
Magnetic Resonance Imaging of Central Nervous System Manifestations of Type 1 Neurofibromatosis: Pictorial Review and Retrospective Study of Their Frequency in a Cohort of Patients
 , , ,
, , , 
Abstract
:1. Introduction
2. Pictorial Review of Central Nervous System Manifestations of Type 1 Neurofibromatosis
2.1. Visual Pathway Lesions
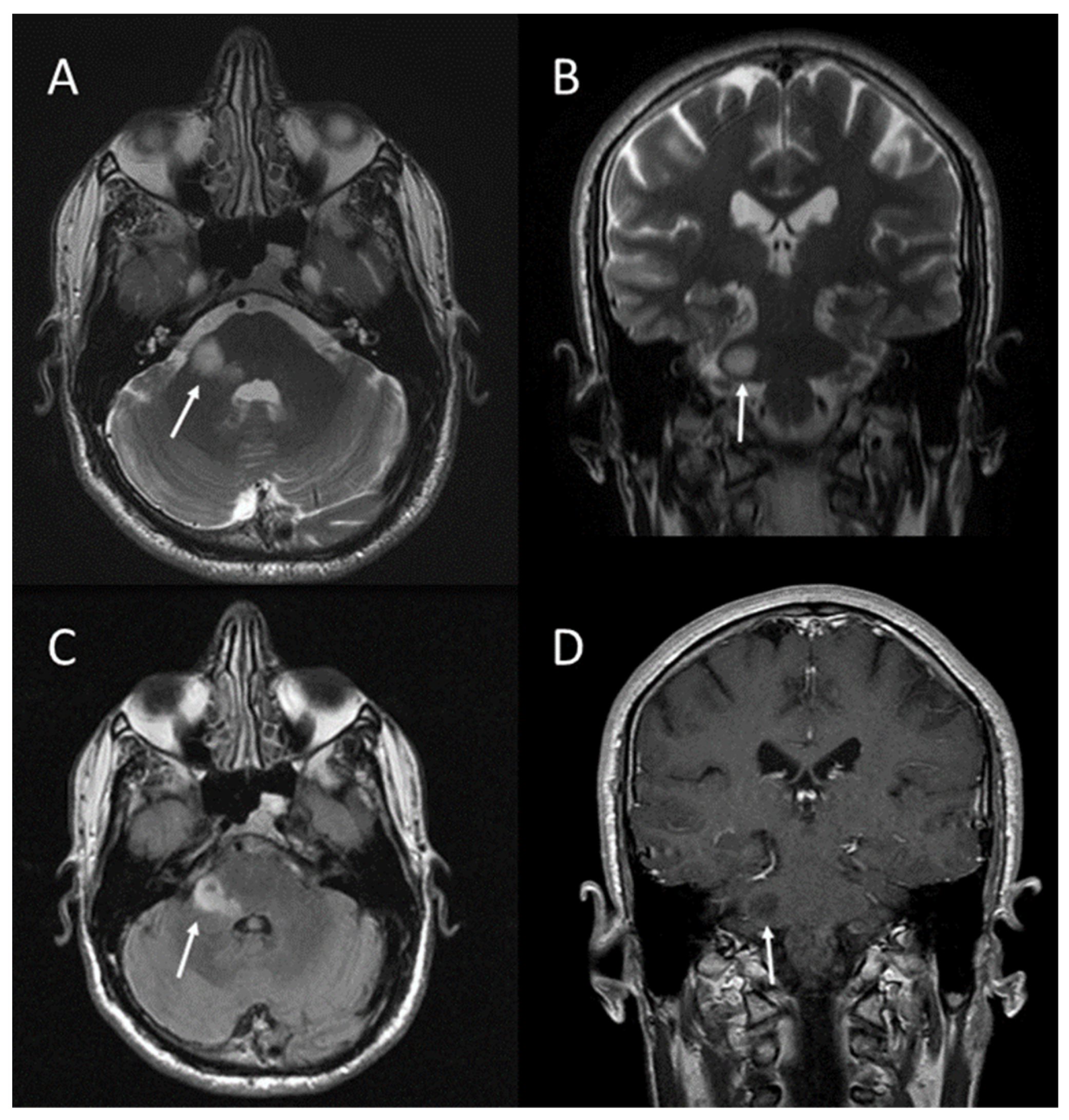
2.2. Brain Tumours
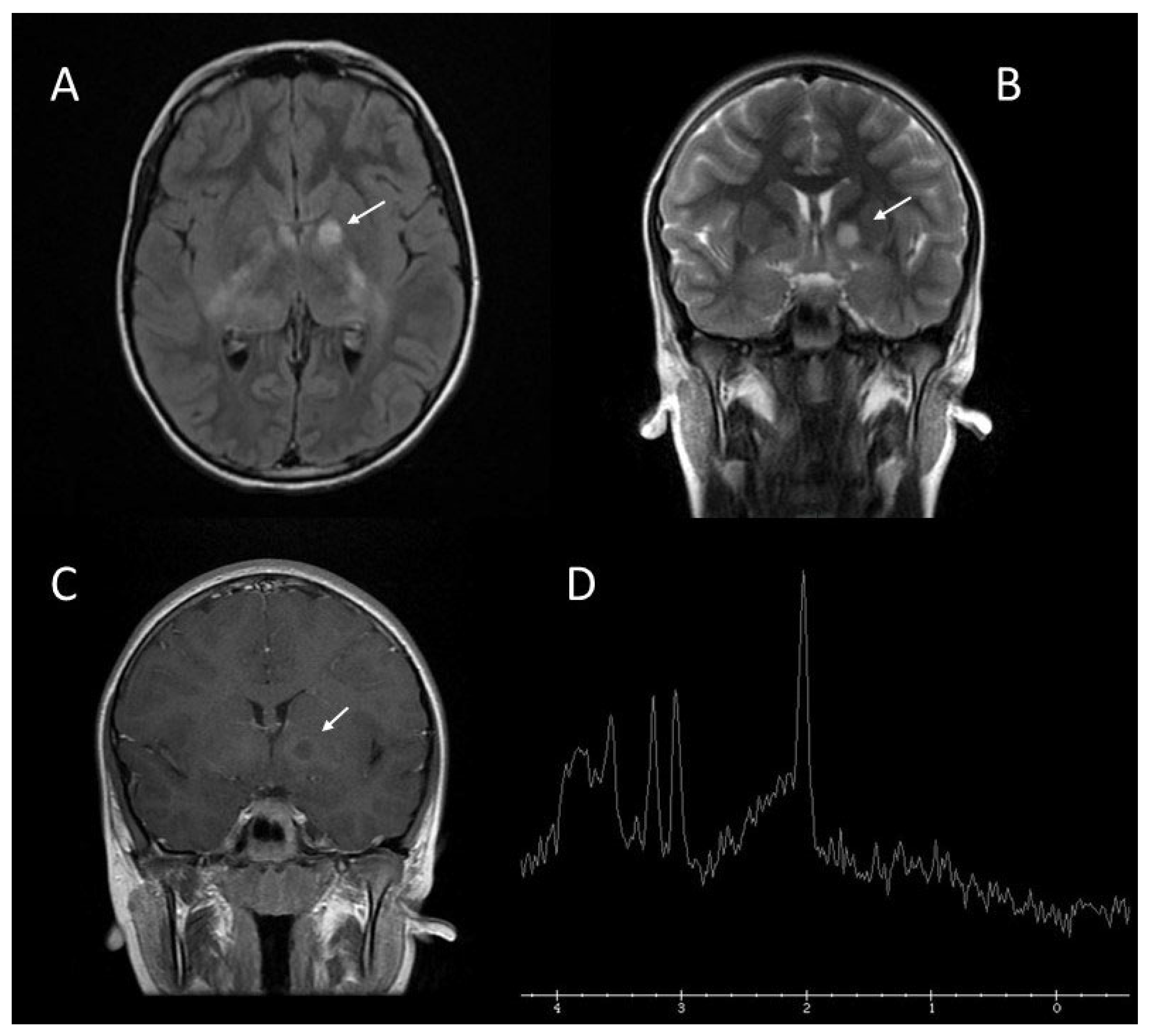
2.3. Brain Abnormalities: UBOs or FASI
2.4. Spinal Tumours
