
Summary of the Therapeutic Options for Patients with Dry and Neovascular AMD

 ,
,
Abstract
:1. Introduction
2. Treatment of Dry AMD
2.1. General Information
2.2. Drugs in Dry AMD
2.2.1. Pegcetacoplan
2.2.2. Avacincaptad Pegol
2.2.3. Brimonidine Tartrate
2.3. Stem Cells
2.4. Other Aids for Dry AMD
3. Treatment of Neovascular AMD
3.1. Photodynamic Therapy
3.2. Anti-VEGF Drugs
3.2.1. General Information
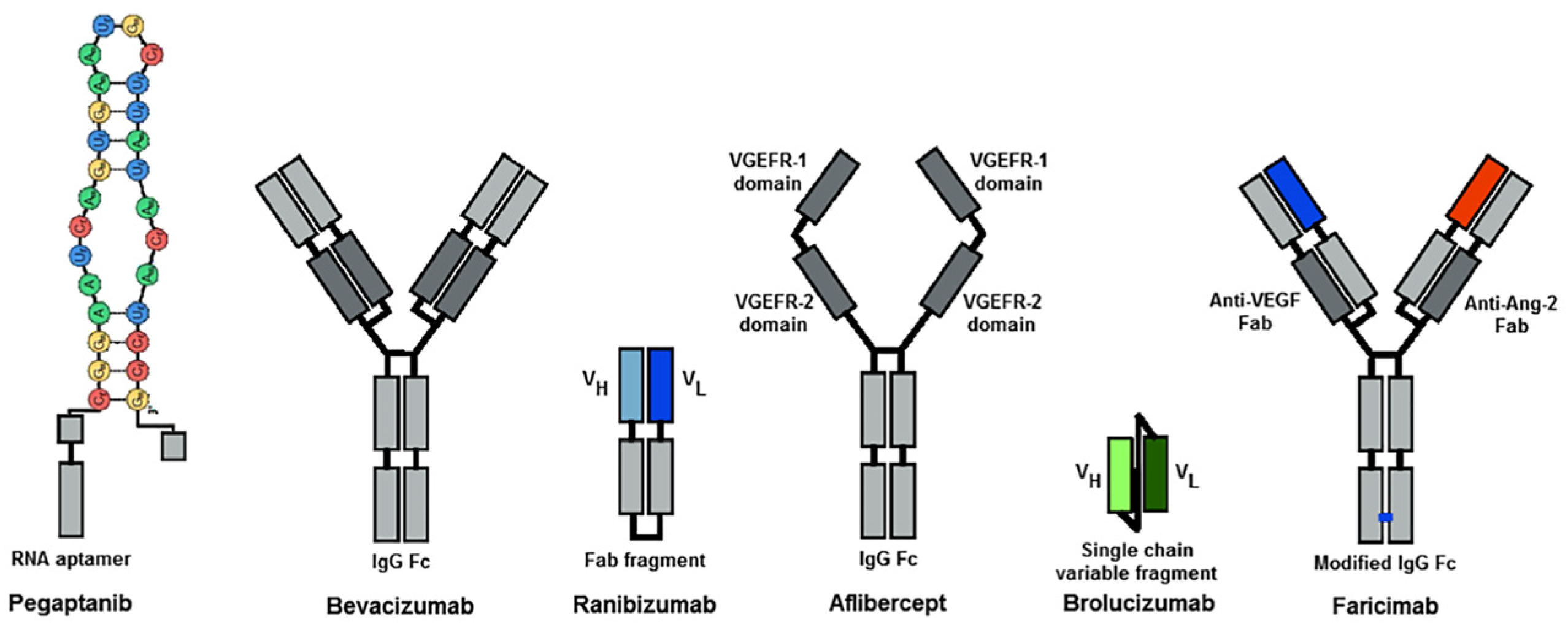
3.2.2. Anti-VEGF Therapies
Pegaptanib
Bevacizumab
Ranibizumab
Aflibercept
Brolcizumab
Faricimab
3.2.3. Other Therapies to Treat nAMD
Umedaptanib Pegol
Hydrogels
Intraocular Port Delivery System
Gene Therapy
3.3. Surgical Treatment
4. Summary
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Congdon, N.; O’Colmain, B.; Klaver, C.C.; Klein, R.; Munoz, B.; Friedman, D.S.; Kempen, J.; Taylor, H.R.; Mitchell, P. Eye Diseases Prevalence Research G: Causes and prevalence of visual impairment among adults in the United States. Arch. Ophthalmol. 2004, 122, 477–485. [Google Scholar] [PubMed]
- Velilla, S.; Garcia-Medina, J.J.; Garcia-Layana, A.; Dolz-Marco, R.; Pons-Vazquez, S.; Pinazo-Duran, M.D.; Gomez-Ulla, F.; Arevalo, J.F.; Diaz-Llopis, M.; Gallego-Pinazo, R. Smoking and age-related macular degeneration: Review and update. J. Ophthalmol. 2013, 2013, 895147. [Google Scholar] [CrossRef] [PubMed]
- Michalska-Małecka, K.; Kabiesz, A.; Nowak, M.; Spiewak, D. Age related macular degeneration—Challenge for future: Pathogenesis and new perspectives for the treatment. Eur. Geriatr. Med. 2015, 6, 69–75. [Google Scholar] [CrossRef]
- Wylęgała, E.T.S.; Piłat, J. Zwyrodnienie Plamki Związane z Wiekiem; Wydawnictwo Medyczne Górnicki: Wrocław, Poland, 2011. [Google Scholar]
- Kortvely, E.; Hauck, S.M.; Duetsch, G.; Gloeckner, C.J.; Kremmer, E.; Alge-Priglinger, C.S.; Deeg, C.A.; Ueffing, M. ARMS2 is a constituent of the extracellular matrix providing a link between familial and sporadic age-related macular degenerations. Investig. Ophthalmol. Vis. Sci. 2010, 51, 79–88. [Google Scholar] [CrossRef]
- Xi, L. Pigment Epithelium-Derived Factor as a Possible Treatment Agent for Choroidal Neovascularization. Oxid. Med. Cell. Longev. 2020, 2020, 8941057. [Google Scholar] [CrossRef]
- Chakravarthy, U.; McKay, G.J.; de Jong, P.T.; Rahu, M.; Seland, J.; Soubrane, G.; Tomazzoli, L.; Topouzis, F.; Vingerling, J.R.; Vioque, J.; et al. ARMS2 increases the risk of early and late age-related macular degeneration in the European Eye Study. Ophthalmology 2013, 120, 342–348. [Google Scholar] [CrossRef] [PubMed]
- Flores, R.; Carneiro, A.; Vieira, M.; Tenreiro, S.; Seabra, M.C. Age-Related Macular Degeneration: Pathophysiology, Management, and Future Perspectives. Ophthalmologica 2021, 244, 495–511. [Google Scholar] [CrossRef] [PubMed]
- Janik-Papis, K.; Ulińska, M.; Krzyżanowska, A.; Stoczyńska, E.; Borucka, A.I.; Woźniak, K.; Zaraś, M.; Jacek, P.; Szaflik, J.P.; Janusz Błasiak, J. Mechanizmy oksydacyjne w patogenezie zwyrodnienia plamki związanego z wiekiem Role of oxidative mechanisms in the pathogenesis of age-related macular degeneratio. Klinika Oczna 2009, 111, 169–173. [Google Scholar]
- Ambati, J.; Ambati, B.K.; Yoo, S.H.; Ianchulev, S.; Adamis, A.P. Age-related macular degeneration: Etiology, pathogenesis, and therapeutic strategies. Surv. Ophthalmol. 2003, 48, 257–293. [Google Scholar] [CrossRef]
- Grossniklaus, H.E.; Green, W.R. Choroidal neovascularization. Am. J. Ophthalmol. 2004, 137, 496–503. [Google Scholar] [CrossRef]
- Keenan, T.D.; Vitale, S.; Agron, E.; Domalpally, A.; Antoszyk, A.N.; Elman, M.J.; Clemons, T.E.; Chew, E.Y. Age-Related Eye Disease Study 2 Research G: Visual Acuity Outcomes after Anti-Vascular Endothelial Growth Factor Treatment for Neovascular Age-Related Macular Degeneration: Age-Related Eye Disease Study 2 Report Number 19. Ophthalmol. Retin. 2020, 4, 3–12. [Google Scholar] [CrossRef]
- Dervenis, N.; Dervenis, P.; Agorogiannis, E. Neovascular age-related macular degeneration: Disease pathogenesis and current state of molecular biomarkers predicting treatment response-a scoping review. BMJ Open Ophthalmol. 2024, 9, e001516. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Lin, Y. Tumor necrosis factor and cancer, buddies or foes? Acta Pharmacol. Sin. 2008, 29, 1275–1288. [Google Scholar] [CrossRef] [PubMed]
- Frater-Schroder, M.; Risau, W.; Hallmann, R.; Gautschi, P.; Bohlen, P. Tumor necrosis factor type alpha, a potent inhibitor of endothelial cell growth in vitro, is angiogenic in vivo. Proc. Natl. Acad. Sci. USA 1987, 84, 5277–5281. [Google Scholar] [CrossRef] [PubMed]
- Knickelbein, J.E.; Chan, C.C.; Sen, H.N.; Ferris, F.L.; Nussenblatt, R.B. Inflammatory Mechanisms of Age-related Macular Degeneration. Int. Ophthalmol. Clin. 2015, 55, 63–78. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, Z. The role of the cytokine TNF-alpha in choroidal neovascularization: A systematic review. Eye 2024, 38, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Gomulka, K.; Liebhart, J. Vascular endothelial growth factor—Structure, function and role in airways inflammation and the clinical course of asthma. Pneumonol. I Alergol. Pol. 2009, 77, 549–553. [Google Scholar]
- Zarbin, M.A. Current concepts in the pathogenesis of age-related macular degeneration. Arch. Ophthalmol. 2004, 122, 598–614. [Google Scholar] [CrossRef] [PubMed]
- Hera, R.; Keramidas, M.; Peoc’h, M.; Mouillon, M.; Romanet, J.P.; Feige, J.J. Expression of VEGF and angiopoietins in subfoveal membranes from patients with age-related macular degeneration. Am. J. Ophthalmol. 2005, 139, 589–596. [Google Scholar] [CrossRef]
