
Optogenetics and Targeted Gene Therapy for Retinal Diseases: Unravelling the Fundamentals, Applications, and Future Perspectives



Abstract
1. Introduction
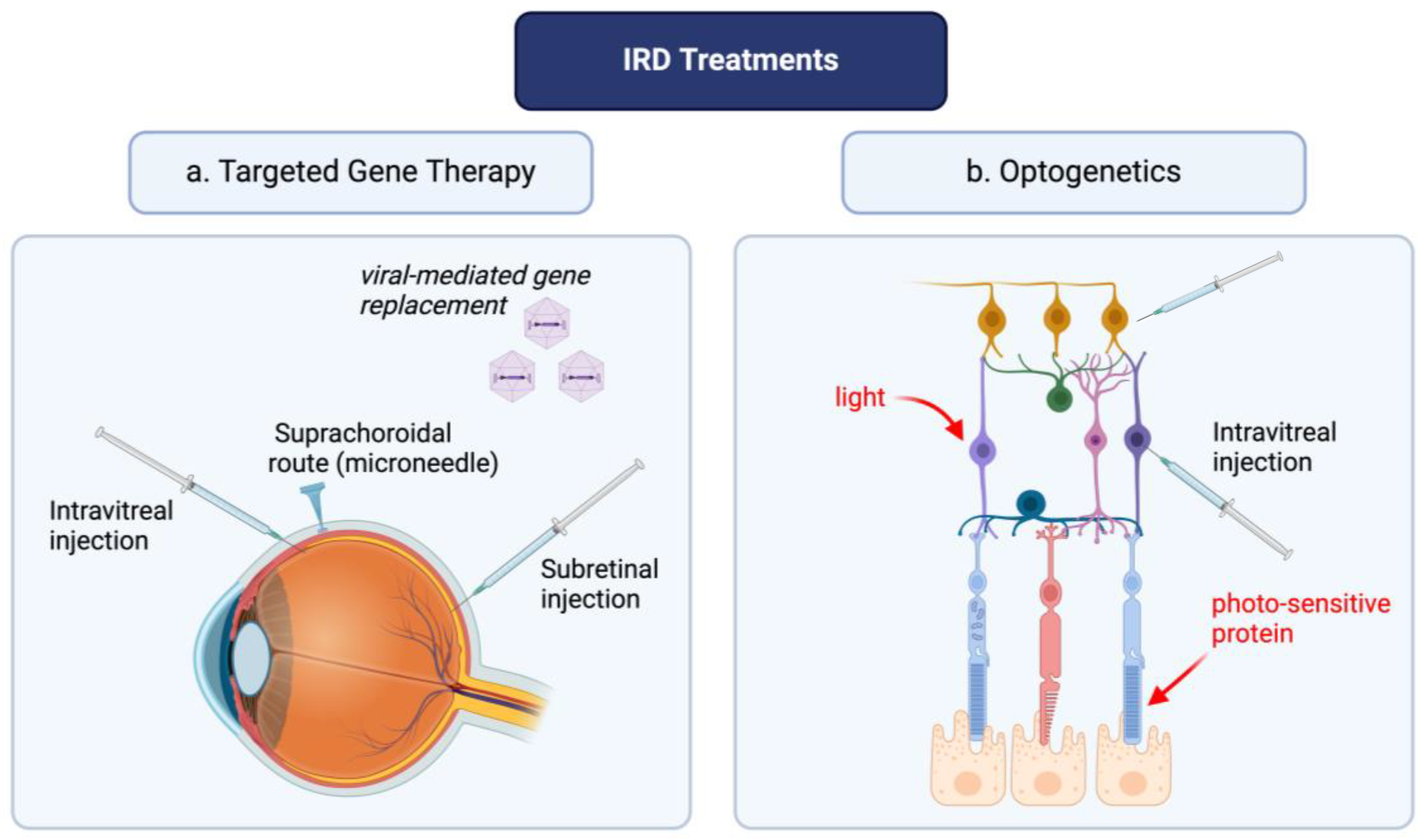
2. Optogenetics and Targeted Gene Therapies: Novel Advances in the Treatment of IRDs
3. Optogenetics
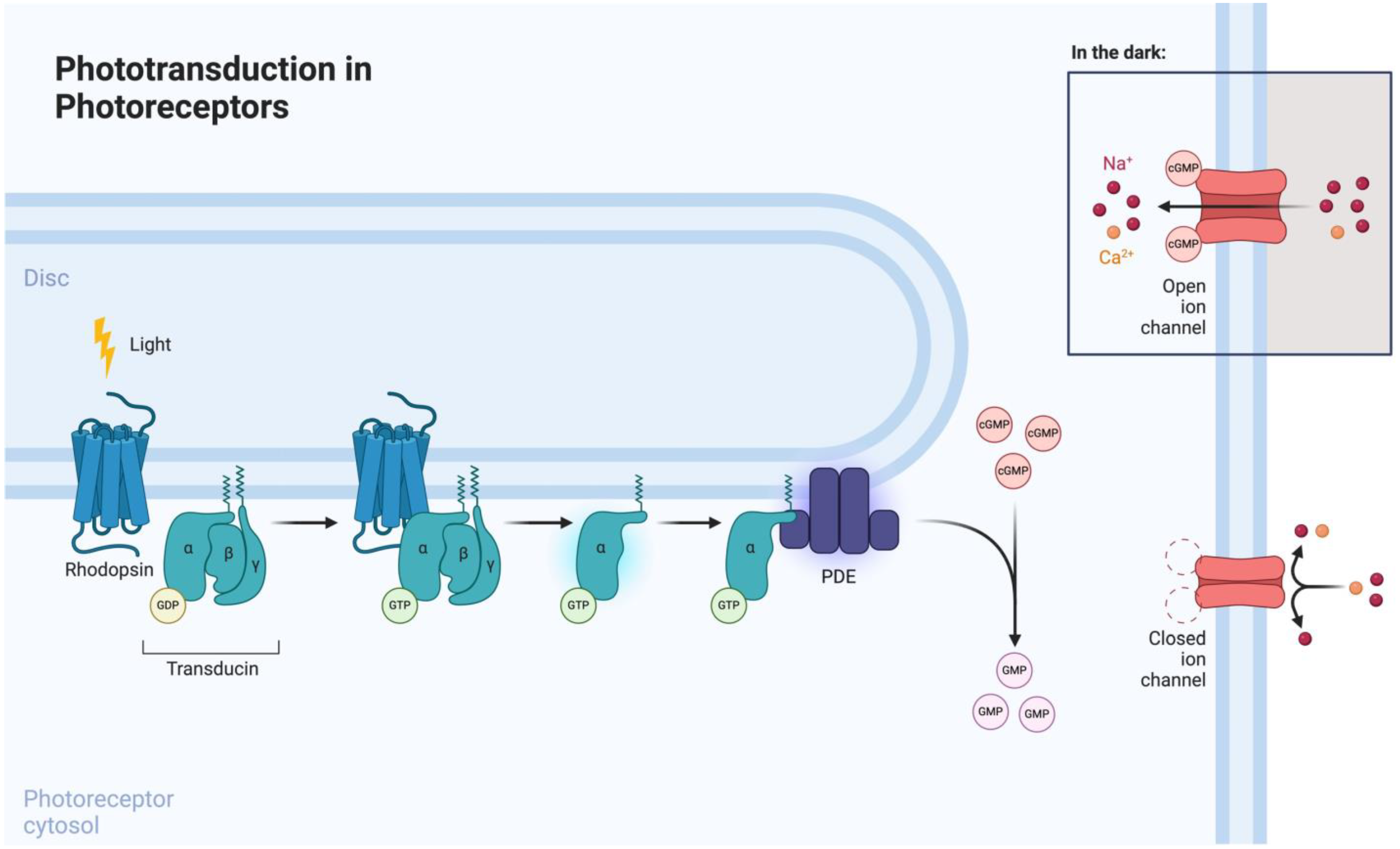
3.1. Phototransduction
3.2. Optogenetic Engineering
Opsins

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | Opsin Examples a | Advantages | Disadvantages | References |
|---|---|---|---|---|
| Light-gated ion channels | ||||
| Blue light opsins | ChR2 GtACRe | Rapid cell depolarization following stimulation (<50 μs) | Limited tissue penetration Requires high stimulus intensity in comparison to physiological rhodopsin and cones | [16,35,36,46] |
| Red-shift opsins | VChR1 ReaChR Chrimson ChrimsonR ChrimsonSA ChRmine frChRmine | Greater tissue penetration | Non-negligeable blue-light sensitivity that may lead to cross-activation | [47,48,49,50,51] |
| Chimeric opsins | Chronos/ChrimsonR CheRiff/ChrimsonR ChR2/ReaChR ChR2/ChrimsonR | Highly specific modulation of red or blue opsins | Low population of blue opsins can limit the excitatory potential | [52,53,54,55,56,57,58,59,60,61] |
| Light-driven ion pumps | ||||
| Hydrogen pumps | BR Arch Mac | Production of higher photocurrent rates and less interference with neurotransmission | [62,63] | |
| Sodium pumps | KR2 (DeNaR) | Efficient for neuron silencing | [64,65] | |
| Chloride pumps | HR NpHR eNpHR 2.0 eNpHR 3.0 | Rapid activation and inactivation kinetics Efficient for neuron silencing | Low levels of generated photocurrent with NpHR Requires high stimulus intensity in comparison to physiological rhodopsin and cones, as well as ChR2 | [66,67,68,69] |
| Light-activated signaling/enzyme opsins | ||||
| Sensory rhodopsins (SR) | SRI SRII | |||
| HKR Rh-GC Rh-PDE | Selective modulation of intracellular signaling pathways | [42,43,44,45] | ||
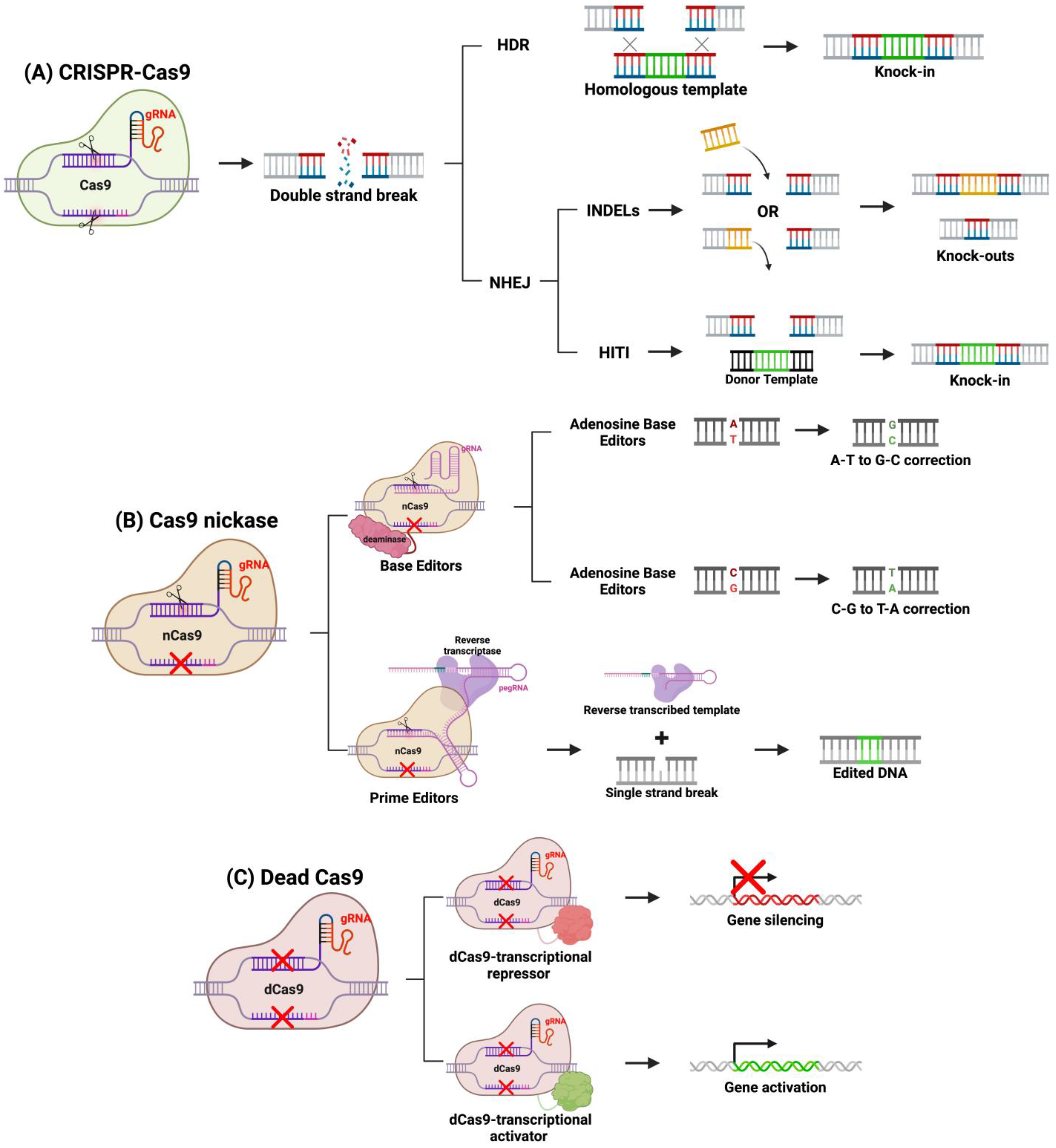
4. Genome Editing with CRISPR-Cas9
System Engineering
5. RNA Interference for IRDs
System Engineering
6. Vectors for Optogenetics and Targeted Gene Therapy
7. Recent Advances in Optogenetics
7.1. Delivery of Optogenetic Actuators to Retinal Ganglion Cells
7.2. Delivery of Optogenetic Actuators to Bipolar Cells
7.3. Delivery of Optogenetic Actuators to Photoreceptors
7.4. Clinical Trials
8. Recent Advances in Targeted Gene Therapy
8.1. Retinitis Pigmentosa
8.2. Leber Congenital Amaurosis
8.3. Stargardt Disease
8.4. Clinical Trials
9. Future Perspectives
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Sahel, J.-A.; Marazova, K.; Audo, I. Clinical characteristics and current therapies for inherited retinal degenerations. Cold Spring Harb. Perspect. Med. 2014, 5, a017111. [Google Scholar] [CrossRef]
- Hanany, M.; Rivolta, C.; Sharon, D. Worldwide carrier frequency and genetic prevalence of autosomal recessive inherited retinal diseases. Proc. Natl. Acad. Sci. USA 2020, 117, 2710–2716. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Cheung, S.; Fasso-Opie, A.; Galvin, O.; Moniz, L.S.; Earle, D.; Durham, T.; Menzo, J.; Li, N.; Duffy, S.; et al. The Impact of Inherited Retinal Diseases in the United States of America (US) and Canada from a Cost-of-Illness Perspective. Clin. Ophthalmol. 2021, 15, 2855–2866. [Google Scholar] [CrossRef] [PubMed]
- Cremers, F.P.M.; Boon, C.J.F.; Bujakowska, K.; Zeitz, C. Special Issue Introduction: Inherited Retinal Disease: Novel Candidate Genes, Genotype-Phenotype Correlations, and Inheritance Models. Genes. 2018, 9, 215. [Google Scholar] [CrossRef] [PubMed]
- Sabbaghi, H.; Madani, S.; Ahmadieh, H.; Daftarian, N.; Suri, F.; Khorrami, F.; Saviz, P.; Shahriari, M.H.; Motevasseli, T.; Fekri, S.; et al. A health terminological system for inherited retinal diseases: Content coverage evaluation and a proposed classification. PLoS ONE 2023, 18, e0281858. [Google Scholar] [CrossRef] [PubMed]
- Ben-Yosef, T. Inherited Retinal Diseases. Int. J. Mol. Sci. 2022, 23, 13467. [Google Scholar] [CrossRef] [PubMed]
- Karali, M.; Testa, F.; Di Iorio, V.; Torella, A.; Zeuli, R.; Scarpato, M.; Romano, F.; Onore, M.E.; Pizzo, M.; Melillo, P.; et al. Genetic epidemiology of inherited retinal diseases in a large patient cohort followed at a single center in Italy. Sci. Rep. 2022, 12, 20815. [Google Scholar] [CrossRef]
- Chen, T.-C.; Huang, D.-S.; Lin, C.-W.; Yang, C.-H.; Yang, C.-M.; Wang, V.Y.; Lin, J.-W.; Luo, A.C.; Hu, F.-R.; Chen, P.-L. Genetic characteristics and epidemiology of inherited retinal degeneration in Taiwan. NPJ Genom. Med. 2021, 6, 16. [Google Scholar] [CrossRef]
- El Shamieh, S.; Maltese, P.E. Editorial: The genetics of inherited retinal diseases in understudied ethnic groups: Novel associations, challenges, and perspectives. Front. Genet. 2022, 13, 990782. [Google Scholar]
- Lin, S.; Vermeirsch, S.; Pontikos, N.; Martin-Gutierrez, M.P.; Daich Varela, M.; Malka, S.; Schiff, E.; Knight, H.; Wright, G.; Jurkute, N.; et al. Spectrum of Genetic Variants in the Most Common Genes Causing Inherited Retinal Disease in a Large Molecularly Characterized United Kingdom Cohort. Ophthalmol. Retin. 2024, 8, 699–709. [Google Scholar] [CrossRef]
- Schlottmann, P.G.; Luna, J.D.; Labat, N.; Yadarola, M.B.; Bainttein, S.; Esposito, E.; Ibañez, A.; Barbaro, E.I.; Álvarez Mendiara, A.; Picotti, C.P.; et al. Nationwide genetic analysis of more than 600 families with inherited eye diseases in Argentina. NPJ Genom. Med. 2023, 8, 8. [Google Scholar] [CrossRef] [PubMed]
- Pontikos, N.; Arno, G.; Jurkute, N.; Schiff, E.; Ba-Abbad, R.; Malka, S.; Gimenez, A.; Georgiou, M.; Wright, G.; Armengol, M.; et al. Genetic Basis of Inherited Retinal Disease in a Molecularly Characterized Cohort of More than 3000 Families from the United Kingdom. Ophthalmology 2020, 127, 1384–1394. [Google Scholar] [CrossRef] [PubMed]
- Nuzbrokh, Y.; Ragi, S.D.; Tsang, S.H. Gene therapy for inherited retinal diseases. Ann. Transl. Med. 2021, 9, 1278. [Google Scholar] [CrossRef] [PubMed]
- Leroy, B.P.; Fischer, M.D.; Flannery, J.G.; MacLaren, R.E.; Dalkara, D.; Scholl, H.P.N.; Chung, D.C.; Spera, C.; Viriato, D.; Banhazi, J. Gene Therapy for Inherited Retinal Disease: Long-Term Durability of Effect. Ophthalmic Res. 2022, 66, 179–196. [Google Scholar] [CrossRef] [PubMed]
- Amato, A.; Arrigo, A.; Aragona, E.; Manitto, M.P.; Saladino, A.; Bandello, F.; Battaglia Parodi, M. Gene Therapy in Inherited Retinal Diseases: An Update on Current State of the Art. Front. Med. 2021, 8, 750586. [Google Scholar] [CrossRef] [PubMed]
- De Silva, S.R.; Moore, A.T. Optogenetic approaches to therapy for inherited retinal degenerations. J. Physiol. 2022, 600, 4623–4632. [Google Scholar] [CrossRef] [PubMed]
- McClements, M.E.; Staurenghi, F.; MacLaren, R.E.; Cehajic-Kapetanovic, J. Optogenetic Gene Therapy for the Degenerate Retina: Recent Advances. Front. Neurosci. 2020, 14, 570909. [Google Scholar] [CrossRef] [PubMed]
- Murro, V.; Banfi, S.; Testa, F.; Iarossi, G.; Falsini, B.; Sodi, A.; Signorini, S.; Iolascon, A.; Russo, R.; Mucciolo, D.P.; et al. A multidisciplinary approach to inherited retinal dystrophies from diagnosis to initial care: A narrative review with inputs from clinical practice. Orphanet J. Rare Dis. 2023, 18, 223. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, M.; Geng, Z.; Khattak, S.; Ji, X.; Wu, D.; Dang, Y. Role of Oxidative Stress in Retinal Disease and the Early Intervention Strategies: A Review. Oxid. Med. Cell Longev. 2022, 2022, 7836828. [Google Scholar] [CrossRef]