2.5. Plexiform Neurofibromas and Sphenoid Dysplasia
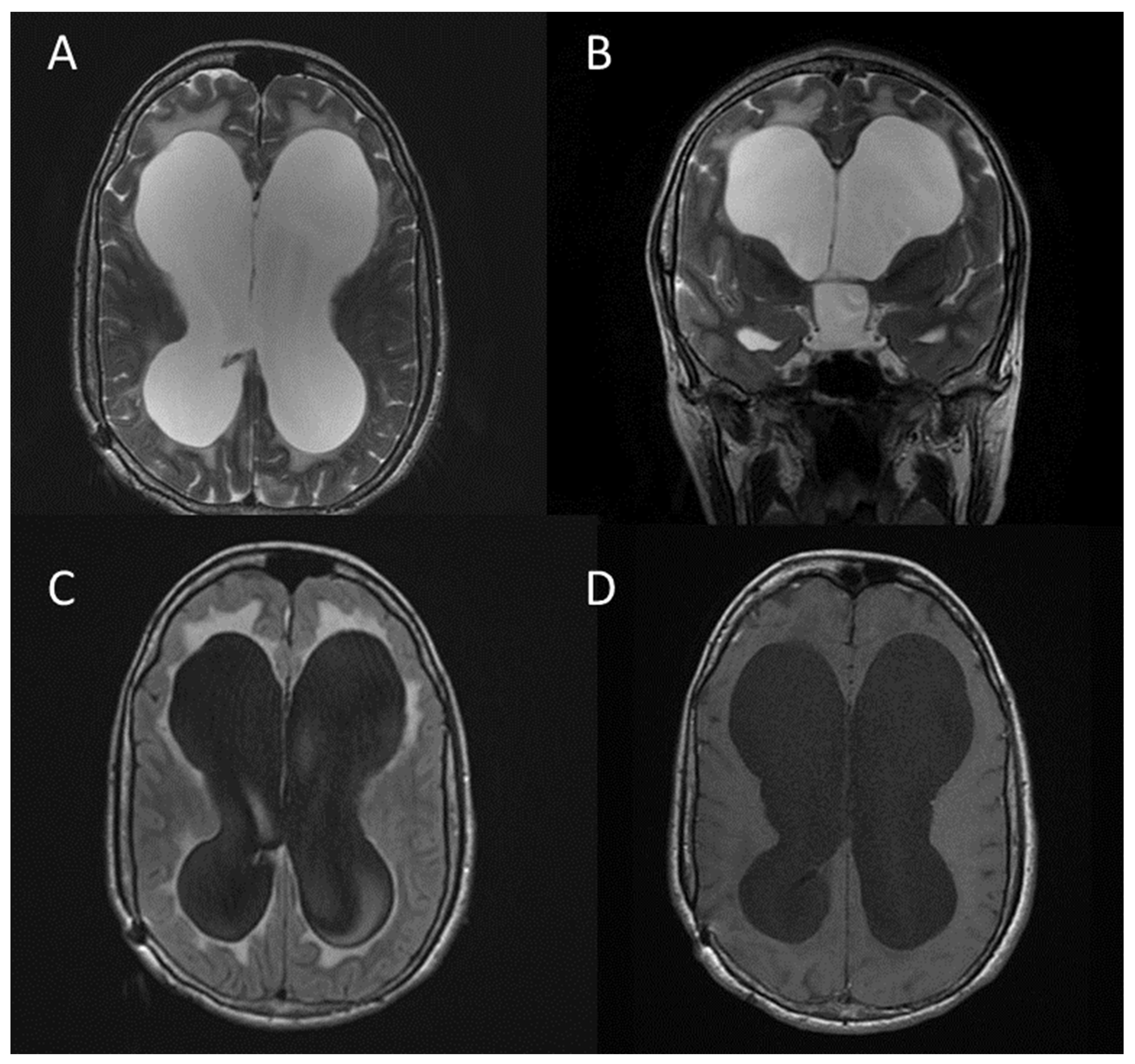
2.6. Other CNS Manifestations of NF1
- Hydrocephalus (usually secondary to aqueduct stenosis) (Figure 10);
- Macrocephaly;
- Meningocoeles;
- Arachnoid cysts;
- Dural ectasia.