- Spaide, R.F.; Jaffe, G.J.; Sarraf, D.; Freund, K.B.; Sadda, S.R.; Staurenghi, G.; Waheed, N.K.; Chakravarthy, U.; Rosenfeld, P.J.; Holz, F.G.; et al. Consensus Nomenclature for Reporting Neovascular Age-Related Macular Degeneration Data: Consensus on Neovascular Age-Related Macular Degeneration Nomenclature Study Group. Ophthalmology 2020, 127, 616–636. [Google Scholar] [CrossRef]
- Finocchio, L.; Zeppieri, M.; Gabai, A.; Toneatto, G.; Spadea, L.; Salati, C. Recent Developments in Gene Therapy for Neovascular Age-Related Macular Degeneration: A Review. Biomedicines 2023, 11, 3221. [Google Scholar] [CrossRef]
- Hilely, A.; Au, A.; Freund, K.B.; Loewenstein, A.; Souied, E.H.; Zur, D.; Sacconi, R.; Borrelli, E.; Peiretti, E.; Iovino, C.; et al. Non-neovascular age-related macular degeneration with subretinal fluid. Br. J. Ophthalmol. 2021, 105, 1415–1420. [Google Scholar] [CrossRef]
- Seddon, J.M. Macular Degeneration Epidemiology: Nature-Nurture, Lifestyle Factors, Genetic Risk, and Gene-Environment Interactions—The Weisenfeld Award Lecture. Investig. Ophthalmol. Vis. Sci. 2017, 58, 6513–6528. [Google Scholar] [CrossRef] [PubMed]
- Hwang, C.K.; Agron, E.; Domalpally, A.; Cukras, C.A.; Wong, W.T.; Chew, E.Y.; Keenan, T.D.L.; Group, A.R. Progression of Geographic Atrophy with Subsequent Exudative Neovascular Disease in Age-Related Macular Degeneration: AREDS2 Report 24. Ophthalmol. Retin. 2021, 5, 108–117. [Google Scholar] [CrossRef]
- Walchuk, C.; Suh, M. Nutrition and the aging retina: A comprehensive review of the relationship between nutrients and their role in age-related macular degeneration and retina disease prevention. Adv. Food Nutr. Res. 2020, 93, 293–332. [Google Scholar]
- Heier, J.S.; Lad, E.M.; Holz, F.G.; Rosenfeld, P.J.; Guymer, R.H.; Boyer, D.; Grossi, F.; Baumal, C.R.; Korobelnik, J.F.; Slakter, J.S.; et al. Pegcetacoplan for the treatment of geographic atrophy secondary to age-related macular degeneration (OAKS and DERBY): Two multicentre, randomised, double-masked, sham-controlled, phase 3 trials. Lancet 2023, 402, 1434–1448. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.S.; Lally, D.R.; Hsu, J.; Wykoff, C.C.; Eichenbaum, D.; Heier, J.S.; Jaffe, G.J.; Westby, K.; Desai, D.; Zhu, L.; et al. Avacincaptad pegol for geographic atrophy secondary to age-related macular degeneration: 18-month findings from the GATHER1 trial. Eye 2023, 37, 3551–3557. [Google Scholar] [CrossRef]
- Khanani, A.M.; Patel, S.S.; Staurenghi, G.; Tadayoni, R.; Danzig, C.J.; Eichenbaum, D.A.; Hsu, J.; Wykoff, C.C.; Heier, J.S.; Lally, D.R.; et al. Efficacy and safety of avacincaptad pegol in patients with geographic atrophy (GATHER2): 12-month results from a randomised, double-masked, phase 3 trial. Lancet 2023, 402, 1449–1458. [Google Scholar] [CrossRef] [PubMed]
- Freeman, W.R.; Bandello, F.; Souied, E.; Guymer, R.H.; Garg, S.J.; Chen, F.K.; Rich, R.; Holz, F.G.; Patel, S.S.; Kim, K.; et al. Randomized Phase IIb Study of Brimonidine Drug Delivery System Generation 2 for Geographic Atrophy in Age-Related Macular Degeneration. Ophthalmol. Retin. 2023, 7, 573–585. [Google Scholar] [CrossRef]
- Vakharia, P.; Eichenbaum, D. Geographic atrophy: Current and future therapeutic agents and practical considerations for retinal specialists. Curr. Opin. Ophthalmol. 2024, 35, 165–169. [Google Scholar] [CrossRef]
- Jaffe, G.J.; Khanani, A.M. Two studies to learn if avacincaptad pegol works and is safe in people with geographic atrophy: A plain language summary of the GATHER1 and GATHER 2 studies. Immunotherapy 2024, 16, 205–221. [Google Scholar] [CrossRef] [PubMed]
- Kashani, A.H.; Lebkowski, J.S.; Rahhal, F.M.; Avery, R.L.; Salehi-Had, H.; Dang, W.; Lin, C.M.; Mitra, D.; Zhu, D.; Thomas, B.B.; et al. A bioengineered retinal pigment epithelial monolayer for advanced, dry age-related macular degeneration. Sci. Transl. Med. 2018, 10, eaao4097. [Google Scholar] [CrossRef]
- Schwartz, S.D.; Regillo, C.D.; Lam, B.L.; Eliott, D.; Rosenfeld, P.J.; Gregori, N.Z.; Hubschman, J.P.; Davis, J.L.; Heilwell, G.; Spirn, M.; et al. Human embryonic stem cell-derived retinal pigment epithelium in patients with age-related macular degeneration and Stargardt’s macular dystrophy: Follow-up of two open-label phase 1/2 studies. Lancet 2015, 385, 509–516. [Google Scholar] [CrossRef] [PubMed]
- Voisin, A.; Penaguin, A.; Gaillard, A.; Leveziel, N. Stem cell therapy in retinal diseases. Neural Regen Res. 2023, 18, 1478–1485. [Google Scholar] [CrossRef] [PubMed]
- Grzybowski, A.; Wang, J.; Mao, F.; Wang, D.; Wang, N. Intraocular vision-improving devices in age-related macular degeneration. Ann. Transl. Med. 2020, 8, 1549. [Google Scholar] [CrossRef] [PubMed]
- Toro, M.D.; Vidal-Aroca, F.; Montemagni, M.; Xompero, C.; Fioretto, G.; Costagliola, C. Three-Month Safety and Efficacy Outcomes for the Smaller-Incision New-Generation Implantable Miniature Telescope (SING IMT). J. Clin. Med. 2023, 12, 518. [Google Scholar] [CrossRef] [PubMed]
- Khoramnia, R.; von Mohrenfels, C.W.; Salgado, J.P.; Schweiger, B.; Engel, M.; Hadeler, J.; Lohmann, C.P. The IOL-Vip system: Principles and clinical application. Der Ophthalmol. Z. Der Dtsch. Ophthalmol. Ges. 2010, 107, 274+276–280. [Google Scholar]
- European Commission-VisudyneINNverteporfin.In. Available online: https://www.ema.europa.eu/en/documents/product-information/visudyne-epar-product-information_en.pdf (accessed on 27 July 2000).