- Ren, X.; Léveillard, T. Modulating antioxidant systems as a therapeutic approach to retinal degeneration. Redox Biol. 2022, 57, 102510. [Google Scholar] [CrossRef]
- García-Ayuso, D.; Di Pierdomenico, J.; Vidal-Sanz, M.; Villegas-Pérez, M.P. Retinal Ganglion Cell Death as a Late Remodeling Effect of Photoreceptor Degeneration. Int. J. Mol. Sci. 2019, 20, 4649. [Google Scholar] [CrossRef] [PubMed]
- Komeima, K.; Rogers, B.S.; Campochiaro, P.A. Antioxidants slow photoreceptor cell death in mouse models of retinitis pigmentosa. J. Cell Physiol. 2007, 213, 809–815. [Google Scholar] [CrossRef] [PubMed]
- Komeima, K.; Rogers, B.S.; Lu, L.; Campochiaro, P.A. Antioxidants reduce cone cell death in a model of retinitis pigmentosa. Proc. Natl. Acad. Sci. USA 2006, 103, 11300–11305. [Google Scholar] [CrossRef] [PubMed]
- Joshi, J.; Rubart, M.; Zhu, W. Optogenetics: Background, Methodological Advances and Potential Applications for Cardiovascular Research and Medicine. Front. Bioeng. Biotechnol. 2019, 7, 466. [Google Scholar] [CrossRef] [PubMed]
- Grassmeyer, J.J.; Munakomi, S. Photopic Vision. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2024. [Google Scholar]
- Perkins, B.D.; Fadool, J.M. Photoreceptor structure and development analyses using GFP transgenes. Methods Cell Biol. 2010, 100, 205–218. [Google Scholar] [CrossRef] [PubMed]
- Pepe, I.M. Recent Advances in Our Understanding of Rhodopsin and Phototransduction. Prog. Retin. Eye Res. 2001, 20, 733–759. [Google Scholar] [CrossRef] [PubMed]
- Terakita, A. The opsins. Genome Biol. 2005, 6, 213. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Fang, H.; Liu, D.; Zhang, Y.; Adu-Amankwaah, J.; Yuan, J.; Tan, R.; Zhu, J. Applications and challenges of rhodopsin-based optogenetics in biomedicine. Front. Neurosci. 2022, 16, 966772. [Google Scholar] [CrossRef] [PubMed]
- Josselyn, S.A. The past, present and future of light-gated ion channels and optogenetics. elife 2018, 7, e42367. [Google Scholar] [CrossRef]
- Zhao, Z.; Fairchild, P.W. Dependence of Light Transmission through Human Skin on Incident Beam Diameter at Different Wavelengths; SPIE: Bellingham, WA, USA, 1998; Volume 3254, pp. 354–360. [Google Scholar]
- Ash, C.; Dubec, M.; Donne, K.; Bashford, T. Effect of wavelength and beam width on penetration in light-tissue interaction using computational methods. Lasers Med. Sci. 2017, 32, 1909–1918. [Google Scholar] [CrossRef]
- Chernov, K.G.; Redchuk, T.A.; Omelina, E.S.; Verkhusha, V.V. Near-infrared fluorescent proteins, biosensors, and optogenetic tools engineered from phytochromes. Chem. Rev. 2017, 117, 6423–6446. [Google Scholar] [CrossRef] [PubMed]
- Deisseroth, K.; Hegemann, P. The form and function of channelrhodopsin. Science 2017, 357, eaan5544. [Google Scholar] [CrossRef]
- Lagali, P.S.; Balya, D.; Awatramani, G.B.; Münch, T.A.; Kim, D.S.; Busskamp, V.; Cepko, C.L.; Roska, B. Light-activated channels targeted to ON bipolar cells restore visual function in retinal degeneration. Nat. Neurosci. 2008, 11, 667–675. [Google Scholar] [CrossRef]
- Bi, A.; Cui, J.; Ma, Y.-P.; Olshevskaya, E.; Pu, M.; Dizhoor, A.M.; Pan, Z.-H. Ectopic expression of a microbial-type rhodopsin restores visual responses in mice with photoreceptor degeneration. Neuron 2006, 50, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Rindner, D.J.; Lur, G. Practical considerations in an era of multicolor optogenetics. Front. Cell Neurosci. 2023, 17, 1160245. [Google Scholar] [CrossRef]
- Oesterhelt, D.; Stoeckenius, W. Rhodopsin-like protein from the purple membrane of Halobacterium halobium. Nat. New Biol. 1971, 233, 149–152. [Google Scholar] [CrossRef]
- Duebel, J.; Marazova, K.; Sahel, J.-A. Optogenetics. Curr. Opin. Ophthalmol. 2015, 26, 226–232. [Google Scholar] [CrossRef]
- Gradinaru, V.; Thompson, K.R.; Deisseroth, K. eNpHR: A Natronomonas halorhodopsin enhanced for optogenetic applications. Brain Cell Biol. 2008, 36, 129–139. [Google Scholar] [CrossRef]
- Gradinaru, V.; Mogri, M.; Thompson, K.R.; Henderson, J.M.; Deisseroth, K. Optical deconstruction of parkinsonian neural circuitry. Science 2009, 324, 354–359. [Google Scholar] [CrossRef]
- Yoshida, K.; Tsunoda, S.P.; Brown, L.S.; Kandori, H. A unique choanoflagellate enzyme rhodopsin exhibits light-dependent cyclic nucleotide phosphodiesterase activity. J. Biol. Chem. 2017, 292, 7531–7541. [Google Scholar] [CrossRef]
- Avelar, G.M.; Schumacher, R.I.; Zaini, P.A.; Leonard, G.; Richards, T.A.; Gomes, S.L. A rhodopsin-guanylyl cyclase gene fusion functions in visual perception in a fungus. Curr. Biol. 2014, 24, 1234–1240. [Google Scholar] [CrossRef] [PubMed]
- Luck, M.; Mathes, T.; Bruun, S.; Fudim, R.; Hagedorn, R.; Nguyen, T.M.T.; Kateriya, S.; Kennis, J.T.; Hildebrandt, P.; Hegemann, P. A photochromic histidine kinase rhodopsin (HKR1) that is bimodally switched by ultraviolet and blue light. J. Biol. Chem. 2012, 287, 40083–40090. [Google Scholar] [CrossRef] [PubMed]
- Sugiura, M.; Tsunoda, S.P.; Hibi, M.; Kandori, H. Molecular Properties of New Enzyme Rhodopsins with Phosphodiesterase Activity. ACS Omega 2020, 5, 10602–10609. [Google Scholar] [CrossRef] [PubMed]
- Vierock, J.; Rodriguez-Rozada, S.; Dieter, A.; Pieper, F.; Sims, R.; Tenedini, F.; Bergs, A.C.; Bendifallah, I.; Zhou, F.; Zeitzschel, N. BiPOLES is an optogenetic tool developed for bidirectional dual-color control of neurons. Nat. Commun. 2021, 12, 4527. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.Y.; Knutsen, P.M.; Muller, A.; Kleinfeld, D.; Tsien, R.Y. ReaChR: A red-shifted variant of channelrhodopsin enables deep transcranial optogenetic excitation. Nat. Neurosci. 2013, 16, 1499–1508. [Google Scholar] [CrossRef] [PubMed]
- Klapoetke, N.C.; Murata, Y.; Kim, S.S.; Pulver, S.R.; Birdsey-Benson, A.; Cho, Y.K.; Morimoto, T.K.; Chuong, A.S.; Carpenter, E.J.; Tian, Z.; et al. Independent optical excitation of distinct neural populations. Nat. Methods 2014, 11, 338–346. [Google Scholar] [CrossRef] [PubMed]
- Oda, K.; Vierock, J.; Oishi, S.; Rodriguez-Rozada, S.; Taniguchi, R.; Yamashita, K.; Wiegert, J.S.; Nishizawa, T.; Hegemann, P.; Nureki, O. Crystal structure of the red light-activated channelrhodopsin Chrimson. Nat. Commun. 2018, 9, 3949. [Google Scholar] [CrossRef] [PubMed]
- Marshel, J.H.; Kim, Y.S.; Machado, T.A.; Quirin, S.; Benson, B.; Kadmon, J.; Raja, C.; Chibukhchyan, A.; Ramakrishnan, C.; Inoue, M. Cortical layer–specific critical dynamics triggering perception. Science 2019, 365, eaaw5202. [Google Scholar] [CrossRef] [PubMed]
- Kishi, K.E.; Kim, Y.S.; Fukuda, M.; Inoue, M.; Kusakizako, T.; Wang, P.Y.; Ramakrishnan, C.; Byrne, E.F.; Thadhani, E.; Paggi, J.M. Structural basis for channel conduction in the pump-like channelrhodopsin ChRmine. Cell 2022, 185, 672–689. [Google Scholar] [CrossRef]
- Christoffel, D.J.; Walsh, J.J.; Heifets, B.D.; Hoerbelt, P.; Neuner, S.; Sun, G.; Ravikumar, V.K.; Wu, H.; Halpern, C.H.; Malenka, R.C. Input-specific modulation of murine nucleus accumbens differentially regulates hedonic feeding. Nat. Commun. 2021, 12, 2135. [Google Scholar] [CrossRef]