3. Materials and Methods
3.1. Population Study
3.2. MR Protocol
3.3. Statistical Analysis
4. Results
4.1. Frequency of Visual Pathway Lesions
4.2. Frequency of Brain Tumours
4.3. Frequency of Brain Abnormalities—FASI or UBOs
4.4. Frequency of Spinal Tumours
4.5. Frequency of Plexiform Neurofibromas
4.6. Frequency of Craniofacial Bone Alterations
4.7. Frequency of Other CNS Abnormalities
- triventricular hydrocephalus in two patients;
- buphthalmos in one patient;
- cerebrovascular anomalies in one patient;
- choroid plexus xanthogranuloma in one patient;
- Hydromyelia in one patient.
4.8. Frequency of Normal MRI Examinations
5. Discussion
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cassina, M.; Frizziero, L.; Opocher, E.; Parrozzani, R.; Sorrentino, U.; Viscardi, E.; Miglionico, G.; Midena, E.; Clementi, M.; Trevisson, E.; et al. Optic pathway glioma in type 1 neurofibromatosis: Review of its pathogenesis, diagnostic assessment, and treatment recommendations. Cancers 2019, 11, 1790. [Google Scholar] [CrossRef] [PubMed]
- Adil, A.; Singh, A.K. Neurofibromatosis Type 1 (Von Recklinghausen); StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Ferner, R.E.; Huson, S.M.; Thomas, N.; Moss, C.; Willshaw, H.; Evans, D.G.; Upadhyaya, M.; Towers, R.; Gleeson, M.; Steiger, C.; et al. Guidelines for the diagnosis and management of individuals with neurofibromatosis. J. Med. Genet. 2007, 44, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Legius, E.; Messiaen, L.; Wolkenstein, P.; Pancza, P.; Avery, R.A.; Berman, Y.; Blakeley, J.; Babovic-Vuksanovic, D.; Cunha, K.S.; Ferner, R.; et al. Revised diagnostic criteria for neurofibromatosis type 1 and Legius syndrome: An international consensus recommendation. Genet. Med. 2021, 23, 1506–1513. [Google Scholar] [CrossRef] [PubMed]
- Mentzel, H.J.; Seidel, J.; Fitzek, C.; Eichhorn, A.; Vogt, S.; Reichenbach, J.R.; Zintl, F.; Kaiser, W.A. Pediatric brain MRI in neurofibromatosis type I. Eur. Radiol. 2005, 15, 814–822. [Google Scholar] [CrossRef] [PubMed]
- Patel, N.B.; Stacy, G.S. Musculoskeletal manifestations of neurofibromatosis type 1. AJR Am. J. Roentgenol. 2012, 199, W99–W106. [Google Scholar] [CrossRef] [PubMed]
- Ahlawat, S.; Blakeley, J.O.; Langmead, S.; Belzberg, A.J.; Fayad, L.M. Current status and recommendations for imaging in neurofibromatosis type 1, neurofibromatosis type 2, and schwannomatosis. Skelet. Radiol. 2020, 49, 199–219. [Google Scholar] [CrossRef] [PubMed]
- Mukonoweshuro, W.; Griffiths, P.; Blaser, S. Neurofibromatosis Type 1: The Role of Neuroradiology. Neuropediatrics 1999, 30, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Van Es, S.; North, K.N.; McHugh, K.; De Silva, M. MRI findings in children with neurofibromatosis type 1: A prospective study. Pediatr. Radiol. 1996, 26, 478–487. [Google Scholar] [CrossRef]
- Eid, H.; Crevier-Sorbo, G.; Aldraihem, A.; Menegotto, F.; Wilson, N. Neurofibromatosis type 1: Description of a novel diagnostic scoring system in pediatric optic nerve glioma. Am. J. Roentgenol. 2019, 212, 892–898. [Google Scholar] [CrossRef]