- Wormald, R.; Evans, J.; Smeeth, L.; Henshaw, K. Photodynamic therapy for neovascular age-related macular degeneration. Cochrane Database Syst. Rev. 2007, 4, CD002030. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.S.; Ngo, W.K.; Chay, I.W.; Ting, D.S.; Sadda, S.R. Neovascular Age-Related Macular Degeneration (nAMD): A Review of Emerging Treatment Options. Clin. Ophthalmol. 2022, 16, 917–933. [Google Scholar] [CrossRef]
- Chen, Y.; Han, F. Profile of ranibizumab: Efficacy and safety for the treatment of wet age-related macular degeneration. Ther. Clin. Risk Manag. 2012, 8, 343–351. [Google Scholar]
- Rejdak, R.; Junemann, A.G. Monografia AMD—Diagnostyka i Leczenie Zwyrodnienia Plamki Związanego z Wiekiem; Via Medica: Gdańsk, Poland, 2016; Volume 2. [Google Scholar]
- Tatsumi, T. Current Treatments for Diabetic Macular Edema. Int. J. Mol. Sci. 2023, 24, 9591. [Google Scholar] [CrossRef] [PubMed]
- Gragoudas, E.S.; Adamis, A.P.; Cunningham, E.T., Jr.; Feinsod, M.; Guyer, D.R. Group VISiONCT: Pegaptanib for neovascular age-related macular degeneration. N. Engl. J. Med. 2004, 351, 2805–2816. [Google Scholar] [CrossRef] [PubMed]
- Tufail, A.; Patel, P.J.; Egan, C.; Hykin, P.; da Cruz, L.; Gregor, Z.; Dowler, J.; Majid, M.A.; Bailey, C.; Mohamed, Q.; et al. Bevacizumab for neovascular age related macular degeneration (ABC Trial): Multicentre randomised double masked study. BMJ 2010, 340, c2459. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.F.; Maguire, M.G.; Fine, S.L.; Ying, G.S.; Jaffe, G.J.; Grunwald, J.E.; Toth, C.; Redford, M.; Ferris, F.L., 3rd; Comparison of Age-Related Macular Degeneration Treatments Trials (CATT) Research Group. Ranibizumab and Bevacizumab for Treatment of Neovascular Age-related Macular Degeneration: Two-Year Results. Ophthalmology 2020, 127, S135–S145. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeld, P.J.; Brown, D.M.; Heier, J.S.; Boyer, D.S.; Kaiser, P.K.; Chung, C.Y.; Kim, R.Y.; Group, M.S. Ranibizumab for neovascular age-related macular degeneration. N. Engl. J. Med. 2006, 355, 1419–1431. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeld, P.J.; Rich, R.M.; Lalwani, G.A. Ranibizumab: Phase III clinical trial results. Ophthalmol. Clin. N. Am 2006, 19, 361–372. [Google Scholar]
- Abraham, P.; Yue, H.; Wilson, L. Randomized, double-masked, sham-controlled trial of ranibizumab for neovascular age-related macular degeneration: PIER study year 2. Am. J. Ophthalmol. 2010, 150, 315–324.e1. [Google Scholar] [CrossRef] [PubMed]
- Lalwani, G.A.; Rosenfeld, P.J.; Fung, A.E.; Dubovy, S.R.; Michels, S.; Feuer, W.; Davis, J.L.; Flynn, H.W., Jr.; Esquiabro, M. A variable-dosing regimen with intravitreal ranibizumab for neovascular age-related macular degeneration: Year 2 of the PrONTO Study. Am. J. Ophthalmol. 2009, 148, 43–58.e1. [Google Scholar] [CrossRef] [PubMed]
- Meyer, C.H.; Eter, N.; Holz, F.G.; SUSTAIN Study Group. Ranibizumab in Patients With Subfoveal Choroidal Neovascularization Secondary to Age-Related Macular Degeneration. Interim Results From the Sustain Trial. Investig. Ophthalmol. Vis. Sci. 2008, 49, 273. [Google Scholar]
- Heier, J.S.; Brown, D.M.; Chong, V.; Korobelnik, J.F.; Kaiser, P.K.; Nguyen, Q.D.; Kirchhof, B.; Ho, A.; Ogura, Y.; Yancopoulos, G.D.; et al. Intravitreal aflibercept (VEGF trap-eye) in wet age-related macular degeneration. Ophthalmology 2012, 119, 2537–2548. [Google Scholar] [CrossRef]
- Gallivan, M.D.; Garcia, K.M.; Torres, A.Z.; Lum, F.; Li, C.; Mbagwu, M.; Leng, T. Emulating VIEW 1 and VIEW 2 Clinical Trial Outcome Data Using the American Academy of Ophthalmology IRIS Registry. Ophthalmic Surg. Lasers Imaging Retin. 2023, 54, 6–14. [Google Scholar] [CrossRef] [PubMed]
- Lanzetta, P.; Korobelnik, J.F.; Heier, J.S.; Leal, S.; Holz, F.G.; Clark, W.L.; Eichenbaum, D.; Iida, T.; Xiaodong, S.; Berliner, A.J.; et al. Intravitreal aflibercept 8 mg in neovascular age-related macular degeneration (PULSAR): 48-week results from a randomised, double-masked, non-inferiority, phase 3 trial. Lancet 2024, 403, 1141–1152. [Google Scholar] [CrossRef] [PubMed]
- Dugel, P.U.; Koh, A.; Ogura, Y.; Jaffe, G.J.; Schmidt-Erfurth, U.; Brown, D.M.; Gomes, A.V.; Warburton, J.; Weichselberger, A.; Holz, F.G.; et al. HAWK and HARRIER: Phase 3, Multicenter, Randomized, Double-Masked Trials of Brolucizumab for Neovascular Age-Related Macular Degeneration. Ophthalmology 2020, 127, 72–84. [Google Scholar] [CrossRef] [PubMed]
- Lally, D.R.; Loewenstein, A.; Arnold, J.J.; Yang, Y.C.; Gedif, K.; Best, C.; Patel, H.; Tadayoni, R.; Heier, J.S. Efficacy and safety of brolucizumab versus aflibercept in eyes with early persistent retinal fluid: 96-week outcomes from the HAWK and HARRIER studies. Eye 2023, 37, 1242–1248. [Google Scholar] [CrossRef] [PubMed]
- Heier, J.S.; Khanani, A.M.; Quezada Ruiz, C.; Basu, K.; Ferrone, P.J.; Brittain, C.; Figueroa, M.S.; Lin, H.; Holz, F.G.; Patel, V.; et al. Efficacy, durability, and safety of intravitreal faricimab up to every 16 weeks for neovascular age-related macular degeneration (TENAYA and LUCERNE): Two randomised, double-masked, phase 3, non-inferiority trials. Lancet 2022, 399, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Khanani, A.M.; Guymer, R.H.; Basu, K.; Boston, H.; Heier, J.S.; Korobelnik, J.F.; Kotecha, A.; Lin, H.; Silverman, D.; Swaminathan, B.; et al. TENAYA and LUCERNE: Rationale and Design for the Phase 3 Clinical Trials of Faricimab for Neovascular Age-Related Macular Degeneration. Ophthalmol. Sci. 2021, 1, 100076. [Google Scholar] [CrossRef] [PubMed]
- Pereira, D.S.; Maturi, R.K.; Akita, K.; Mahesh, V.; Bhisitkul, R.B.; Nishihata, T.; Sakota, E.; Ali, Y.; Nakamura, E.; Bezwada, P.; et al. Clinical proof of concept for anti-FGF2 therapy in exudative age-related macular degeneration (nAMD): Phase 2 trials in treatment-naive and anti-VEGF pretreated patients. Eye 2024, 38, 1140–1148. [Google Scholar] [CrossRef] [PubMed]