- Bauer, J.; Weiler, S.; Fernholz, M.H.; Laubender, D.; Scheuss, V.; Hübener, M.; Bonhoeffer, T.; Rose, T. Limited functional convergence of eye-specific inputs in the retinogeniculate pathway of the mouse. Neuron 2021, 109, 2457–2468. [Google Scholar] [CrossRef] [PubMed]
- Anisimova, M.; van Bommel, B.; Wang, R.; Mikhaylova, M.; Wiegert, J.S.; Oertner, T.G.; Gee, C.E. Spike-timing-dependent plasticity rewards synchrony rather than causality. Cereb. Cortex 2023, 33, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Hooks, B.M.; Lin, J.Y.; Guo, C.; Svoboda, K. Dual-channel circuit mapping reveals sensorimotor convergence in the primary motor cortex. J. Neurosci. 2015, 35, 4418–4426. [Google Scholar] [CrossRef] [PubMed]
- Rindner, D.J.; Proddutur, A.; Lur, G. Cell-type-specific integration of feedforward and feedback synaptic inputs in the posterior parietal cortex. Neuron 2022, 110, 3760–3773. [Google Scholar] [CrossRef] [PubMed]
- Joffe, M.E.; Maksymetz, J.; Luschinger, J.R.; Dogra, S.; Ferranti, A.S.; Luessen, D.J.; Gallinger, I.M.; Xiang, Z.; Branthwaite, H.; Melugin, P.R. Acute restraint stress redirects prefrontal cortex circuit function through mGlu5 receptor plasticity on somatostatin-expressing interneurons. Neuron 2022, 110, 1068–1083. [Google Scholar] [CrossRef] [PubMed]
- Xia, S.; Yu, J.; Huang, X.; Sesack, S.R.; Huang, Y.H.; Schlüter, O.M.; Cao, J.-L.; Dong, Y. Cortical and thalamic interaction with amygdala-to-accumbens synapses. J. Neurosci. 2020, 40, 7119–7132. [Google Scholar] [CrossRef] [PubMed]
- Prasad, A.A.; Xie, C.; Chaichim, C.; Nguyen, J.H.; McClusky, H.E.; Killcross, S.; Power, J.M.; McNally, G.P. Complementary roles for ventral pallidum cell types and their projections in relapse. J. Neurosci. 2020, 40, 880–893. [Google Scholar] [CrossRef]
- Birdsong, W.T.; Jongbloets, B.C.; Engeln, K.A.; Wang, D.; Scherrer, G.; Mao, T. Synapse-specific opioid modulation of thalamo-cortico-striatal circuits. elife 2019, 8, e45146. [Google Scholar] [CrossRef]
- Chiu, C.Q.; Martenson, J.S.; Yamazaki, M.; Natsume, R.; Sakimura, K.; Tomita, S.; Tavalin, S.J.; Higley, M.J. Input-specific NMDAR-dependent potentiation of dendritic GABAergic inhibition. Neuron 2018, 97, 368–377. [Google Scholar] [CrossRef]
- Husson, S.J.; Liewald, J.F.; Schultheis, C.; Stirman, J.N.; Lu, H.; Gottschalk, A. Microbial light-activatable proton pumps as neuronal inhibitors to functionally dissect neuronal networks in C. elegans. PLoS ONE 2012, 7, e40937. [Google Scholar] [CrossRef]
- Chow, B.Y.; Han, X.; Dobry, A.S.; Qian, X.; Chuong, A.S.; Li, M.; Henninger, M.A.; Belfort, G.M.; Lin, Y.; Monahan, P.E. High-performance genetically targetable optical neural silencing by light-driven proton pumps. Nature 2010, 463, 98–102. [Google Scholar] [CrossRef]
- Inoue, K.; Ono, H.; Abe-Yoshizumi, R.; Yoshizawa, S.; Ito, H.; Kogure, K.; Kandori, H. A light-driven sodium ion pump in marine bacteria. Nat. Commun. 2013, 4, 1678. [Google Scholar] [CrossRef]
- Hososhima, S.; Kandori, H.; Tsunoda, S.P. Ion transport activity and optogenetics capability of light-driven Na+-pump KR2. PLoS ONE 2021, 16, e0256728. [Google Scholar] [CrossRef]
- Zhang, F.; Wang, L.-P.; Brauner, M.; Liewald, J.F.; Kay, K.; Watzke, N.; Wood, P.G.; Bamberg, E.; Nagel, G.; Gottschalk, A. Multimodal fast optical interrogation of neural circuitry. Nature 2007, 446, 633–639. [Google Scholar] [CrossRef]
- Chen, Y.-H.; Wu, J.-L.; Hu, N.-Y.; Zhuang, J.-P.; Li, W.-P.; Zhang, S.-R.; Li, X.-W.; Yang, J.-M.; Gao, T.-M. Distinct projections from the infralimbic cortex exert opposing effects in modulating anxiety and fear. J. Clin. Investig. 2021, 131, e145692. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Boyden, E.S. Multiple-color optical activation, silencing, and desynchronization of neural activity, with single-spike temporal resolution. PLoS ONE 2007, 2, e299. [Google Scholar] [CrossRef] [PubMed]
- Busskamp, V.; Duebel, J.; Balya, D.; Fradot, M.; Viney, T.J.; Siegert, S.; Groner, A.C.; Cabuy, E.; Forster, V.; Seeliger, M.; et al. Genetic reactivation of cone photoreceptors restores visual responses in retinitis pigmentosa. Science 2010, 329, 413–417. [Google Scholar] [CrossRef]
- Joung, J.K.; Sander, J.D. TALENs: A widely applicable technology for targeted genome editing. Nat. Rev. Mol. Cell Biol. 2013, 14, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Chou, S.-T.; Leng, Q.; Mixson, A.J. Zinc Finger Nucleases: Tailor-made for Gene Therapy. Drugs Future 2012, 37, 183–196. [Google Scholar] [CrossRef]
- Pulman, J.; Sahel, J.-A.; Dalkara, D. New Editing Tools for Gene Therapy in Inherited Retinal Dystrophies. CRISPR J. 2022, 5, 377–388. [Google Scholar] [CrossRef]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A Programmable Dual-RNA–Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Jiang, F.; Doudna, J.A. CRISPR–Cas9 Structures and Mechanisms. Annu. Rev. Biophys. 2017, 46, 505–529. [Google Scholar] [CrossRef]
- Makarova, K.S.; Wolf, Y.I.; Iranzo, J.; Shmakov, S.A.; Alkhnbashi, O.S.; Brouns, S.J.J.; Charpentier, E.; Cheng, D.; Haft, D.H.; Horvath, P.; et al. Evolutionary classification of CRISPR–Cas systems: A burst of class 2 and derived variants. Nat. Rev. Microbiol. 2020, 18, 67–83. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B. CRISPR/Cas gene therapy. J. Cell. Physiol. 2021, 236, 2459–2481. [Google Scholar] [CrossRef] [PubMed]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [PubMed]
- Cox, D.B.T.; Platt, R.J.; Zhang, F. Therapeutic genome editing: Prospects and challenges. Nat. Med. 2015, 21, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Yan, A.L.; Du, S.W.; Palczewski, K. Genome editing, a superior therapy for inherited retinal diseases. Vis. Res. 2023, 206, 108192. [Google Scholar] [CrossRef] [PubMed]
- Du, W.; Li, J.; Tang, X.; Yu, W.; Zhao, M. CRISPR/SaCas9-based gene editing rescues photoreceptor degeneration throughout a rhodopsin-associated autosomal dominant retinitis pigmentosa mouse model. Exp. Biol. Med. 2023, 248, 1818–1828. [Google Scholar] [CrossRef]
- Suzuki, K.; Tsunekawa, Y.; Hernandez-Benitez, R.; Wu, J.; Zhu, J.; Kim, E.J.; Hatanaka, F.; Yamamoto, M.; Araoka, T.; Li, Z.; et al. In vivo genome editing via CRISPR/Cas9 mediated homology-independent targeted integration. Nature 2016, 540, 144–149. [Google Scholar] [CrossRef]
- Hu, S.; Du, J.; Chen, N.; Jia, R.; Zhang, J.; Liu, X.; Yang, L. In Vivo CRISPR/Cas9-Mediated Genome Editing Mitigates Photoreceptor Degeneration in a Mouse Model of X-Linked Retinitis Pigmentosa. Investig. Ophthalmol. Vis. Sci. 2020, 61, 31. [Google Scholar] [CrossRef]
- Da Costa, B.L.; Li, Y.; Levi, S.R.; Tsang, S.H.; Quinn, P.M.J. Generation of CRB1 RP Patient-Derived iPSCs and a CRISPR/Cas9-Mediated Homology-Directed Repair Strategy for the CRB1 c.2480G>T Mutation. In Retinal Degenerative Diseases XIX; Ash, J.D., Pierce, E., Anderson, R.E., Bowes Rickman, C., Hollyfield, J.G., Grimm, C., Eds.; Advances in Experimental Medicine and Biology; Springer International Publishing: Cham, Switzerland, 2023; Volume 1415, pp. 571–576. Available online: https://link.springer.com/10.1007/978-3-031-27681-1_83 (accessed on 15 May 2024).