- Di Pietro, S.; Palmucci, S.; Di Mari, A.; Monaco, E.; Pennisi, I.; Belfiore, G.; Foti, P.V.; Ruggieri, M.; Basile, A. Central Nervous System manifestations of Type 1 Neurofibromatosis: Spectrum of MRI findings. In Proceedings of the European Congress of Radiology-ECR, Online, 2–6 March 2022. [Google Scholar]
- Costa, A.D.A.; Gutmann, D.H. Brain tumors in neurofibromatosis type 1. Neuro-Oncol. Adv. 2020, 2 (Suppl. S1), i85–i97. [Google Scholar] [CrossRef]
- Guillamo, J.S.; Creange, A.; Kalifa, C.; Grill, J.; Rodriguez, D.; Doz, F.; Barbarot, S.; Zerah, M.; Sanson, M.; Bastuji-Garin, S.; et al. Prognostic factors of CNS tumours in Neurofibromatosis 1 (NF1): A retrospective study of 104 patients. Brain 2003, 126, 152–160. [Google Scholar] [CrossRef] [PubMed]
- Evans, D.G.R.; Salvador, H.; Chang, V.Y.; Erez, A.; Voss, S.D.; Schneider, K.W.; Scott, H.S.; Plon, S.E.; Tabori, U. Cancer and central nervous system tumor surveillance in pediatric neurofibromatosis 1. Clin. Cancer Res. 2017, 23, e46–e53. [Google Scholar] [CrossRef] [PubMed]
- Filho, J.R.L.F.; Munis, M.P.; Souza, A.S.; Sanches, R.A.; Goloni-Bertollo, E.M.; Pavarino-Bertelli, E.C. Unidentified bright objects on brain MRI in children as a diagnostic criterion for neurofibromatosis type 1. Pediatr. Radiol. 2008, 38, 305–310. [Google Scholar] [CrossRef] [PubMed]
- DeBella, K.; Poskitt, K.; Szudek, J.; Friedman, J.M. Use of ‘unidentified bright objects’ on MRI for diagnosis of neurofibromatosis 1 in children. Neurology 2000, 54, 1646–1650. [Google Scholar] [CrossRef] [PubMed]
- Di Paolo, D.P.; Zimmerman, R.A.; Rorke, L.B.; Zackai, E.H.; Bilaniuk, L.T.; Yachnis, A.T. Neurofibromatosis type 1: Pathologic substrate of high-signal-intensity foci in the brain. Radiology 1995, 195, 721–724. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, P.D.; Blaser, S.; Mukonoweshuro, W.; Armstrong, D.; Milo-Mason, G.; Cheung, S. Neurofibromatosis bright objects in children with neurofibromatosis type 1: A proliferative potential? Pediatrics 1999, 104, e49. [Google Scholar] [CrossRef]
- Wilkinson, I.D.; Griffiths, P.D.; Wales, J.K.H. Proton magnetic resonance spectroscopy of brain lesions in children with neurofibromatosis type 1. Magn. Reson. Imaging 2001, 19, 1081–1089. [Google Scholar] [CrossRef] [PubMed]
- Gonen, O.; Wang, Z.J.; Viswanathan, A.K.; Molloy, P.T.; Zimmerman, R.A. Three-Dimensional Multivoxel Proton MR Spectroscopy of the Brain in Children with Neurofibromatosis Type 1. Am. J. Neuroradiol. 1999, 20, 1333–1341. [Google Scholar] [PubMed]
- Restrepo, C.S.; Riascos, R.F.; Hatta, A.A.; Rojas, R. Neurofibromatosis type 1: Spinal manifestations of a systemic disease. J. Comput. Assist. Tomogr. 2005, 29, 532–539. [Google Scholar] [CrossRef]
- Thakkar, S.D.; Feigen, U.; Mautner, V.F. Spinal tumours in neurofibromatosis type 1: An MRI study of frequency, multiplicity and variety. Neuroradiology 1999, 41, 625–629. [Google Scholar] [CrossRef]
- Khong, P.L.; Goh, W.H.S.; Wong, V.C.N.; Fung, C.W.; Ooi, G.C. MR imaging of spinal tumors in children with neurofibromatosis I. Am. J. Roentgenol. 2003, 180, 413–417. [Google Scholar] [CrossRef] [PubMed]