- Ng, E.W.; Shima, D.T.; Calias, P.; Cunningham, E.T., Jr.; Guyer, D.R.; Adamis, A.P. Pegaptanib, a targeted anti-VEGF aptamer for ocular vascular disease. Nat. Rev. Drug Discov. 2006, 5, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Apte, R.S. Pegaptanib sodium for the treatment of age-related macular degeneration. Expert Opin. Pharmacother. 2008, 9, 499–508. [Google Scholar] [CrossRef]
- Doggrell, S.A. Pegaptanib: The first antiangiogenic agent approved for neovascular macular degeneration. Expert. Opin. Pharmacother. 2005, 6, 1421–1423. [Google Scholar] [CrossRef]
- Zhu, Q.; Ziemssen, F.; Henke-Fahle, S.; Tatar, O.; Szurman, P.; Aisenbrey, S.; Schneiderhan-Marra, N.; Xu, X. Tubingen Bevacizumab Study G, Grisanti S: Vitreous levels of bevacizumab and vascular endothelial growth factor-A in patients with choroidal neovascularization. Ophthalmology 2008, 115, 1750–1755. [Google Scholar] [CrossRef]
- Moisseiev, E.; Waisbourd, M.; Ben-Artsi, E.; Levinger, E.; Barak, A.; Daniels, T.; Csaky, K.; Loewenstein, A.; Barequet, I.S. Pharmacokinetics of bevacizumab after topical and intravitreal administration in human eyes. Graefes Arch. Clin. Exp. Ophthalmol. 2014, 252, 331–337. [Google Scholar] [CrossRef] [PubMed]
- Avery, R.L.; Castellarin, A.A.; Steinle, N.C.; Dhoot, D.S.; Pieramici, D.J.; See, R.; Couvillion, S.; Nasir, M.A.; Rabena, M.D.; Maia, M.; et al. Systemic Pharmacokinetics and Pharmacodynamics of Intravitreal Aflibercept, Bevacizumab, and Ranibizumab. Retina 2017, 37, 1847–1858. [Google Scholar] [CrossRef] [PubMed]
- Veritti, D.; Sarao, V.; Gorni, G.; Lanzetta, P. Anti-VEGF Drugs Dynamics: Relevance for Clinical Practice. Pharmaceutics 2022, 14, 265. [Google Scholar] [CrossRef] [PubMed]
- Ghazi, N.G. Bevacizumab for neovascular age-related macular degeneration (ABC trial): Multicenter randomized double-masked study. Expert. Rev. Clin. Pharmacol. 2010, 3, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Ziemssen, F.; Sobolewska, B. Therapeutic efficacy of bevacizumab for age-related macular degeneration: What are the implications of CATT for routine management? Drugs Aging 2011, 28, 853–865. [Google Scholar] [CrossRef] [PubMed]
- Drzyzga, L.; Spiewak, D.; Dorecka, M.; Wygledowska-Promienska, D. Available Therapeutic Options for Corneal Neovascularization: A Review. Int. J. Mol. Sci. 2024, 25, 5479. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Lu, T.; Tuomi, L.; Jumbe, N.; Lu, J.; Eppler, S.; Kuebler, P.; Damico-Beyer, L.A.; Joshi, A. Pharmacokinetics of ranibizumab in patients with neovascular age-related macular degeneration: A population approach. Investig. Ophthalmol. Vis. Sci. 2013, 54, 1616–1624. [Google Scholar] [CrossRef] [PubMed]
- Bakri, S.J.; Snyder, M.R.; Reid, J.M.; Pulido, J.S.; Ezzat, M.K.; Singh, R.J. Pharmacokinetics of intravitreal ranibizumab (Lucentis). Ophthalmology 2007, 114, 2179–2182. [Google Scholar] [CrossRef]
- Krohne, T.U.; Liu, Z.; Holz, F.G.; Meyer, C.H. Intraocular pharmacokinetics of ranibizumab following a single intravitreal injection in humans. Am. J. Ophthalmol. 2012, 154, 682–686.e2. [Google Scholar] [CrossRef]
- Bressler, N.M.; Chang, T.S.; Suñer, I.J.; Fine, J.T.; Dolan, C.M.; Ward, J.; Ianchulev, T. Vision-related function after ranibizumab treatment by better- or worse-seeing eye: Clinical trial results from MARINA and ANCHOR. Ophthalmology 2010, 117, 747–756. [Google Scholar] [CrossRef] [PubMed]
- Regillo, C.D.; Brown, D.M.; Abraham, P.; Yue, H.; Ianchulev, T.; Schneider, S.; Shams, N. Randomized, double-masked, sham-controlled trial of ranibizumab for neovascular age-related macular degeneration: PIER Study year 1. Am. J. Ophthalmol. 2008, 145, 239–248. [Google Scholar] [CrossRef] [PubMed]
- Holz, F.G.; Amoaku, W.; Donate, J.; Guymer, R.H.; Kellner, U.; Schlingemann, R.O.; Weichselberger, A.; Staurenghi, G.; Group, S.S. Safety and efficacy of a flexible dosing regimen of ranibizumab in neovascular age-related macular degeneration: The SUSTAIN study. Ophthalmology 2011, 118, 663–671. [Google Scholar] [CrossRef] [PubMed]
- Stewart, M.W. The study of intravitreal drug pharmacokinetics: Does it matter? and if so, how? Expert Opin. Drug Metab. Toxicol. 2018, 14, 5–7. [Google Scholar] [CrossRef] [PubMed]
- Stewart, M.W. What are the half-lives of ranibizumab and aflibercept (VEGF Trap-eye) in human eyes? Calculations with a mathematical model. Eye Rep. 2011, 1, e5. [Google Scholar] [CrossRef]
- Do, D.V.; Rhoades, W.; Nguyen, Q.D. Pharmacokinetic Study of Intravitreal Aflibercept in Humans with Neovascular Age-Related Macular Degeneration. Retina 2020, 40, 643–647. [Google Scholar] [CrossRef]
- Kherani, A.; Brunck, L.R.; Katz, T.A.; Galic, J. First-dose effects with intravitreal aflibercept in wet age-related macular degeneration: A post-hoc analysis of VIEW-1 and VIEW-2 phase 3 studies. Can. J. Ophthalmol. J. Can. D’ophtalmol. 2021, 56, 268–269. [Google Scholar] [CrossRef]
- Holz, F.G.; Dugel, P.U.; Weissgerber, G.; Hamilton, R.; Silva, R.; Bandello, F.; Larsen, M.; Weichselberger, A.; Wenzel, A.; Schmidt, A.; et al. Single-Chain Antibody Fragment VEGF Inhibitor RTH258 for Neovascular Age-Related Macular Degeneration: A Randomized Controlled Study. Ophthalmology 2016, 123, 1080–1089. [Google Scholar] [CrossRef] [PubMed]
- Ferro Desideri, L.; Traverso, C.E.; Nicolo, M.; Munk, M.R. Faricimab for the Treatment of Diabetic Macular Edema and Neovascular Age-Related Macular Degeneration. Pharmaceutics 2023, 15, 1413. [Google Scholar] [CrossRef]
- Vabysmo. 2022. Available online: http://www.ema.europa.eu/medicines/human/EPAR/vabysmo (accessed on 15 September 2022).