- Burnight, E.R.; Wiley, L.A.; Mullin, N.K.; Adur, M.K.; Lang, M.J.; Cranston, C.M.; Jiao, C.; Russell, S.R.; Sohn, E.H.; Han, I.C.; et al. CRISPRi-Mediated Treatment of Dominant Rhodopsin-Associated Retinitis Pigmentosa. CRISPR J. 2023, 6, 502–513. [Google Scholar] [CrossRef]
- Böhm, S.; Splith, V.; Riedmayr, L.M.; Rötzer, R.D.; Gasparoni, G.; Nordström, K.J.V.; Wagner, J.E.; Hinrichsmeyer, K.S.; Walter, J.; Wahl-Schott, C.; et al. A gene therapy for inherited blindness using dCas9-VPR–mediated transcriptional activation. Sci. Adv. 2020, 6, eaba5614. [Google Scholar] [CrossRef]
- Riedmayr, L.M.; Hinrichsmeyer, K.S.; Karguth, N.; Böhm, S.; Splith, V.; Michalakis, S.; Becirovic, E. dCas9-VPR-mediated transcriptional activation of functionally equivalent genes for gene therapy. Nat. Protoc. 2022, 17, 781–818. [Google Scholar] [CrossRef]
- Gaudelli, N.M.; Komor, A.C.; Rees, H.A.; Packer, M.S.; Badran, A.H.; Bryson, D.I.; Liu, D.R. Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature 2017, 551, 464–471. [Google Scholar] [CrossRef]
- Komor, A.C.; Kim, Y.B.; Packer, M.S.; Zuris, J.A.; Liu, D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 2016, 533, 420–424. [Google Scholar] [CrossRef] [PubMed]
- Kurt, I.C.; Zhou, R.; Iyer, S.; Garcia, S.P.; Miller, B.R.; Langner, L.M.; Grünewald, J.; Joung, J.K. CRISPR C-to-G base editors for inducing targeted DNA transversions in human cells. Nat. Biotechnol. 2021, 39, 41–46. [Google Scholar] [CrossRef]
- Kabra, M.; Shahi, P.K.; Wang, Y.; Sinha, D.; Spillane, A.; Newby, G.A.; Saxena, S.; Tong, Y.; Chang, Y.; Abdeen, A.A.; et al. Nonviral base editing of KCNJ13 mutation preserves vision in a model of inherited retinal channelopathy. J. Clin. Investig. 2023, 133, e171356. [Google Scholar] [CrossRef]
- Costa, B.L.D.; Levi, S.R.; Eulau, E.; Tsai, Y.-T.; Quinn, P.M.J. Prime Editing for Inherited Retinal Diseases. Front. Genome Ed. 2021, 3, 775330. [Google Scholar] [CrossRef] [PubMed]
- Anzalone, A.V.; Randolph, P.B.; Davis, J.R.; Sousa, A.A.; Koblan, L.W.; Levy, J.M.; Chen, P.J.; Wilson, C.; Newby, G.A.; Raguram, A.; et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 2019, 576, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.; Jo, D.H.; Cho, C.S.; Shin, J.H.; Seo, J.H.; Yu, G.; Gopalappa, R.; Kim, D.; Cho, S.-R.; Kim, J.H.; et al. Application of prime editing to the correction of mutations and phenotypes in adult mice with liver and eye diseases. Nat. Biomed. Eng. 2021, 6, 181–194. [Google Scholar] [CrossRef]
- Mohan, K.; Dubey, S.K.; Jung, K.; Dubey, R.; Wang, Q.J.; Prajapati, S.; Roney, J.; Abney, J.; Kleinman, M.E. Long-Term Evaluation of Retinal Morphology and Function in Rosa26-Cas9 Knock-In Mice. Int. J. Mol. Sci. 2023, 24, 5186. [Google Scholar] [CrossRef]
- Gemayel, M.C.; Bhatwadekar, A.D.; Ciulla, T. RNA therapeutics for retinal diseases. Expert Opin. Biol. Ther. 2021, 21, 603–613. [Google Scholar] [CrossRef] [PubMed]
- Dana, H.; Chalbatani, G.M.; Mahmoodzadeh, H.; Karimloo, R.; Rezaiean, O.; Moradzadeh, A.; Mehmandoost, N.; Moazzen, F.; Mazraeh, A.; Marmari, V.; et al. Molecular Mechanisms and Biological Functions of siRNA. Int. J. Biomed. Sci. 2017, 13, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Carrella, S.; Di Guida, M.; Brillante, S.; Piccolo, D.; Ciampi, L.; Guadagnino, I.; Garcia Piqueras, J.; Pizzo, M.; Marrocco, E.; Molinari, M.; et al. miR-181a/b downregulation: A mutation-independent therapeutic approach for inherited retinal diseases. EMBO Mol. Med. 2022, 14, e15941. [Google Scholar] [CrossRef] [PubMed]
- Anasagasti, A.; Lara-López, A.; Milla-Navarro, S.; Escudero-Arrarás, L.; Rodríguez-Hidalgo, M.; Zabaleta, N.; González Aseguinolaza, G.; De La Villa, P.; Ruiz-Ederra, J. Inhibition of MicroRNA 6937 Delays Photoreceptor and Vision Loss in a Mouse Model of Retinitis Pigmentosa. Pharmaceutics 2020, 12, 913. [Google Scholar] [CrossRef] [PubMed]
- Carthew, R.W.; Sontheimer, E.J. Origins and Mechanisms of miRNAs and siRNAs. Cell 2009, 136, 642–655. [Google Scholar] [CrossRef]
- Hu, B.; Zhong, L.; Weng, Y.; Peng, L.; Huang, Y.; Zhao, Y.; Liang, X.-J. Therapeutic siRNA: State of the art. Signal Transduct. Target. Ther. 2020, 5, 101. [Google Scholar] [CrossRef]
- Cheng, S.-Y.; Caiazzi, J.; Biscans, A.; Alterman, J.F.; Echeverria, D.; McHugh, N.; Hassler, M.; Jolly, S.; Giguere, D.; Cipi, J.; et al. Single intravitreal administration of a tetravalent siRNA exhibits robust and efficient gene silencing in mouse and pig photoreceptors. Mol. Ther.-Nucleic Acids 2024, 35, 102088. [Google Scholar] [CrossRef] [PubMed]
- Kleinman, M.E.; Kaneko, H.; Cho, W.G.; Dridi, S.; Fowler, B.J.; Blandford, A.D.; Albuquerque, R.J.; Hirano, Y.; Terasaki, H.; Kondo, M.; et al. Short-interfering RNAs Induce Retinal Degeneration via TLR3 and IRF3. Mol. Ther. 2012, 20, 101–108. [Google Scholar] [CrossRef]
- Cideciyan, A.V.; Sudharsan, R.; Dufour, V.L.; Massengill, M.T.; Iwabe, S.; Swider, M.; Lisi, B.; Sumaroka, A.; Marinho, L.F.; Appelbaum, T.; et al. Mutation-independent rhodopsin gene therapy by knockdown and replacement with a single AAV vector. Proc. Natl. Acad. Sci. USA 2018, 115, E8547–E8556. [Google Scholar] [CrossRef]
- Fenner, B.J.; Tan, T.-E.; Barathi, A.V.; Tun, S.B.B.; Yeo, S.W.; Tsai, A.S.H.; Lee, S.Y.; Cheung, C.M.G.; Chan, C.M.; Mehta, J.S.; et al. Gene-Based Therapeutics for Inherited Retinal Diseases. Front. Genet. 2022, 12, 794805. [Google Scholar] [CrossRef]
- Kovacs, K.D.; Ciulla, T.A.; Kiss, S. Advancements in ocular gene therapy delivery: Vectors and subretinal, intravitreal, and suprachoroidal techniques. Expert. Opin. Biol. Ther. 2022, 22, 1193–1208. [Google Scholar] [CrossRef]
- Wu, Z.; Yang, H.; Colosi, P. Effect of genome size on AAV vector packaging. Mol. Ther. 2010, 18, 80–86. [Google Scholar] [CrossRef]
- Ebner, L.J.A.; Grimm, C. AAV Serotypes and Their Suitability for Retinal Gene Therapy. In Retinal Degenerative Diseases XIX; Ash, J.D., Pierce, E., Anderson, R.E., Bowes Rickman, C., Hollyfield, J.G., Grimm, C., Eds.; Advances in Experimental Medicine and Biology; Springer International Publishing: Cham, Switzerland, 2023; Volume 1415, pp. 131–134. Available online: https://link.springer.com/10.1007/978-3-031-27681-1_20 (accessed on 4 May 2024).
- Chien, Y.; Hsiao, Y.-J.; Chou, S.-J.; Lin, T.-Y.; Yarmishyn, A.A.; Lai, W.-Y.; Lee, M.-S.; Lin, Y.-Y.; Lin, T.-W.; Hwang, D.-K.; et al. Nanoparticles-mediated CRISPR-Cas9 gene therapy in inherited retinal diseases: Applications, challenges, and emerging opportunities. J. Nanobiotechnol. 2022, 20, 511. [Google Scholar] [CrossRef]
- Bordet, T.; Behar-Cohen, F. Ocular gene therapies in clinical practice: Viral vectors and nonviral alternatives. Drug Discov. Today 2019, 24, 1685–1693. [Google Scholar] [CrossRef]
- Lopes, V.S.; Williams, D.S. Gene Therapy for the Retinal Degeneration of Usher Syndrome Caused by Mutations in MYO7A. Cold Spring Harb. Perspect. Med. 2015, 5, a017319. [Google Scholar] [CrossRef]
- Al-Khuzaei, S.; Broadgate, S.; Foster, C.R.; Shah, M.; Yu, J.; Downes, S.M.; Halford, S. An Overview of the Genetics of ABCA4 Retinopathies, an Evolving Story. Genes 2021, 12, 1241. [Google Scholar] [CrossRef]
- Mellen, R.W.; Calabro, K.R.; McCullough, K.T.; Crosson, S.M.; Cova, A.D.L.; Fajardo, D.; Xu, E.; Boye, S.L.; Boye, S.E. Development of an AAV-CRISPR-Cas9-based treatment for dominant cone-rod dystrophy 6. Mol. Ther.-Methods Clin. Dev. 2023, 30, 48–64. [Google Scholar] [CrossRef]
- Bali, B.; Gruber-Dujardin, E.; Kusch, K.; Rankovic, V.; Moser, T. Analyzing efficacy, stability, and safety of AAV-mediated optogenetic hearing restoration in mice. Life Sci. Alliance 2022, 5, e202101338. [Google Scholar] [CrossRef]
- Mendoza, S.D.; El-Shamayleh, Y.; Horwitz, G.D. AAV-mediated delivery of optogenetic constructs to the macaque brain triggers humoral immune responses. J. Neurophysiol. 2017, 117, 2004–2013. [Google Scholar] [CrossRef]
- Zárate, R.V.; Arancibia, D.; Fernández, A.; Signorelli, J.R.; Larrondo, L.F.; Andrés, M.E.; Zamorano, P. Optimization of the Light-On system in a lentiviral platform to a light-controlled expression of genes in neurons. Electron. J. Biotechnol. 2021, 51, 50–57. [Google Scholar] [CrossRef]
- Arancibia, D.; Pol, I.; Vargas-Fernández, M.; Zárate, R.V.; Signorelli, J.R.; Zamorano, P. OPTO-BLUE: An Integrated Bidirectional Optogenetic Lentiviral Platform for Controlled Light-Induced Gene Expression. Int. J. Mol. Sci. 2023, 24, 9537. [Google Scholar] [CrossRef]
- Yi, Z.; All, A.H.; Liu, X. Upconversion Nanoparticle-Mediated Optogenetics. Adv. Exp. Med. Biol. 2021, 1293, 641–657. [Google Scholar] [CrossRef]
- Sun, D.; Sun, W.; Gao, S.-Q.; Lehrer, J.; Wang, H.; Hall, R.; Lu, Z.-R. Intravitreal Delivery of PEGylated-ECO Plasmid DNA Nanoparticles for Gene Therapy of Stargardt Disease. Pharm. Res. 2024, 41, 807–817. [Google Scholar] [CrossRef]
- Lin, Y.; Yao, Y.; Zhang, W.; Fang, Q.; Zhang, L.; Zhang, Y.; Xu, Y. Applications of upconversion nanoparticles in cellular optogenetics. Acta Biomater. 2021, 135, 1–12. [Google Scholar] [CrossRef]
- Chen, S.; Weitemier, A.Z.; Zeng, X.; He, L.; Wang, X.; Tao, Y.; Huang, A.J.Y.; Hashimotodani, Y.; Kano, M.; Iwasaki, H.; et al. Near-infrared deep brain stimulation via upconversion nanoparticle–mediated optogenetics. Science 2018, 359, 679–684. [Google Scholar] [CrossRef]
- Wu, C.; Su, B.; Xin, N.; Tang, J.; Xiao, J.; Luo, H.; Wei, D.; Luo, F.; Sun, J.; Fan, H. An upconversion nanoparticle-integrated fibrillar scaffold combined with a NIR-optogenetic strategy to regulate neural cell performance. J. Mater. Chem. B 2023, 11, 430–440. [Google Scholar] [CrossRef]
- Gu, L.; Shivalingaiah, S.; Ficinski, M.; Wong, E.; Mohanty, S. Non-viral delivery and optimized optogenetic stimulation of retinal ganglion cells led to behavioral restoration of vision. Nat. Preced. 2012. [Google Scholar] [CrossRef]
- Hsieh, F.-Y.; Han, H.-W.; Chen, X.-R.; Yang, C.-S.; Wei, Y.; Hsu, S. Non-viral delivery of an optogenetic tool into cells with self-healing hydrogel. Biomaterials 2018, 174, 31–40. [Google Scholar] [CrossRef]
- Johannsmeier, S.; Torres, M.; Ripken, T.; Heinemann, D.; Heisterkamp, A. Hydrogels for Efficient Light Delivery in Optogenetic Applications; SPIE: Bellingham, WA, USA, 2018; Volume 10482, pp. 27–35. [Google Scholar]
- Emiliani, V.; Entcheva, E.; Hedrich, R.; Hegemann, P.; Konrad, K.R.; Lüscher, C.; Mahn, M.; Pan, Z.-H.; Sims, R.R.; Vierock, J.; et al. Optogenetics for light control of biological systems. Nat. Rev. Methods Primers 2022, 2, 55. [Google Scholar] [CrossRef]
- Kwan, W.C.; Brunton, E.K.; Begeng, J.M.; Richardson, R.T.; Ibbotson, M.R.; Tong, W. Timing is Everything: Stochastic Optogenetic Stimulation Reduces Adaptation in Retinal Ganglion Cells. In Proceedings of the 2023 45th Annual International Conference of the IEEE Engineering in Medicine & Biology Society (EMBC), Sydney, Australia, 24–27 July 2023; IEEE: Sydney, Australia, 2023; pp. 1–4. Available online: https://ieeexplore.ieee.org/document/10340849/ (accessed on 30 May 2024).