- Tchernev, G.; Chokoeva, A.A.; Patterson, J.W.; Bakardzhiev, I.; Wollina, U.; Tana, C. Plexiform Neurofibroma. Medicine 2016, 95, e2663. [Google Scholar] [CrossRef] [PubMed]
- Korf, B.R. Plexiform neurofibromas. Am. J. Med. Genet. 1999, 89, 31–37. [Google Scholar] [CrossRef]
- Meersschaut, V.A.; Kros, J.M.; Catsman-Berrevoets, C.E.; Lequin, M.H. Congenital bilateral plexiform neurofibromas of the cavernous sinuses. Pediatr. Radiol. 2003, 33, 272–274. [Google Scholar] [CrossRef] [PubMed]
- Naran, S.; Swanson, J.W.; Ligh, C.A.; Shubinets, V.; Taylor, J.A.; Bartlett, S.P. Sphenoid dysplasia in neurofibromatosis: Patterns of presentation and outcomes of treatment. Plast. Reconstr. Surg. 2018, 142, 518E–526E. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, P.S.; Rothner, A.D.; Emch, T.M.; Friedman, N.R.; Moodley, M. Cerebral vasculopathy in children with neurofibromatosis type 1. J. Child Neurol. 2013, 28, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Serafini, N.B.; Serafini, C.B.; Vinhas, A.S.; Godinho, M.B. Moyamoya syndrome associated with neurofibromatosis type 1 in a pediatric patient. An. Bras. Dermatol. 2017, 92, 870–873. [Google Scholar] [CrossRef]
- Kaas, B.; Huisman, T.A.G.M.; Tekes, A.; Bergner, A.; Blakeley, J.O.; Jordan, L.C. Spectrum and prevalence of vasculopathy in pediatric neurofibromatosis type 1. J. Child Neurol. 2013, 28, 561–569. [Google Scholar] [CrossRef] [PubMed]
- Ferner, R.E.; Gutmann, D.H. Neurofibromatosis type 1 (NF1): Diagnosis and management. Handb. Clin. Neurol. 2013, 115, 939–955. [Google Scholar]
- Friedman, J.M. Neurofibromatosis 1. In GeneReviews; University of Washington: Seattle, WA, USA, 2022. [Google Scholar]
- Chauvel-Picard, J.; Lion-Francois, L.; Beuriat, P.-A.; Paulus, C.; Szathmari, A.; Mottolese, C.; Gleizal, A.; Di Rocco, F. Craniofacial bone alterations in patients with neurofibromatosis type 1. Childs Nerv. Syst. 2020, 36, 2391–2399. [Google Scholar] [CrossRef]
- Scala, M.; Schiavetti, I.; Madia, F.; Chelleri, C.; Piccolo, G.; Accogli, A.; Riva, A.; Salpietro, V.; Bocciardi, R.; Morcaldi, G.; et al. Genotype-phenotype correlations in neurofibromatosis type 1: A single-center cohort study. Cancers 2021, 13, 1879. [Google Scholar] [CrossRef] [PubMed]
- Roth, J.; Constantini, S.; Cinalli, G. Neurofibromatosis type 1-related hydrocephalus: Causes and treatment considerations. Childs. Nerv. Syst. 2020, 36, 2385–2390. [Google Scholar] [CrossRef] [PubMed]
- Chaudhry, I.A.; Morales, J.; Shamsi, F.A.; Al-Rashed, W.; Elzaridi, E.; Arat, Y.O.; Jacquemin, C.; Oystreck, D.T.; Bosley, T.M. Orbitofacial neurofibromatosis: Clinical characteristics and treatment outcome. Eye 2012, 26, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Rosser, T.L.; Vezina, G.; Packer, R.J. Cerebrovascular abnormalities in a population of children with neurofibromatosis type 1. Neurology 2005, 64, 553–555. [Google Scholar] [CrossRef]