- Urbańska, K.; Szarpak, J.; Biel, N.; Woźniak, M.; Kawecka, W. Endophthalmitis—A rare but dangerous complication of intravitreal anti-VEGF injections. Ophthalmol. J. 2023, 8, 52–55. [Google Scholar] [CrossRef]
- Pereira, D.S.; Akita, K.; Bhisitkul, R.B.; Nishihata, T.; Ali, Y.; Nakamura, E.; Nakamura, Y. Safety and tolerability of intravitreal umedaptanib pegol (anti-FGF2) for neovascular age-related macular degeneration (nAMD): A phase 1, open-label study. Eye 2024, 38, 1149–1154. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Borrell, M.A.; Venerus, D.C.; Mieler, W.F.; Kang-Mieler, J.J. Characterization of Biodegradable Microsphere-Hydrogel Ocular Drug Delivery System for Controlled and Extended Release of Ranibizumab. Transl. Vis. Sci. Technol. 2019, 8, 12. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Tawakol, A.P.; Rudeen, K.M.; Mieler, W.F.; Kang-Mieler, J.J. Treatment Efficacy and Biocompatibility of a Biodegradable Aflibercept-Loaded Microsphere-Hydrogel Drug Delivery System. Transl. Vis. Sci. Technol. 2020, 9, 13. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Hong, H.K.; Song, J.S.; Jeong, S.I.; Chung, J.Y.; Woo, S.J.; Park, K.D. Intravitreal injectable hydrogel rods with long-acting bevacizumab delivery to the retina. Acta Biomater. 2023, 171, 273–288. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Lau, L.C.; Lo, A.C.; Chau, Y. Injectable Chemically Crosslinked Hydrogel for the Controlled Release of Bevacizumab in Vitreous: A 6-Month In Vivo Study. Transl. Vis. Sci. Technol. 2015, 4, 5. [Google Scholar] [CrossRef] [PubMed]
- Lynch, C.R.; Kondiah, P.P.D.; Choonara, Y.E.; du Toit, L.C.; Ally, N.; Pillay, V. Hydrogel Biomaterials for Application in Ocular Drug Delivery. Front. Bioeng. Biotechnol. 2020, 8, 228. [Google Scholar] [CrossRef] [PubMed]