- Hososhima, S.; Ueno, S.; Okado, S.; Inoue, K.; Konno, M.; Yamauchi, Y.; Inoue, K.; Terasaki, H.; Kandori, H.; Tsunoda, S.P. A light-gated cation channel with high reactivity to weak light. Sci. Rep. 2023, 13, 7625. [Google Scholar] [CrossRef]
- Gauvain, G.; Akolkar, H.; Chaffiol, A.; Arcizet, F.; Khoei, M.A.; Desrosiers, M.; Jaillard, C.; Caplette, R.; Marre, O.; Bertin, S.; et al. Optogenetic therapy: High spatiotemporal resolution and pattern discrimination compatible with vision restoration in non-human primates. Commun. Biol. 2021, 4, 125. [Google Scholar] [CrossRef]
- Yan, B.; Viswanathan, S.; Brodie, S.E.; Deng, W.-T.; Coleman, K.E.; Hauswirth, W.W.; Nirenberg, S. A clinically viable approach to restoring visual function using optogenetic gene therapy. Mol. Ther.-Methods Clin. Dev. 2023, 29, 406–417. [Google Scholar] [CrossRef]
- Ferrari, U.; Deny, S.; Sengupta, A.; Caplette, R.; Trapani, F.; Sahel, J.-A.; Dalkara, D.; Picaud, S.; Duebel, J.; Marre, O. Towards optogenetic vision restoration with high resolution. PLoS Comput. Biol. 2020, 16, e1007857. [Google Scholar] [CrossRef] [PubMed]
- McGregor, J.E.; Godat, T.; Dhakal, K.R.; Parkins, K.; Strazzeri, J.M.; Bateman, B.A.; Fischer, W.S.; Williams, D.R.; Merigan, W.H. Optogenetic restoration of retinal ganglion cell activity in the living primate. Nat. Commun. 2020, 11, 1703. [Google Scholar] [CrossRef] [PubMed]
- Ganjawala, T.H.; Lu, Q.; Fenner, M.D.; Abrams, G.W.; Pan, Z.-H. Improved CoChR Variants Restore Visual Acuity and Contrast Sensitivity in a Mouse Model of Blindness under Ambient Light Conditions. Mol. Ther. 2019, 27, 1195–1205. [Google Scholar] [CrossRef]
- Watanabe, Y.; Sugano, E.; Tabata, K.; Hatakeyama, A.; Sakajiri, T.; Fukuda, T.; Ozaki, T.; Suzuki, T.; Sayama, T.; Tomita, H. Development of an optogenetic gene sensitive to daylight and its implications in vision restoration. NPJ Regen. Med. 2021, 6, 64. [Google Scholar] [CrossRef] [PubMed]
- Berry, M.H.; Holt, A.; Salari, A.; Veit, J.; Visel, M.; Levitz, J.; Aghi, K.; Gaub, B.M.; Sivyer, B.; Flannery, J.G.; et al. Restoration of high-sensitivity and adapting vision with a cone opsin. Nat. Commun. 2019, 10, 1221. [Google Scholar] [CrossRef]
- Batabyal, S.; Gajjeraman, S.; Pradhan, S.; Bhattacharya, S.; Wright, W.; Mohanty, S. Sensitization of ON-bipolar cells with ambient light activatable multi-characteristic opsin rescues vision in mice. Gene Ther. 2021, 28, 162–176. [Google Scholar] [CrossRef]
- Kralik, J.; Van Wyk, M.; Stocker, N.; Kleinlogel, S. Bipolar cell targeted optogenetic gene therapy restores parallel retinal signaling and high-level vision in the degenerated retina. Commun. Biol. 2022, 5, 1116. [Google Scholar] [CrossRef]
- Gaub, B.M.; Berry, M.H.; Holt, A.E.; Isacoff, E.Y.; Flannery, J.G. Optogenetic Vision Restoration Using Rhodopsin for Enhanced Sensitivity. Mol. Ther. 2015, 23, 1562–1571. [Google Scholar] [CrossRef]
- Lu, Q.; Ganjawala, T.H.; Krstevski, A.; Abrams, G.W.; Pan, Z.-H. Comparison of AAV-Mediated Optogenetic Vision Restoration between Retinal Ganglion Cell Expression and ON Bipolar Cell Targeting. Mol. Ther.-Methods Clin. Dev. 2020, 18, 15–23. [Google Scholar] [CrossRef]
- Katada, Y.; Yoshida, K.; Serizawa, N.; Lee, D.; Kobayashi, K.; Negishi, K.; Okano, H.; Kandori, H.; Tsubota, K.; Kurihara, T. Highly sensitive visual restoration and protection via ectopic expression of chimeric rhodopsin in mice. iScience 2023, 26, 107716. [Google Scholar] [CrossRef]
- Idzhilova, O.S.; Kolotova, D.E.; Smirnova, G.R.; Abonakour, A.; Dolgikh, D.A.; Petrovskaya, L.E.; Kirpichnikov, M.P.; Ostrovsky, M.A.; Malyshev, A.Y. Nonselective Expression of Short-Wavelength Cone Opsin Improves Learning in Mice with Retinal Degeneration in a Visually Guided Task. Dokl. Biol. Sci. 2023, 510, 167–171. [Google Scholar] [CrossRef]
- Nikonov, S.; Aravand, P.; Lyubarsky, A.; Nikonov, R.; Luo, A.J.; Wei, Z.; Maguire, A.M.; Phelps, N.T.; Shpylchak, I.; Willett, K.; et al. Restoration of Vision and Retinal Responses After Adeno-Associated Virus–Mediated Optogenetic Therapy in Blind Dogs. Trans. Vis. Sci. Tech. 2022, 11, 24. [Google Scholar] [CrossRef]
- Khabou, H.; Garita-Hernandez, M.; Chaffiol, A.; Reichman, S.; Jaillard, C.; Brazhnikova, E.; Bertin, S.; Forster, V.; Desrosiers, M.; Winckler, C.; et al. Noninvasive gene delivery to foveal cones for vision restoration. JCI Insight 2018, 3, e96029. [Google Scholar] [CrossRef]
- Chaffiol, A.; Caplette, R.; Jaillard, C.; Brazhnikova, E.; Desrosiers, M.; Dubus, E.; Duhamel, L.; Macé, E.; Marre, O.; Benoit, P.; et al. A New Promoter Allows Optogenetic Vision Restoration with Enhanced Sensitivity in Macaque Retina. Mol. Ther. 2017, 25, 2546–2560. [Google Scholar] [CrossRef]
- Simon, C.-J.; Sahel, J.-A.; Duebel, J.; Herlitze, S.; Dalkara, D. Opsins for vision restoration. Biochem. Biophys. Res. Commun. 2020, 527, 325–330. [Google Scholar] [CrossRef]
- Gilhooley, M.J.; Lindner, M.; Palumaa, T.; Hughes, S.; Peirson, S.N.; Hankins, M.W. A systematic comparison of optogenetic approaches to visual restoration. Mol. Ther.-Methods Clin. Dev. 2022, 25, 111–123. [Google Scholar] [CrossRef]
- Wright, W.W.; Gajjeraman, S.; Batabyal, S.; Pradhan, S.; Bhattacharya, S.; Mahapatra, V.; Tripathy, A.; Mohanty, S.K. Restoring vision in mice with retinal degeneration using multicharacteristic opsin. Neurophoton. 2017, 4, 041505. [Google Scholar] [CrossRef]
- Gaub, B.M.; Berry, M.H.; Visel, M.; Holt, A.; Isacoff, E.Y.; Flannery, J.G. Optogenetic Retinal Gene Therapy with the Light Gated GPCR Vertebrate Rhodopsin. In Retinal Gene Therapy; Boon, C.J.F., Wijnholds, J., Eds.; Methods in Molecular Biology; Springer: New York, NY, USA, 2018; Volume 1715, pp. 177–189. Available online: http://link.springer.com/10.1007/978-1-4939-7522-8_12 (accessed on 12 May 2024).
- Cehajic-Kapetanovic, J.; Eleftheriou, C.; Allen, A.E.; Milosavljevic, N.; Pienaar, A.; Bedford, R.; Davis, K.E.; Bishop, P.N.; Lucas, R.J. Restoration of Vision with Ectopic Expression of Human Rod Opsin. Curr. Biol. 2015, 25, 2111–2122. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, V.H.; Lam, B.L.; Zak, V.; Mohanty, S.; Bataybal, S.; Chang, J.; Ayyagari, A.; Chavala, S.H.; Piltz-Seymour, J.; Koester, J.; et al. MCO-010 intravitreal optogenetic therapy in Stargardt disease. 6-month outcomes from the Phase 2 STARLIGHT trial. Investig. Ophthalmol. Vis. Sci. 2023, 64, 3546. [Google Scholar]
- Nanoscope Therapeutics Announces Positive Top-Line Results from Randomized Controlled Trial of MCO-010 for Retinitis Pigmentosa. 2024. Available online: https://nanostherapeutics.com/2024/03/26/nanoscope-therapeutics-announces-top-line-results-from-ph2-trial-of-mco-010-for-retinitis-pigmentosa/ (accessed on 5 May 2024).
- PackGene Biotech lnc. Zhongmou Therapeutics Unveils Promising Clinical Data for Innovative Retinitis Pigmentosa Gene Therapy. 2024. Available online: https://www.packgene.com/frontier/240401/ (accessed on 17 June 2024).
- Eramian, D. Nanoscope Therapeutics, Inc. Positive Data from Nanoscope’s Phase 1/2a Trial of Gene Therapy to Restore Vision in Patients Blinded by Retinitis Pigmentosa to Be Featured at 2021. 2021. Available online: https://nanostherapeutics.com/2021/10/08/positive-data-from-nanoscopes-phase-1-2a-trial-of-gene-therapy-to-restore-vision-in-patients-blinded-by-retinitis-pigmentosa-to-be-featured-at-2021-american-society-of-retina-specialists-meet/ (accessed on 17 June 2024).