- Ferner, R.E.; Chaudhuri, R.; Bingham, J.; Cox, T.; Hughes, R. MRI in neurofibromatosis 1. The nature and evolution of increased intensity T2 weighted-lesions and their relationship to intellectual impairment. J. Neurol. Neurosurg. Psychiatry 1993, 56, 492–495. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1 | Six or more café-au-lait macules > 5 mm in prepubertal individuals and over 15 mm in post-pubertal individuals |
| 2 | Freckling in the axillary or inguinal region |
| 3 | Two or more neurofibromas of any type or one plexiform neurofibroma |
| 4 | Optic pathway glioma |
| 5 | Two or more iris Lisch nodules or two or more choroidal abnormalities |
| 6 | A distinctive osseous lesion such as sphenoid dysplasia, anterolateral bowing of the tibia, or pseudarthrosis of a long bone |
| 7 | A heterozygous pathogenic NF1 variant with a variant allele fraction of 50% in apparently normal tissue such as white blood cells |
| Finding | % of Patients Affected | MRI Features |
|---|---|---|
| Visual pathway lesions (optic pathway gliomas) | 5–15% | Enlargement of optic nerves or chiasma (diameter greater than 3.9 mm); |
| On T2-weighted images: compact low signal core with higher intensity circumferential component; | ||
| On T1-weighted images: isointense signal, with enhancement after gadolinium administration. | ||
| Brain tumours | Variable—based on brain location and grade of tumour | In children, low-grade gliomas are predominant, and the cerebellum and the brainstem are the most involved regions. In adults, high-grade gliomas are more commonly, frequently arising in the cerebral hemispheres; |
| On T2-weighted images: hyperintense signal; | ||
| On T1-weighted images: isointense or slightly hypointense signal, with enhancement after gadolinium administration. | ||
| Brain abnormalities: UBOs or FASI | 43–93% | Areas of increased signal intensity on T2-weighted MRI sequence, not visualized on T1-weighted images and do not show mass effect or contrast enhancement; |
| Rarely seen in patients older than 20 years; | ||
| The cerebellum, the brainstem, and the basal ganglia are the regions most involved. | ||
| Spinal tumours: peripheral nerve sheath tumours and intramedullary tumours | 40–96% | Benign neurofibromas represent the majority of spinal tumours, while intramedullary lesions are rarely observed; |
| Neurofibromas usually show hyperintense signal on T2-weighted images with central hypointense target, and hypointense signal on T1-weighted images, with heterogeneous contrast enhancement. | ||
| Features suggestive of malignant nerve sheath tumour: enlarging tumour, tumour size > 5 cm, ill-defined margins, lack of a central hypointense target on T2-weighted images, heterogeneity with central necrosis. | ||
| Plexiform neurofibromas | Up to 30% | Observed most frequently in the craniomaxillofacial region; |
| On T2-weighted images: heterogeneous masses with high signal intensity, often with a central area of low signal; | ||
| On T1-weighted images: slightly hyperintense to muscle, with variable contrast enhancement. |