- Timmons, K.; Heckmann, L.C.; Ren, Y.; Gunderson, I.; Makkouk, F.; Berger, B.B. Ranibizumab Injection (Susvimo) Implant Septum Dislodgement in a Patient With Neovascular Age-Related Macular Degeneration. JAMA Ophthalmol. 2022, 140, 832. [Google Scholar] [CrossRef]
- Campochiaro, P.A.; Avery, R.; Brown, D.M.; Heier, J.S.; Ho, A.C.; Huddleston, S.M.; Jaffe, G.J.; Khanani, A.M.; Pakola, S.; Pieramici, D.J.; et al. Gene therapy for neovascular age-related macular degeneration by subretinal delivery of RGX-314: A phase 1/2a dose-escalation study. Lancet 2024, 403, 1563–1573. [Google Scholar] [CrossRef]
- Gelfman, C.M.; Grishanin, R.; Bender, K.O.; Nguyen, A.; Greengard, J.; Sharma, P.; Nieves, J.; Kiss, S.; Gasmi, M. Comprehensive Preclinical Assessment of ADVM-022, an Intravitreal Anti-VEGF Gene Therapy for the Treatment of Neovascular AMD and Diabetic Macular Edema. J. Ocul. Pharmacol. Ther. 2021, 37, 181–190. [Google Scholar] [CrossRef]
- Khanani, A.M.; Thomas, M.J.; Aziz, A.A.; Weng, C.Y.; Danzig, C.J.; Yiu, G.; Kiss, S.; Waheed, N.K.; Kaiser, P.K. Review of gene therapies for age-related macular degeneration. Eye 2022, 36, 303–311. [Google Scholar] [CrossRef]
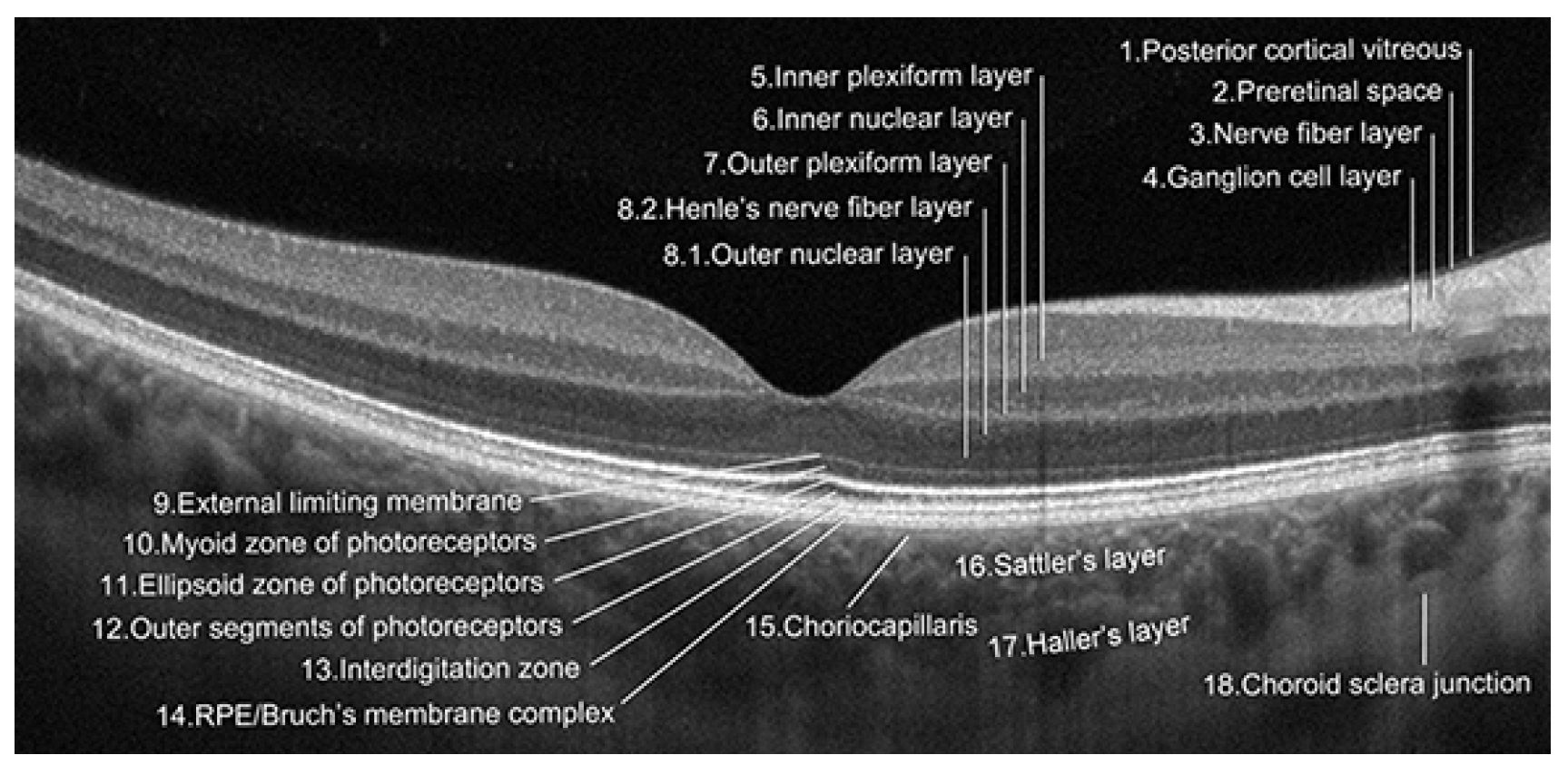


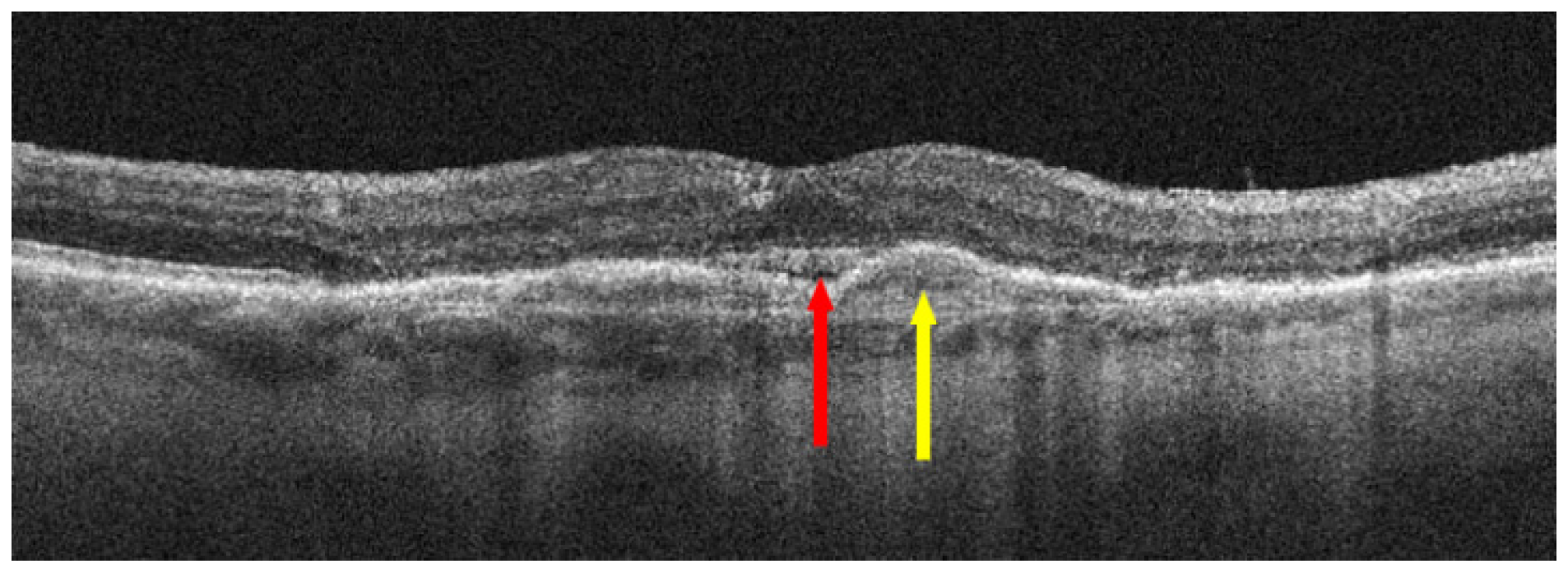
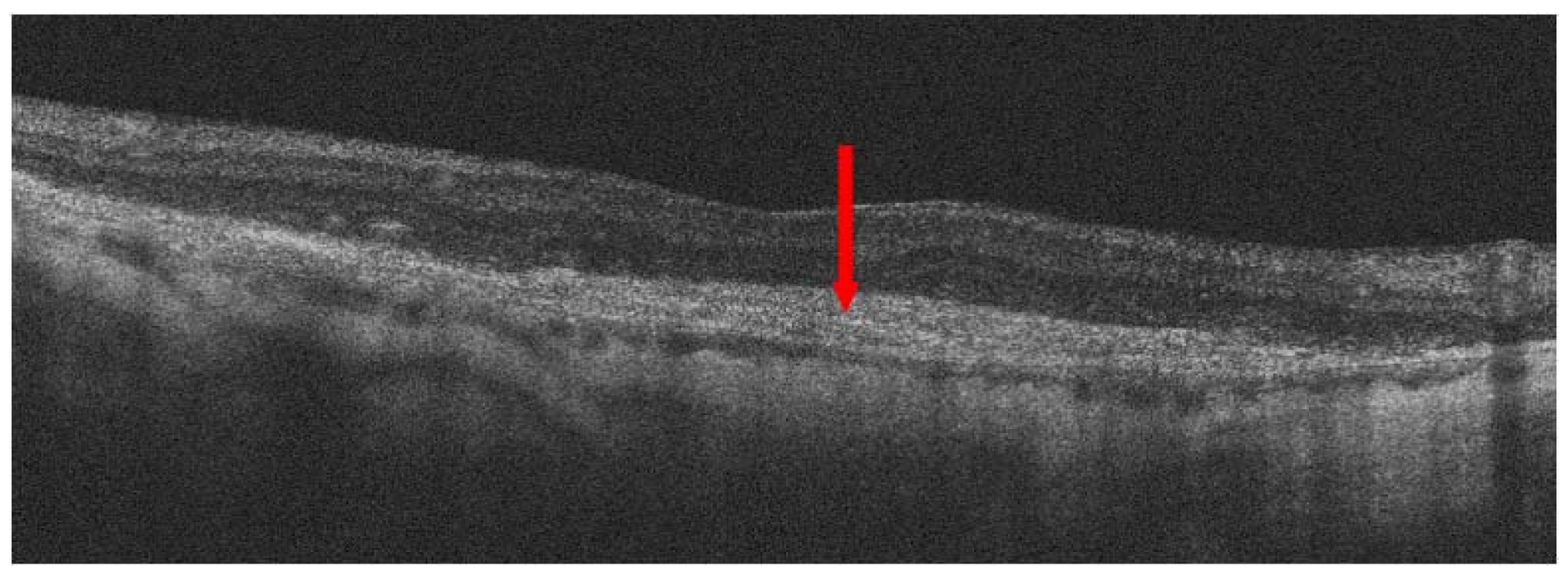
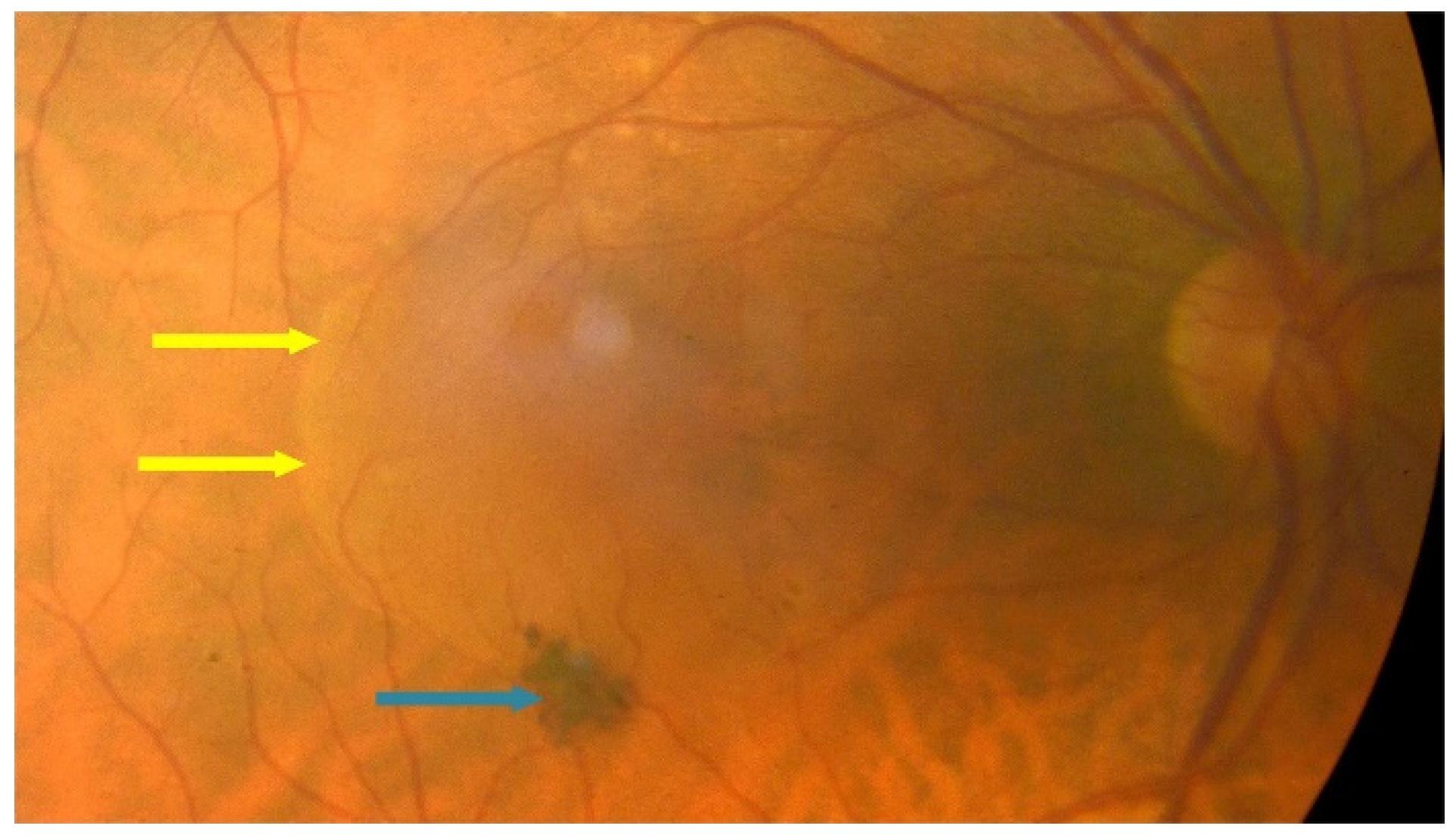
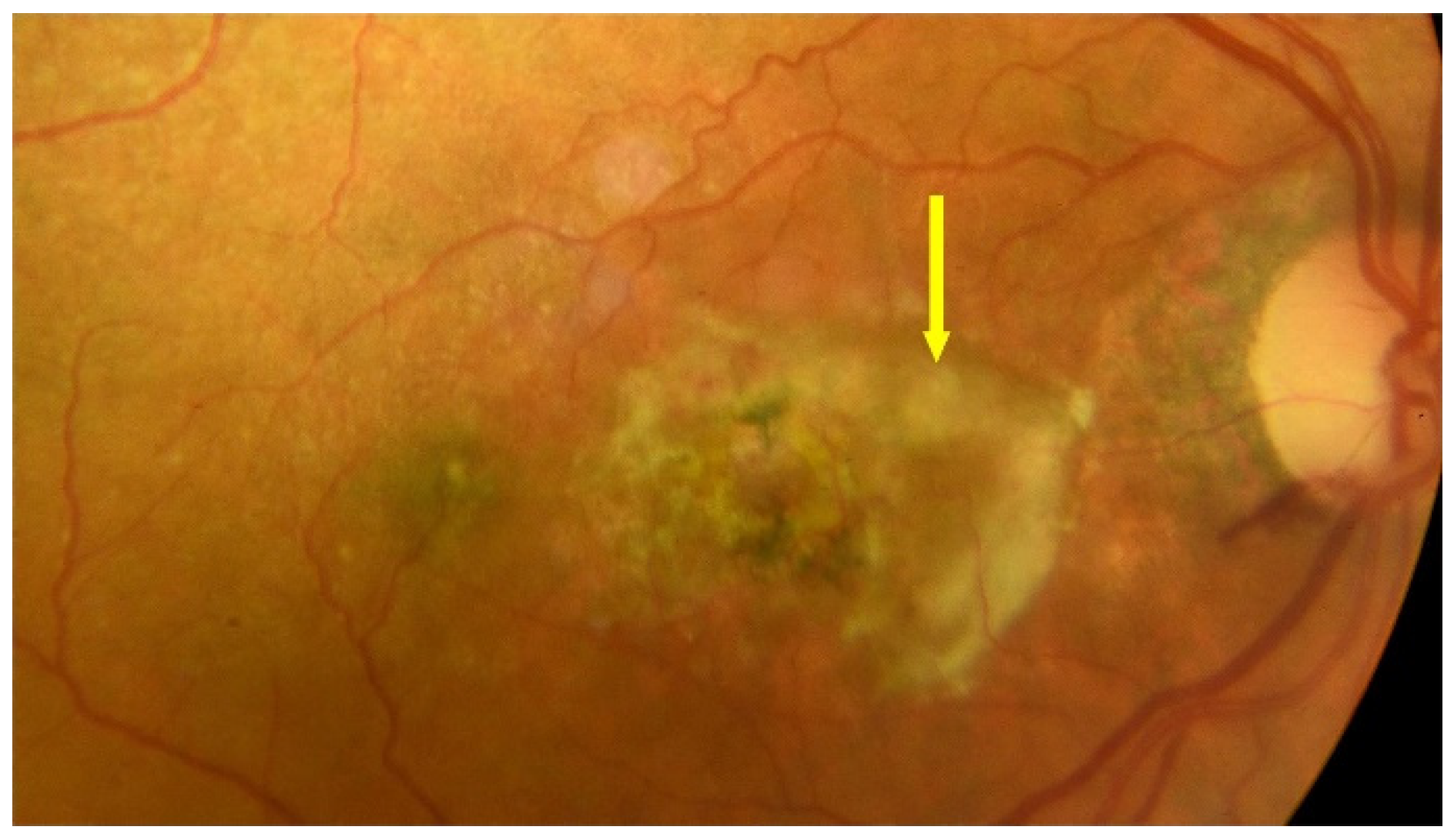
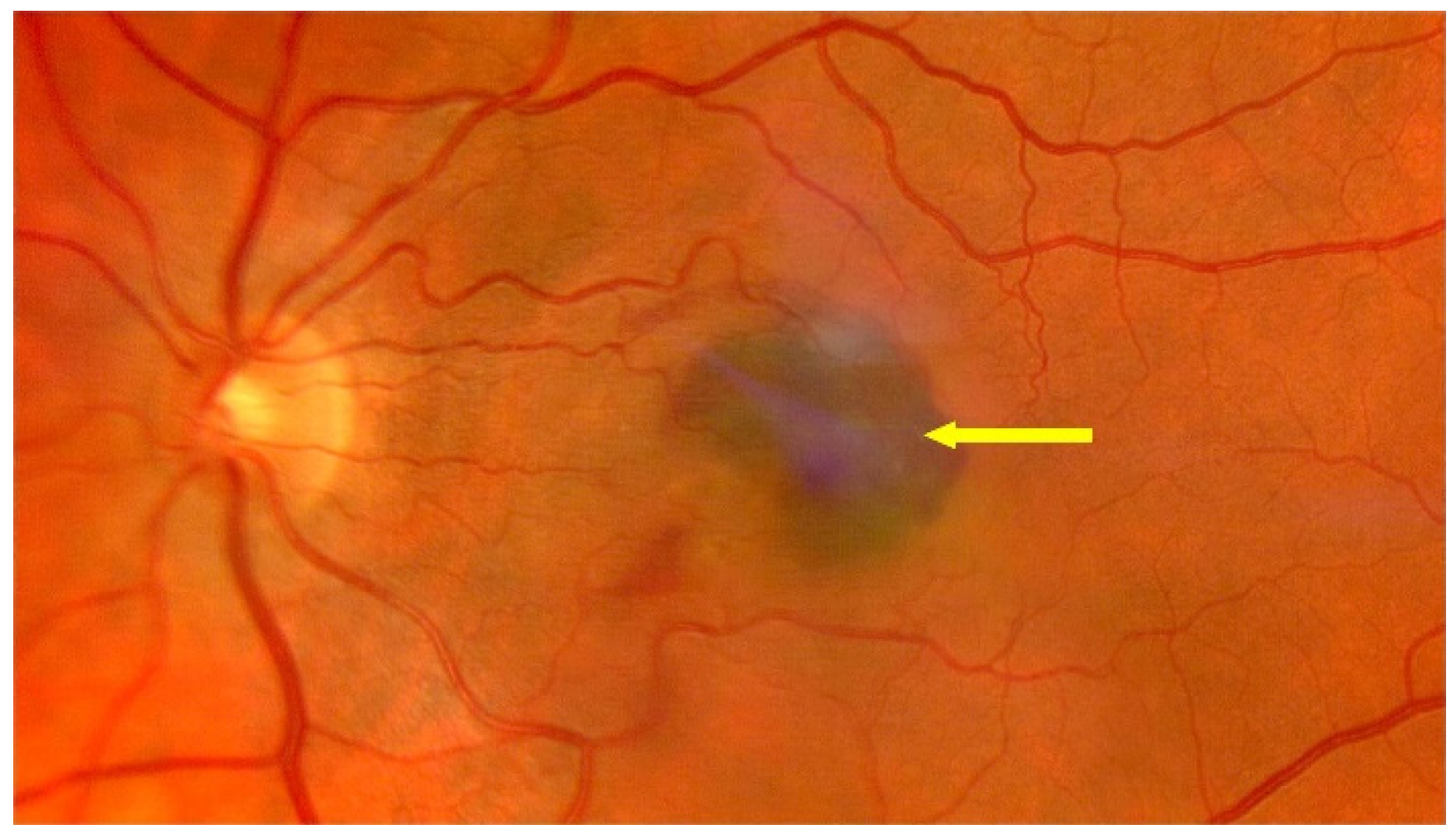

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name/Gene ID | Description | Location | Aliases | MIM |
|---|---|---|---|---|
| 3075CFH ID: 3075 | complement factor H [Homo sapiens (human)] | Chromosome 1, NC_000001.11 (196652043..196747504) | AHUS1, AMBP1, ARMD4, ARMS1L3, FH, FHL1, HF, HF1, HF2, HUS, CFH | 134370 |