- Sahel, J.-A.; Boulanger-Scemama, E.; Pagot, C.; Arleo, A.; Galluppi, F.; Martel, J.N.; Esposti, S.D.; Delaux, A.; De Saint Aubert, J.-B.; De Montleau, C.; et al. Partial recovery of visual function in a blind patient after optogenetic therapy. Nat. Med. 2021, 27, 1223–1229. [Google Scholar] [CrossRef] [PubMed]
- Dhurandhar, D.; Sahoo, N.; Mariappan, I.; Narayanan, R. Gene therapy in retinal diseases: A review. Indian J. Ophthalmol. 2021, 69, 2257. [Google Scholar] [CrossRef] [PubMed]
- Su, J.; She, K.; Song, L.; Jin, X.; Li, R.; Zhao, Q.; Xiao, J.; Chen, D.; Cheng, H.; Lu, F.; et al. In vivo base editing rescues photoreceptors in a mouse model of retinitis pigmentosa. Mol. Ther.-Nucleic Acids 2023, 31, 596–609. [Google Scholar] [CrossRef] [PubMed]
- Qin, H.; Zhang, W.; Zhang, S.; Feng, Y.; Xu, W.; Qi, J.; Zhang, Q.; Xu, C.; Liu, S.; Zhang, J.; et al. Vision rescue via unconstrained in vivo prime editing in degenerating neural retinas. J. Exp. Med. 2023, 220, e20220776. [Google Scholar] [CrossRef] [PubMed]
- Vagni, P.; Perlini, L.E.; Chenais, N.A.L.; Marchetti, T.; Parrini, M.; Contestabile, A.; Cancedda, L.; Ghezzi, D. Gene Editing Preserves Visual Functions in a Mouse Model of Retinal Degeneration. Front. Neurosci. 2019, 13, 945. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Qiao, J.; Jia, R.; Zhang, F.; Meng, X.; Li, Y.; Yang, L. Allele-specific gene-editing approach for vision loss restoration in RHO-associated retinitis pigmentosa. eLife 2023, 12, e84065. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.-H.; Tsai, Y.-T.; Huang, I.-W.; Cheng, C.-H.; Hsu, C.-W.; Cui, X.; Ryu, J.; Quinn, P.M.J.; Caruso, S.M.; Lin, C.-S.; et al. CRISPR genome surgery in a novel humanized model for autosomal dominant retinitis pigmentosa. Mol. Ther. 2022, 30, 1407–1420. [Google Scholar] [CrossRef]
- Orlans, H.O.; McClements, M.E.; Barnard, A.R.; Martinez-Fernandez De La Camara, C.; MacLaren, R.E. Mirtron-mediated RNA knockdown/replacement therapy for the treatment of dominant retinitis pigmentosa. Nat. Commun. 2021, 12, 4934. [Google Scholar] [CrossRef]
- Tornabene, P.; Ferla, R.; Llado-Santaeularia, M.; Centrulo, M.; Dell’Anno, M.; Esposito, F.; Marrocco, E.; Pone, E.; Minopoli, R.; Iodice, C.; et al. Therapeutic homology-independent targeted integration in retina and liver. Nat. Commun. 2022, 13, 1963. [Google Scholar] [CrossRef] [PubMed]
- Cui, T.; Cai, B.; Tian, Y.; Liu, X.; Liang, C.; Gao, Q.; Li, B.; Ding, Y.; Li, R.; Zhou, Q.; et al. Therapeutic In Vivo Gene Editing Achieved by a Hypercompact CRISPR-Cas12f1 System Delivered with All-in-One Adeno-Associated Virus. Adv. Sci. 2024, 11, e2308095. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Mookherjee, S.; Chaitankar, V.; Hiriyanna, S.; Kim, J.-W.; Brooks, M.; Ataeijannati, Y.; Sun, X.; Dong, L.; Li, T.; et al. Nrl knockdown by AAV-delivered CRISPR/Cas9 prevents retinal degeneration in mice. Nat. Commun. 2017, 8, 14716. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Ming, C.; Fu, X.; Duan, Y.; Hoang, D.A.; Rutgard, J.; Zhang, R.; Wang, W.; Hou, R.; Zhang, D.; et al. Gene and mutation independent therapy via CRISPR-Cas9 mediated cellular reprogramming in rod photoreceptors. Cell Res. 2017, 27, 830–833. [Google Scholar] [CrossRef] [PubMed]
- Nolan, N.D.; Cui, X.; Robbings, B.M.; Demirkol, A.; Pandey, K.; Wu, W.H.; Hu, H.F.; Jenny, L.A.; Lin, C.-S.; Hass, D.T.; et al. CRISPR editing of anti-anemia drug target rescues independent preclinical models of retinitis pigmentosa. Cell Rep. Med. 2024, 5, 101459. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.-K.; Jo, D.H.; Lee, S.-N.; Cho, C.S.; Jeong, Y.K.; Jung, Y.; Yu, J.; Kim, J.H.; Woo, J.-S.; Bae, S. High-purity production and precise editing of DNA base editing ribonucleoproteins. Sci. Adv. 2021, 7, eabg2661. [Google Scholar] [CrossRef] [PubMed]
- She, K.; Liu, Y.; Zhao, Q.; Jin, X.; Yang, Y.; Su, J.; Li, R.; Song, L.; Xiao, J.; Yao, S.; et al. Dual-AAV split prime editor corrects the mutation and phenotype in mice with inherited retinal degeneration. Signal Transduct. Target. Ther. 2023, 8, 57. [Google Scholar] [CrossRef] [PubMed]
- Banskota, S.; Raguram, A.; Suh, S.; Du, S.W.; Davis, J.R.; Choi, E.H.; Wang, X.; Nielsen, S.C.; Newby, G.A.; Randolph, P.B.; et al. Engineered virus-like particles for efficient in vivo delivery of therapeutic proteins. Cell 2022, 185, 250–265. [Google Scholar] [CrossRef] [PubMed]
- Choi, E.H.; Suh, S.; Foik, A.T.; Leinonen, H.; Newby, G.A.; Gao, X.D.; Banskota, S.; Hoang, T.; Du, S.W.; Dong, Z.; et al. In vivo base editing rescues cone photoreceptors in a mouse model of early-onset inherited retinal degeneration. Nat. Commun. 2022, 13, 1830. [Google Scholar] [CrossRef] [PubMed]
- Jo, D.H.; Jang, H.-K.; Cho, C.S.; Han, J.H.; Ryu, G.; Jung, Y.; Bae, S.; Kim, J.H. Visual function restoration in a mouse model of Leber congenital amaurosis via therapeutic base editing. Mol. Ther.-Nucleic Acids 2023, 31, 16–27. [Google Scholar] [CrossRef]
- Suh, S.; Choi, E.H.; Leinonen, H.; Foik, A.T.; Newby, G.A.; Yeh, W.-H.; Dong, Z.; Kiser, P.D.; Lyon, D.C.; Liu, D.R.; et al. Restoration of visual function in adult mice with an inherited retinal disease via adenine base editing. Nat. Biomed. Eng. 2020, 5, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Maeder, M.L.; Stefanidakis, M.; Wilson, C.J.; Baral, R.; Barrera, L.A.; Bounoutas, G.S.; Bumcrot, D.; Chao, H.; Ciulla, D.M.; DaSilva, J.A.; et al. Development of a gene-editing approach to restore vision loss in Leber congenital amaurosis type 10. Nat. Med. 2019, 25, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Riedmayr, L.M.; Hinrichsmeyer, K.S.; Thalhammer, S.B.; Mittas, D.M.; Karguth, N.; Otify, D.Y.; Böhm, S.; Weber, V.J.; Bartoschek, M.D.; Splith, V.; et al. mRNA trans-splicing dual AAV vectors for (epi)genome editing and gene therapy. Nat. Commun. 2023, 14, 6578. [Google Scholar] [CrossRef] [PubMed]
- McClements, M.E.; Barnard, A.R.; Singh, M.S.; Charbel Issa, P.; Jiang, Z.; Radu, R.A.; MacLaren, R.E. An AAV Dual Vector Strategy Ameliorates the Stargardt Phenotype in Adult Abca4−/− Mice. Hum. Gene Ther. 2019, 30, 590–600. [Google Scholar] [CrossRef] [PubMed]
- Chou, S.; Yang, P.; Ban, Q.; Yang, Y.; Wang, M.; Chien, C.; Chen, S.; Sun, N.; Zhu, Y.; Liu, H.; et al. Dual Supramolecular Nanoparticle Vectors Enable CRISPR/Cas9-Mediated Knockin of Retinoschisin 1 Gene—A Potential Nonviral Therapeutic Solution for X-Linked Juvenile Retinoschisis. Adv. Sci. 2020, 7, 1903432. [Google Scholar] [CrossRef] [PubMed]
- Chang, B.; Hawes, N.L.; Hurd, R.E.; Davisson, M.T.; Nusinowitz, S.; Heckenlively, J.R. Retinal degeneration mutants in the mouse. Vis. Res. 2002, 42, 517–525. [Google Scholar] [CrossRef] [PubMed]
- Cideciyan, A.V. Leber congenital amaurosis due to RPE65 mutations and its treatment with gene therapy. Prog. Retin. Eye Res. 2010, 29, 398–427. [Google Scholar] [CrossRef] [PubMed]
- Wimmer, T.; Sawinski, H.; Urban, A.M.; Motlik, J.; Stieger, K. Rapid and Reliable Quantification of Prime Editing Targeting within the Porcine ABCA4 Gene Using a BRET-Based Sensor. Nucleic Acid. Ther. 2023, 33, 226–232. [Google Scholar] [CrossRef] [PubMed]
- Regent, F.; Chen, H.Y.; Kelley, R.A.; Qu, Z.; Swaroop, A.; Li, T. A simple and efficient method for generating human retinal organoids. Mol. Vis. 2020, 26, 97–105. [Google Scholar]
- Chichagova, V.; Hilgen, G.; Ghareeb, A.; Georgiou, M.; Carter, M.; Sernagor, E.; Lako, M.; Armstrong, L. Human iPSC differentiation to retinal organoids in response to IGF1 and BMP4 activation is line- and method-dependent. Stem Cells 2020, 38, 195–201. [Google Scholar] [CrossRef]
- Daich Varela, M.; Sen, S.; De Guimaraes, T.A.C.; Kabiri, N.; Pontikos, N.; Balaskas, K.; Michaelides, M. Artificial intelligence in retinal disease: Clinical application, challenges, and future directions. Graefes Arch. Clin. Exp. Ophthalmol. 2023, 261, 3283–3297. [Google Scholar] [CrossRef] [PubMed]
- Sulak, R.; Liu, X.; Smedowski, A. The concept of gene therapy for glaucoma: The dream that has not come true yet. Neural Regen. Res. 2024, 19, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Prosseda, P.P.; Alvarado, J.A.; Wang, B.; Kowal, T.J.; Ning, K.; Stamer, W.D.; Hu, Y.; Sun, Y. Optogenetic stimulation of phosphoinositides reveals a critical role of primary cilia in eye pressure regulation. Sci. Adv. 2020, 6, eaay8699. [Google Scholar] [CrossRef] [PubMed]
- Kowal, T.J.; Prosseda, P.P.; Ning, K.; Wang, B.; Alvarado, J.; Sendayen, B.E.; Jabbehdari, S.; Stamer, W.D.; Hu, Y.; Sun, Y. Optogenetic modulation of intraocular pressure in a glucocorticoid-induced ocular hypertension mouse model. Transl. Vis. Sci. Technol. 2021, 10, 10. [Google Scholar] [CrossRef] [PubMed]
- Ballios, B.G.; Pierce, E.A.; Huckfeldt, R.M. Gene editing technology: Towards precision medicine in inherited retinal diseases. Semin. Ophthalmol. 2021, 36, 176–184. [Google Scholar] [CrossRef]
- Koulisis, N.; Nagiel, A. Precision Therapy for Inherited Retinal Disease. Clin. Lab. Med. 2020, 40, 189–204. [Google Scholar] [CrossRef]

| Characteristic | miRNA | siRNA | shRNA | ASOs | References |
|---|---|---|---|---|---|
| Nucleotide characteristics | 20–25 bp, double-stranded RNA | 21–23 bp, double-stranded RNA | 19–22 bp, double-stranded RNA with a 4–11 bp hairpin loop | 15–30 bp, single stranded DNA or RNA | [95,96] |
| Intracellular processing | Cleaved by nuclear and cytoplasmic RNase | Cleaved by cytoplasmic RNase | Expression from cDNA | No intracellular processing | [95] |
| Complex formed | form RISC complexes | Form RISC complexes | Form RISC complexes | Bind mRNA directly | [95] |
| Effect on mRNA | Interfere with translation and activate degradation | Interfere with translation and activate degradation | Interfere with translation or activate degradation | Interfere with translation, modify splicing, or activate degradation | [95] |
| Delivery method | Naked miRNA or vectors | Naked siRNA or vectors | Exogenous DNA expression vectors | Naked ASOs or vectors | [97,98] |
| Vector Type | Advantages | Disadvantages | References |
|---|---|---|---|
| Adenovirus |
|
| [105,106] |
| Adeno-associated viruses |
|
| [105,107,108] |
| Lentiviruses |
|
| [105,109] |
| Nanoparticles |
|
| [109] |
