; Goff, B.A.; King, M.-C. Tumor-Specific P53 Sequences in Blood and Peritoneal Fluid of Women with Epithelial Ovarian Cancer. Am. J. Obs. Gynecol. 2005, 193, 662–667. [Google Scholar] [CrossRef] [PubMed]
- Guardant360®CDx FDA Approval Letter. Available online: https://www.accessdata.fda.gov/cdrh_docs/pdf20/P200010S008A.pdf (accessed on 5 May 2021).
- Powrózek, T.; Krawczyk, P.; Kucharczyk, T.; Milanowski, J. Septin 9 Promoter Region Methylation in Free Circulating DNA—Potential Role in Noninvasive Diagnosis of Lung Cancer: Preliminary Report. Med. Oncol. 2014, 31, 917. [Google Scholar] [CrossRef] [PubMed]
- University of California, San Francisco. Circulating Tumor DNA for Risk Stratification in Lung Cancer Screening; clinicaltrials.gov, 2020. Available online: https://clinicaltrials.gov/ct2/show/NCT03774758 (accessed on 5 May 2021).
- University College, London. The SUMMIT Study: Cancer Screening Study with or without Low Dose Lung CT to Validate a Multi-Cancer Early Detection Test; clinicaltrials.gov; University College: London, UK, 2021. Available online: https://clinicaltrials.gov/ct2/show/NCT03934866 (accessed on 5 May 2021).
- Cescon, D.W.; Bratman, S.V.; Chan, S.M.; Siu, L.L. Circulating Tumor DNA and Liquid Biopsy in Oncology. Nat. Cancer 2020, 1, 276–290. [Google Scholar] [CrossRef] [PubMed]
- Grunblatt, E.; Wu, N.; Zhang, H.; Liu, X.; Norton, J.P.; Ohol, Y.; Leger, P.; Hiatt, J.B.; Eastwood, E.C.; Thomas, R.; et al. MYCN Drives Chemoresistance in Small Cell Lung Cancer While USP7 Inhibition Can Restore Chemosensitivity. Genes Dev. 2020, 34, 1210–1226. [Google Scholar] [CrossRef]
- Uprety, D.; Remon, J.; Adjei, A.A. All That Glitters Is Not Gold: The Story of Rovalpituzumab Tesirine in SCLC. J. Thorac. Oncol. 2021, 16, 1429–1433. [Google Scholar] [CrossRef]

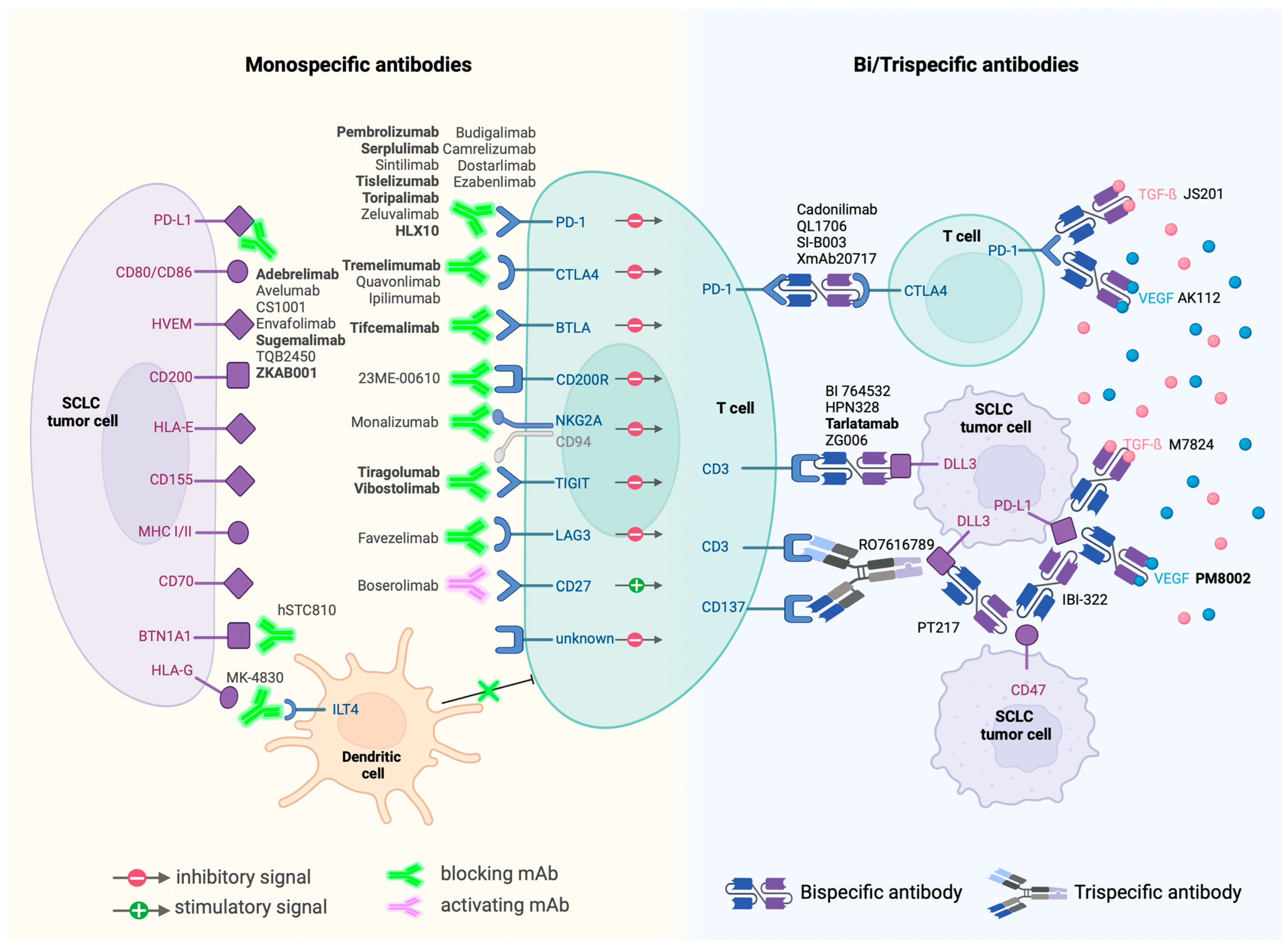
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).