| 5654HTRA1 ID: 5654 | HtrA serine peptidase 1 [Homo sapiens (human)] | Chromosome 10, NC_000010.11 (122461553..122514907) | ARMD7, CADASIL2, CARASIL, HtrA, L56, ORF480, PRSS11 | 602194 |
| 387715ARMS2 ID: 387715 | age-related maculopathy susceptibility 2 [Homo sapiens (human)] | Chromosome 10, NC_000010.11 (122454653..122457352) | ARMD8 | 611313 |
| 7422VEGFA ID: 7422 | vascular endothelial growth factor A [Homo sapiens (human)] | Chromosome 6, NC_000006.12 (43770211..43786487) | L-VEGF, MVCD1, VEGF, VPF | 192240 |
| 718C3 ID: 718 | complement C3 [Homo sapiens (human)] | Chromosome 19, NC_000019.10 (6677704..6720650, complement) | AHUS5, ARMD9, ASPa, C3b, CPAMD1, HEL-S-62p, C3 | 120700 |
| 348APOE ID: 348 | apolipoprotein E [Homo sapiens (human)] | Chromosome 19, NC_000019.10 (44905796..44909393) | AD2, APO-E, ApoE4, LDLCQ5, LPG | 107741 |
| 1401CRP ID: 1401 | C-reactive protein [Homo sapiens (human)] | Chromosome 1, NC_000001.11 (159712289..159714589, complement) | PTX1 | 123260 |
| 5176SERPINF1 ID: 5176 | serpin family F member 1 [Homo sapiens (human)] | Chromosome 17, NC_000017.11 (1762060..1777565) | EPC-1, OI12, OI6, PEDF, PIG35 | 172860 |
| 1524CX3CR1 ID: 1524 | C-X3-C motif chemokine receptor 1 [Homo sapiens (human)] | Chromosome 3, NC_000003.12 (39263494..39292966, complement) | CCRL1, CMKBRL1, CMKDR1, GPR13, GPRV28, V28 | 601470 |
| 3791KDR ID: 3791 | kinase insert domain receptor [Homo sapiens (human)] | Chromosome 4, NC_000004.12 (55078481..55125595, complement) | CD309, FLK1, VEGFR, VEGFR2 | 191306 |
| 3576CXCL8 ID: 3576 | C-X-C motif chemokine ligand 8 [Homo sapiens (human)] | Chromosome 4, NC_000004.12 (73740569..73743716) | GCP-1, GCP1, IL8, LECT, LUCT, LYNAP, MDNCF, MONAP, NAF, NAP-1, NAP1, SCYB8 | 146930 |