| TR (ms) | TE (ms) | Thickness (mm) | Gap (mm) | FOV | Matrix | NEX | |
|---|---|---|---|---|---|---|---|
| Sagittal 2D FSE T1 | 380 | 12 | 4 | 0.5 | 25 | 256 × 384 | 1 |
| Coronal 2D T2 FRFSE | 6420 | 101.2 | 4 | 0.5 | 25 | 224 × 320 | 4 |
| Axial 2D T2 FRFSE | 5600 | 123.5 | 4 | 0.5 | 25 | 256 × 384 | 4 |
| Axial 2D T2 FLAIR | 10,002 | 136.3 | 4 | 0.5 | 25 | 224 × 256 | 1 |
| Axial 2D DWI b1000 | 6850 | 81.9 | 4 | 0.5 | 25 | 128 × 128 | 2 |
| Axial 2D FSE T1 | 400 | 12.4 | 4 | 0.5 | 25 | 256 × 384 | 2 |
| Axial 2D T2 GRE | 400 | 14.4 | 4 | 0.5 | 25 | 256 × 320 | 1 |
| Axial 3D b-SSFP | 6.6 | 2.5 | 0.8 | −0.4 | 25 | 256 × 448 | 4 |
| Axial 2D FSE T2 FS | 2080 | 95.9 | 3 | 0 | 16–18 | 256 × 256 | 4 |
| Axial 2D FSE T2 | 1880 | 102.4 | 3 | 0 | 16–18 | 256 × 256 | 4 |
| Axial 2D FSE T1 FS | 360 | 10.6 | 3 | 0 | 16–18 | 224 × 256 | 2 |
| Axial 3D IR FSPGR T1 | 8.5 | 3.1 | 2 | 0 | 25 | 224 × 256 | 2 |
| 3D TOF MRA | 23 | 2.8 | 1.4 | −0.7 | 16 | 256 × 320 | 1 |
| Sagittal 2D FSE T1 | 500 | 11 | 3 | 0.3 | 36–38 | 256 × 320 | 2 |
| Sagittal 2D FRFSE T2 FS | 5700 | 107.3 | 3 | 0.3 | 36–38 | 256 × 320 | 2 |
| Finding | % |
|---|---|
| Visual pathway lesions | 20.3 |
| With bilateral involvement | 9.4 |
| Brain tumours | 5.4 |
| Supratentorial region | 2.7 |
| Infratentorial region | 2.7 |
| FASI/UBOs | 58.1 |
| Spinal tumours | 33.8 |
| Peripheral nerve sheath tumours | 33.8 |
| Intramedullary tumours | 0 |
| Plexiform neurofibromas | 16.2 |
| Craniofacial bone alterations | 4.1 |
| Craniosynostosis | 2.7 |
| Sphenoid dysplasia | 1.3 |
| Other CNS abnormalities | 8.1 |
| Triventricular hydrocephalus | 2.7 |
| Buphthalmos | 1.3 |
| Cerebrovascular anomalies | 1.3 |
| Choroid plexus xanthogranuloma | 1.3 |
| Hydromyelia | 1.3 |
| Normal MRI examinations | 12.1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Pietro, S.; Reali, L.; Tona, E.; Belfiore, G.; Praticò, A.D.; Ruggieri, M.; David, E.; Foti, P.V.; Santonocito, O.G.; Basile, A.; et al. Magnetic Resonance Imaging of Central Nervous System Manifestations of Type 1 Neurofibromatosis: Pictorial Review and Retrospective Study of Their Frequency in a Cohort of Patients. J. Clin. Med. 2024, 13, 3311. https://doi.org/10.3390/jcm13113311
Di Pietro S, Reali L, Tona E, Belfiore G, Praticò AD, Ruggieri M, David E, Foti PV, Santonocito OG, Basile A, et al. Magnetic Resonance Imaging of Central Nervous System Manifestations of Type 1 Neurofibromatosis: Pictorial Review and Retrospective Study of Their Frequency in a Cohort of Patients. Journal of Clinical Medicine. 2024; 13(11):3311. https://doi.org/10.3390/jcm13113311
Chicago/Turabian StyleDi Pietro, Stefano, Linda Reali, Emanuela Tona, Giuseppe Belfiore, Andrea Domenico Praticò, Martino Ruggieri, Emanuele David, Pietro Valerio Foti, Orazio Giuseppe Santonocito, Antonio Basile, and et al. 2024. "Magnetic Resonance Imaging of Central Nervous System Manifestations of Type 1 Neurofibromatosis: Pictorial Review and Retrospective Study of Their Frequency in a Cohort of Patients" Journal of Clinical Medicine 13, no. 11: 3311. https://doi.org/10.3390/jcm13113311