| Opsin Source | Optogenetic Tools | Vectors (Delivery Route) | Models | Results | References |
|---|---|---|---|---|---|
| Targeting retinal ganglion cells | |||||
| Microbial | ChR2- H134R | Heterozygous transgenic mice | Reduction of adaptation in RGCs and increased mean firing rate. | [126] | |
| GtCCR4 | AAV7m8 | rd1 mice | GtCCR4 expression in abnormal RGCs of rd1 mice restored light sensitivity. | [127] | |
| ChR-tdT | AAV2.7m8 (IV) | Macaques | Visual acuity restored to estimated 20/249 based on MEA recordings. | [128] | |
| Chronos | AAV2 (IV) | rd1 mice, S334ter-3 rats (IHC safety) Cynomolgus macaques (safety) | Dose-dependent ERG response. Effective and well-tolerated across various doses and light intensities. | [129] | |
| ReaChR CatCh (ChR2-L132C) | AAV2 (IV) | rd1 mice Macaques | Visual acuity restored at estimated 20/72. | [130] | |
| ChrimsonR | AAV2 (IV) | Macaques | Optogenetic responses remained after 1 year post transfection. | [131] | |
| CoChR-LC CoChR-3M | AV2 (IV) AAV2.7m8 (IV) | Opn4−/− Gnat1−/− Cnga3−/− mice | Contrast sensitivity and visual acuity restored in ambient light and maintained 1 year post-injection. | [132] | |
| Animal | mVChR1/ChR2/CoChR chimera (ex3mV1) | AAV2 (IV) | RCS rats | VEP recorded up to 17 months post-injection. | [133] |
| MW-opsin | AAV2/2-4YF (IV) | rd1 mice | Restoration of key aspect of natural vision with quicker response kinetics. Significant increase in VEP amplitude. | [134] | |
| Targeting bipolar cells | |||||
| Microbial | ChCR2 mutant + Chrimson (MCO1) | AAV2 (IV) | rd10 mice | Vision restored with dual-wavelength ChRs in ambient light. Improvement in visually guided behaviors. | [135] |
| Animal | MW-opsin melanopsin | AAV2.7m8 | rd1 mice | Significant increase in sensitivity than microbial alternatives. Adapted wide range of light intensities. | [136] |
| Rhodopsin | AAV2.4YF (IV) AAV8.BP2 (IV) AAV2.7m8 (IV) | rd1 mice | AAV8.BP2 and AAV2.7m8 showed greater transduction. Consistent responses with WT-like amplitude. Restored innate light avoidance. | [137] | |
| Combined targeting of retinal ganglion cells and bipolar cells | |||||
| Microbial | CoChR-L112C | AAV2 (IV) | Opn4−/− Gnat1−/− Cnga3−/− mice | At equivalent light intensity, the expression in RGCs yields higher visual acuity than ON BC. | [138] |
| Non-selective targeting | |||||
| Animal | coGHCR | AAV2 (IV) | rd1 mice RHOP23 mice | Restored light sensitivity and object recognition in low-light environments. Protective effects against retinal degeneration. | [139] |
| SWOpsin | AAV2 | rd1_KO mice | Significantly higher correct decision rate in treated mice. | [140] | |
| Targeting photoreceptors | |||||
| Animal | eNpHR (enhanced halorhodopsin cDNA) | AAV9 (SR) | rcd1 dogs (PDE6 ß-mutant) | Partial vision restoration when targeting outer retina on larger animals. | [141] |
| Microbial | eNpHR | AAV2.1 | rd1 mice Rho−/− Cnga3−/− mice | ON, OFF, and ON/OFF light responses observed at the RGC level. Restored optomotor reflexes and light avoidance. | [69] |
| Red-shifted cruxhalorhodopsin | AAV2.7m8 (IV) AAV9.7m8 (SR) | Macaques | Efficient noninvasive foveal targeting permitting robust light responses. | [142] | |
| NCT Number (Start-End Year) | Study Phase | Target | Optogenetic Tool | Results/Notes | References |
|---|---|---|---|---|---|
| NCT05417126 (2022–2023) | Phase IIa | ON BCs Cones | vMCO-010 (ChR2 mutant + Chrimson) | Clinical meaningful improvements in best-corrected visual acuity. No serious adverse events. | [149] |
| NCT04945772 (2021–2024) | Phase IIb | ON BCs Cones | vMCO-010 (ChR2 mutant + Chrimson) | MCO-010 met primary and key secondary endpoints. No serious adverse events. | [150] |
| NCT05294978 (2021–2024) | N/A | ON BCs Cones | N/A | No results reported. OCT test to estimate IRD patients with remaining cone photoreceptors. | |
| NCT06292650 (2020–2029) | N/A | RGCs | CatCh (ChR2-L132C) | Improved functional visual abilities, light sensitivity, and overall visual performance in various lighting conditions. | [151] |
| NCT04919473 (2019–2020) | Phase I/IIa | ON BCs | vMCO-010 (ChR2 mutant + Chrimson) | Significant vision improvement in 11 patients. Treatment well tolerated. | [152] |
| NCT03326336 (2018–2025) | Phase I/IIa | RGCs | ChrimsonR | Partial recovery of visual function in a blind patient using light-stimulating goggles. | [153] |
| NCT04278131 (2018–2025) | Phase I/II | RGCs | Chronos | Dose-dependent significant vision improvement in all 12 patients. | [129] |
| IRD | Inheritance Mode | Genes | Animal Models | Tools | Vector | Preclinical Phase Studies | References |
|---|---|---|---|---|---|---|---|
| Retinitis pigmentosa | Autosomal recessive | Pde6b | Rd10 mice | ABEs | AAV | In vivo base editing rescues photoreceptors in a mouse model of retinitis pigmentosa | [155] |
| PEs | AAV | Vision rescue via unconstrained in vivo prime editing in degenerating neural retinas | [156] | ||||
| HDR | Gene Editing Preserves Visual Functions in a Mouse Model of Retinal Degeneration | [157] | |||||
| Mertk | RCS rat | HITI | AAV8 | In vivo genome editing via CRISPR/Cas9 mediated homology-independent targeted integration | [81] | ||
| Autosomal dominant | RHO-T17M | Rhowt/hu mice | BE | AAV2/8 | Allele-specific gene-editing approach for vision loss restoration in RHO-associated retinitis pigmentosa | [158] | |
| RHO | hRHOC110R/hRHOWT | AR | AAV2/8 | CRISPR genome surgery in a novel humanized model for autosomal dominant retinitis pigmentosa | [159] | ||
| RhoP23H/P23H | AR | Dual AAV9 | CRISPR/SaCas9-based gene editing rescues photoreceptor degeneration throughout a rhodopsin-associated autosomal dominant retinitis pigmentosa mouse model | [80] | |||
| RhoP23H/+ mice | mirtron | AAV | Mirtron-mediated RNA knockdown/replacement therapy for the treatment of dominant retinitis pigmentosa | [160] | |||
| Pro23His Pig | KRAB | AAV | CRISPRi-Mediated Treatment of Dominant Rhodopsin-Associated Retinitis Pigmentosa | [84] | |||
| RhoP23H/+ mice | HITI | AAV8 | Therapeutic homology-independent targeted integration in retina and liver | [161] | |||
| Nr2e3 | RhoP23H/+ mice | CasMINI | AAV8 | Therapeutic In Vivo Gene Editing Achieved by a Hypercompact CRISPR-Cas12f1 System Delivered with All-in-One Adeno-Associated Virus | [162] | ||
| Nrl | Rd10, Rho−/−, RHO-P347S mice | AAV8 | Nrl knockdown by AAV-delivered CRISPR/Cas9 prevents retinal degeneration in mice | [163] | |||
| Rd10 | AAV | Gene and mutation independent therapy via CRISPR-Cas9 mediated cellular reprogramming in rod photoreceptors | [164] | ||||
| Dominant and recessive | PHD2/Egln1 | RhoC110R/+ mice (adRP) Pde6bH620Q/H620Q mice (arRP) | AAV8 | CRISPR editing of anti-anemia drug target rescues independent preclinical models of retinitis pigmentosa | [165] | ||
| X-linked | Rpgr | Rpgr KO mice | HDR | AAV2/8 | In Vivo CRISPR/Cas9-Mediated Genome Editing Mitigates Photoreceptor Degeneration in a Mouse Model of X-Linked Retinitis Pigmentosa | [82] | |
| Leber congenital amaurosis | Autosomal recessive | Rpe65 (LCA2) | Rd12 mice | PE3 | AAVs | Application of prime editing to the correction of mutations and phenotypes in adult mice with liver and eye diseases | [93] |
| ABE-RNP | LV | High-purity production and precise editing of DNA base editing ribonucleoproteins | [166] | ||||
| PE2 | AAV | Dual-AAV split prime editor corrects the mutation and phenotype in mice with inherited retinal degeneration | [167] | ||||
| ABEs | eVLPs | Engineered virus-like particles for efficient in vivo delivery of therapeutic proteins | [168] | ||||
| NG-ABE | LV | In vivo base editing rescues cone photoreceptors in a mouse model of early-onset inherited retinal degeneration | [169] | ||||
| ABEs | AAV | Visual function restoration in a mouse model of Leber congenital amaurosis via therapeutic base editing | [170] | ||||
| Rd12 mice rd12 Gnat1−/− mice | ABEs | LV | Restoration of visual function in adult mice with an inherited retinal disease via adenine base editing | [171] | |||
| CEP290 | HuCEP290 knock-in mice and monkeys | EDIT-101 | AAV | Development of a gene-editing approach to restore vision loss in Leber congenital amaurosis type 10 | [172] | ||
| Stargardt disease | Autosomal recessive | ABCA4 | Abca4−/− Rdh8−/− mice | REVeRT | AAV | mRNA trans-splicing dual AAV vectors for (epi)genome editing and gene therapy | [173] |
| Abca4−/− mice | AAV | An AAV Dual Vector Strategy Ameliorates the Stargardt Phenotype in Adult Abca4−/− Mice | [174] | ||||
| Cone-rod dystrophy 6 | Autosomal dominant | GUCY2D | RetGC1 (hR838S, hWT) mouse | AR | AAV | Development of an AAV-CRISPR-Cas9-based treatment for dominant cone-rod dystrophy 6 | [112] |
| X-linked juvenile retinoschisis | X-linked | RS1 | BALB/c-strain mice | HITI | SMNP | Dual Supramolecular Nanoparticle Vectors Enable CRISPR/Cas9-Mediated Knockin of Retinoschisin 1 Gene—A Potential Nonviral Therapeutic Solution for X-Linked Juvenile Retinoschisis | [175] |
| NCT Number | Gene | Vector | Interventions | Study Title | Phases |
|---|---|---|---|---|---|
| Achromatopsia | |||||
| NCT02935517 | CNGA3 | AAV8 | AGTC-402 | Safety and Efficacy Trial of AAV Gene Therapy in Patients With CNGA3 Achromatopsia (A Clarity Clinical Trial) | Phase 1, Phase 2 |
| NCT02610582 | CNGA3 | AAV2 | rAAV.hCNGA3 | Safety and Efficacy of rAAV.hCNGA3 Gene Therapy in Patients With CNGA3-linked Achromatopsia | Phase 1, Phase 2 |
| NCT02599922 | CNGB3 | AAV2 | rAAV2tYF-PR1.7-hCNGB3 | Safety and Efficacy Trial of AAV Gene Therapy in Patients With CNGB3 Achromatopsia (A Clarity Clinical Trial) | Phase 1, Phase 2 |
| NCT03001310 | CNGB3 | AAV2/8 | AAV2/8-hCARp.hCNGB3 | Gene Therapy for Achromatopsia (CNGB3) (CNGB3) | Phase 1, Phase 2 |
| Bietti Crystalline Dystrophy | |||||
| NCT05399069 | CYP4V2 | AAV8 | VGR-R01 | Safety and Tolerability of VGR-R01 in Patients With Bietti Crystalline Dystrophy | Early Phase 1 |
| NCT04722107 | CYP4V2 | AAV2/8 | rAAV2/8-hCYP4V2 | Safety Study of rAAV2/8-hCYP4V2 in Patients With Bietti’s Crystalline Dystrophy (BCD) | Early Phase 1 |
| Choroideremia | |||||
| NCT03507686 | CHM/REP1 | AAV2 | BIIB111 (AAV2-REP1) | A Safety Study of Retinal Gene Therapy for Choroideremia With Administration of BIIB111 | Phase 2 |
| NCT03496012 | CHM/REP1 | AAV2 | BIIB111 (AAV2-REP1) | Efficacy and Safety of BIIB111 for the Treatment of Choroideremia | Phase 3 |
| NCT02553135 | CHM/REP1 | AAV2 | AAV2-REP1 | Choroideremia Gene Therapy Clinical Trial | Phase 2 |
| NCT02341807 | CHM/REP1 | AAV2 | AAV2-hCHM | Safety and Dose-escalation Study of AAV2-hCHM in Participants With CHM (Choroideremia) Gene Mutations | Phase 1, Phase 2 |
| NCT02671539 | CHM/REP1 | AAV2 | rAAV2.REP1 | THOR—Tübingen Choroideremia Gene Therapy Trial | Phase 2 |
| NCT02077361 | CHM/REP1 | AAV2 | rAAV2.REP1 | An Open Label Clinical Trial of Retinal Gene Therapy for Choroideremia | Phase 1, Phase 2 |
| NCT02407678 | CHM/REP1 | AAV2 | AAV-mediated REP1 gene replacement | REP1 Gene Replacement Therapy for Choroideremia | Phase 2 |
| NCT01461213 | CHM/REP1 | AAV2 | rAAV2.REP1 | Gene Therapy for Blindness Caused by Choroideremia | Phase 1, Phase 2 |
| Leber Congenital Amaurosis | |||||
| NCT03920007 | GLUCY2D | AAV5 | ATSN-101 | Study of Subretinally Injected ATSN-101 Administered in Patients With Leber Congenital Amaurosis Caused by Biallelic Mutations in GUCY2D | Phase 1, Phase 2 |
| NCT00749957 | RPE65 | AAV2 | rAAV2-CB-hRPE65 | Phase 1/2 Safety and Efficacy Study of AAV-RPE65 Vector to Treat Leber Congenital Amaurosis | Phase 1, Phase 2 |
| NCT00821340 | RPE65 | AAV2 | rAAV2-hRPE65 | Clinical Trial of Gene Therapy for Leber Congenital Amaurosis Caused by RPE65 Mutations | Phase 1 |
| NCT05906953 | RPE65 | AAV2 | HG004 | Safety and Efficacy Trial of HG004 for Leber Congenital Amaurosis Related to Rpe65 Gene Mutations (STAR) | Phase 1, Phase 2 |
| NCT02781480 | RPE65 | AAV2/5 | AAV RPE65 | Clinical Trial of Gene Therapy for the Treatment of Leber Congenital Amaurosis (LCA) | Phase 1, Phase 2 |
| NCT01496040 | RPE65 | AAV4 | rAAV2/4.hRPE65 | Clinical Gene Therapy Protocol for the Treatment of Retinal Dystrophy Caused by Defects in RPE65 | Phase 1, Phase 2 |
| NCT00999609 | RPE65 | AAV2 | AAV2-hRPE65v2,voretigene neparvovec-rzyl | Safety and Efficacy Study in Subjects With Leber Congenital Amaurosis | Phase 3 |
| NCT00516477 | RPE65 | AAV2 | AAV2-hRPE65v2 (Luxterna; voretigene neparvovec-rzyl) | Safety Study in Subjects With Leber Congenital Amaurosis | Phase 1 |
| NCT00643747 | RPE65 | AAV2 | tgAAG76 (rAAV2/2.hRPE65p.hRPE65) | Safety Study of RPE65 Gene Therapy to Treat Leber Congenital Amaurosis | Phase 1, Phase 2 |
| NCT00481546 | RPE65 | AAV2 | rAAV2-CBSB-hRPE65 | Phase I Trial of Gene Vector to Patients With Retinal Disease Due to RPE65 Mutations | Phase 1 |
| NCT06088992 | RPE65 | AAV9 | HG004 | Leber Congenital Amaurosis Inherited Blindness of Gene Therapy Trial(LIGHT) | Early Phase 1 |
| NCT03872479 | IVS26/CEP290 | AAV5 | EDIT-101 | Single Ascending Dose Study in Participants With LCA10 | Phase 1, Phase 2 |
| NCT03913143 | CEP290 | ASO | QR-110 (sepofarsen) | A Study to Evaluate Efficacy, Safety, Tolerability and Exposure After a Repeat-dose of Sepofarsen (QR-110) in LCA10 (ILLUMINATE) | Phase 2, Phase 3 |
| NCT03913130 | CEP290 | ASO | QR-110 (sepofarsen) | Extension Study to Study PQ-110-001 (NCT03140969) | Phase 1, Phase 2 |
| NCT04855045 | CEP290 | ASO | QR-110 (sepofarsen) | An Open-label, Dose Escalation and Double-masked, Randomized, Controlled Trial Evaluating Safety and Tolerability of Sepofarsen in Children (<8 Years of Age) With LCA10 Caused by Mutations in the CEP290 Gene. | Phase 2, Phase 3 |
| NCT03140969 | CEP290 | ASO | QR-110 (sepofarsen) | Study to Evaluate QR-110 in Leber’s Congenital Amaurosis (LCA) Due to the c.2991 + 1655A > G Mutation (p.Cys998X) in the CEP290 Gene | Phase 1, Phase 2 |
| NCT05616793 | LCA5 | AAV8 | AAV8.hLCA5 | Safety and Tolerability Subretinal OPGx-001 for LCA5-Associated Inherited Retinal Degeneration (LCA5-IRD) | Phase 1, Phase 2 |
| Leber Hereditary Optic Neuropathy | |||||
| NCT01267422 | MT-ND4 | AAV2 | rAAV2-ND4 | Safety and Efficacy Study of rAAV2-ND4 Treatment of Leber Hereditary Optic Neuropathy (LHON) | NA |
| NCT02161380 | MT-ND4 | AAV2 | scAAV2-P1ND4v2 | Safety Study of an Adeno-associated Virus Vector for Gene Therapy of Leber’s Hereditary Optic Neuropathy | Phase 1 |
| NCT03406104 | MT-ND4 | AAV2 | GS010 | RESCUE and REVERSE Long-term Follow-up | Phase 3 |
| NCT02064569 | MT-ND4 | AAV2 | GS010 | Safety Evaluation of Gene Therapy in Leber Hereditary Optic Neuropathy (LHON) Patients | Phase 1, Phase 2 |
| NCT02652780 | MT-ND4 (G11778A) | AAV2 | GS010 | Efficacy Study of GS010 for Treatment of Vision Loss From 7 Months to 1 Year From Onset in LHON Due to the ND4 Mutation (REVERSE) | Phase 3 |
| NCT02652767 | MT-ND4 (G11778A) | AAV2 | GS010 | Efficacy Study of GS010 for the Treatment of Vision Loss up to 6 Months From Onset in LHON Due to the ND4 Mutation | Phase 3 |
| NCT03293524 | MT-ND4 | AAV2 | GS010 | Efficacy & Safety Study of Bilateral IVT Injection of GS010 in LHON Subjects Due to the ND4 Mutation for up to 1 Year | Phase 3 |
| NCT03153293 | MT-ND4 | AAV2 | rAAV2-ND4 | A Single Intravitreal Injection of rAAV2-ND4 for the Treatment of Leber’s Hereditary Optic Neuropathy | Phase 2, Phase 3 |
| Retinitis Pigmentosa | |||||
| NCT03328130 | PDE6B | AAV2/5 | AAV2/5-hPDE6B | Safety and Efficacy Study in Patients With Retinitis Pigmentosa Due to Mutations in PDE6B Gene | Phase 1, Phase 2 |
| NCT01482195 | MERTK | AAV2 | rAAV2-VMD2-hMERTK | Trial of Subretinal Injection of (rAAV2-VMD2-hMERTK) | Phase 1 |
| NCT03374657 | RLBP1 | AAV8 | CPK850 | A First-in-human, Proof of Concept Study of CPK850 in Patients With RLBP1 Retinitis Pigmentosa | Phase 1, Phase 2 |
| NCT06388200 | RHO/NR2E3 | AAV | OCU400-301 | A Phase 3 Study Of OCU400 Gene Therapy for the Treatment Of Retinitis Pigmentosa | Phase 3 |
| NCT05805007 | RHO | AAV | ZVS203e | Safety and Tolerability Study of Gene Editing Drug ZVS203e in Participants With Retinitis Pigmentosa | Early Phase 1 |
| NCT04611503 | PDE6A | AAV | rAAV.hPDE6A | PDE6A Gene Therapy for Retinitis Pigmentosa | Phase 1, Phase 2 |
| NCT06291935 | CNGA1 | AAV2 | VG901 (AAV2.NN-CNGA1) | Safety and Tolerability of Intravitreal Administration of VG901 in Patients With Retinitis Pigmentosa Due to Mutations in the CNGA1 Gene | Phase 1 |
| NCT04123626 | P23H (RHO) | ASO | QR-1123 | A Study to Evaluate the Safety and Tolerability of QR-1123 in Subjects With Autosomal Dominant Retinitis Pigmentosa Due to the P23H Mutation in the RHO Gene | Phase 1, Phase 2 |
| NCT05176717 | USH2A (exon 3) | ASO | Ultevursen (QR-421a) | Study to Evaluate the Efficacy Safety and Tolerability of QR-421a in Subjects With RP Due to Mutations in Exon 13 of the USH2A Gene With Early to Moderate Vision Loss (Celeste) | Phase 2, Phase 3 |
| NCT05158296 | USH2A (exon 3) | ASO | Ultevursen (QR-421a) | Study to Evaluate the Efficacy Safety and Tolerability of Ultevursen in Subjects With RP Due to Mutations in Exon 13 of the USH2A Gene (Sirius) | Phase 2, Phase 3 |
| NCT03780257 | USH2A (exon 3) | ASO | Ultevursen (QR-421a) | Study to Evaluate Safety and Tolerability of QR-421a in Subjects With RP Due to Mutations in Exon 13 of the USH2A Gene | Phase 1, Phase 2 |
| NCT04517149 | RPGR | (AAV) R100 | 4D-125 | 4D-125 in Patients With X-Linked Retinitis Pigmentosa (XLRP) | Phase 1, Phase 2 |
| NCT06333249 | RPGR | AAV2 | AGTC-501 (rAAV2tYF-GRK1-RPGR) | A Study Comparing Two Doses of AGTC-501 in Male Subjects With X-linked Retinitis Pigmentosa Caused by RPGR Mutations (SKYLINE) | Phase 2 |
| NCT06275620 | RPGR | AAV2 | AGTC-501 (rAAV2tYF-GRK1-RPGR) | A Study Comparing Two Doses of AGTC-501 in Male Participants With X-linked Retinitis Pigmentosa Caused by RPGR Mutations (DAWN) | Phase 2 |
| NCT04850118 | RPGR | AAV2 | rAAV2tYF-GRK1-hRPGRco | A Clinical Trial Evaluating the Safety and Efficacy of a Single Subretinal Injection of AGTC-501 in Participants With XLRP | Phase 2, Phase 3 |
| NCT03316560 | RPGR | AAV2 | rAAV2tYF-GRK1-RPGR | Safety and Efficacy of rAAV2tYF-GRK1-RPGR in Subjects With X-linked Retinitis Pigmentosa Caused by RPGR Mutations | Phase 1, Phase 2 |
| NCT03116113 | RPGR | AAV8 | BIIB112 (AAV8-RPGR) | A Clinical Trial of Retinal Gene Therapy for X-linked Retinitis Pigmentosa Using BIIB112 | Phase 1, Phase 2 |
| NCT05874310 | RPGR | AAV | FT-002 | Gene Therapy for Subjects With RPGR Mutation-associated X-linked Retinitis Pigmentosa | Early Phase 1 |
| NCT04794101 | RPGR | AAV5 | AAV5-hRKp.RPGR | Follow-up Gene Therapy Trial for the Treatment of X-linked Retinitis Pigmentosa Associated With Variants in the RPGR Gene | Phase 3 |
| NCT04671433 | RPGR | AAV5 | AAV5-hRKp.RPGR | Gene Therapy Trial for the Treatment of X-linked Retinitis Pigmentosa Associated With Variants in the RPGR Gene | Phase 3 |
| NCT03252847 | RPGR | AAV2/5 | AAV2/5-RPGR | Gene Therapy for X-linked Retinitis Pigmentosa (XLRP)—Retinitis Pigmentosa GTPase Regulator (RPGR) | Phase 1, Phase 2 |
| Stargardt Disease 1 | |||||
| NCT06300476 | ABCA4 | AAV | JWK006 | Safety and Efficacy of a Single Subretinal Injection of JWK006 Gene Therapy in Subjects With Stargardt Disease(STGD1) | Phase 1, Phase 2 |
| X-Linked Retinoschisis | |||||
| NCT06289452 | RS1 | AAV8 | IVB102 | Safety and Efficacy Study of IVB102 Injection in Subjects With X-linked Retinoschisis | Early Phase 1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kulbay, M.; Tuli, N.; Akdag, A.; Kahn Ali, S.; Qian, C.X. Optogenetics and Targeted Gene Therapy for Retinal Diseases: Unravelling the Fundamentals, Applications, and Future Perspectives. J. Clin. Med. 2024, 13, 4224. https://doi.org/10.3390/jcm13144224
Kulbay M, Tuli N, Akdag A, Kahn Ali S, Qian CX. Optogenetics and Targeted Gene Therapy for Retinal Diseases: Unravelling the Fundamentals, Applications, and Future Perspectives. Journal of Clinical Medicine. 2024; 13(14):4224. https://doi.org/10.3390/jcm13144224
Chicago/Turabian StyleKulbay, Merve, Nicolas Tuli, Arjin Akdag, Shigufa Kahn Ali, and Cynthia X. Qian. 2024. "Optogenetics and Targeted Gene Therapy for Retinal Diseases: Unravelling the Fundamentals, Applications, and Future Perspectives" Journal of Clinical Medicine 13, no. 14: 4224. https://doi.org/10.3390/jcm13144224
APA StyleKulbay, M., Tuli, N., Akdag, A., Kahn Ali, S., & Qian, C. X. (2024). Optogenetics and Targeted Gene Therapy for Retinal Diseases: Unravelling the Fundamentals, Applications, and Future Perspectives. Journal of Clinical Medicine, 13(14), 4224. https://doi.org/10.3390/jcm13144224