| 6499SKIC2 ID: 6499 | SKI2 subunit of superkiller complex [Homo sapiens (human)] | Chromosome 6, NC_000006.12 (31959175..31969751) | 170A, DDX13, HLP, SKI2, SKI2W, SKIV2, SKIV2L, SKIV2L1, THES2 | 600478 |
| 710SERPING1 ID: 710 | serpin family G member 1 [Homo sapiens (human)] | Chromosome 11, NC_000011.10 (57597685..57614848) | C1IN, C1INH, C1NH, HAE1, HAE2 | 606860 |
| 1295COL8A1 ID: 1295 | collagen type VIII alpha 1 chain [Homo sapiens (human)] | Chromosome 3, NC_000003.12 (99638594..99799217) | C3orf7 | 120251 |
| 20296Ccl2 ID: 20296 | C-C motif chemokine ligand 2 [Mus musculus (house mouse)] | Chromosome 11, NC_000077.7 (81926403..81928278) | HC11, JE, MCAF, MCP-1, SMC-CF, Scya2, Sigje | |
| CFB ID: 629 | complement factor B [Homo sapiens (human)] | Chromosome 6, NC_000006.12 (31946095..31952084) | AHUS4, ARMD14, BF, BFD, CFABD, FB, FBI12, GBG, H2-Bf, PBF2, CFB | 138470 |
| CFI ID: 3426 | complement factor I [Homo sapiens (human)] | Chromosome 4, NC_000004.12 (109730982..109801999, complement) | AHUS3, ARMD13, C3BINA, C3b-INA, FI, IF, KAF | 217030 |
| C2 ID: 717 | complement C2 [Homo sapiens (human)] | Chromosome 6, NC_000006.12 (31897783..31945672) | ARMD14, CO2 | 613927 |
| FLT1 ID: 2321 | fms related receptor tyrosine kinase 1 [Homo sapiens (human)] | Chromosome 13, NC_000013.11 (28300346..28495128, complement) | FLT, FLT-1, VEGFR-1, VEGFR1 | 165070 |
| CETP ID: 1071 | cholesteryl ester transfer protein [Homo sapiens (human)] | Chromosome 16, NC_000016.10 (56961950..56983845) | BPIFF, HDLCQ10 | 118470 |
| TLR3 ID: 7098 | toll like receptor 3 [Homo sapiens (human)] | Chromosome 4, NC_000004.12 (186069156..186088073) | CD283, IIAE2, IMD83 | 603029 |
| CFHR3 ID: 10878 | complement factor H related 3 [Homo sapiens (human)] | Chromosome 1, NC_000001.11 (196774840..196795407) | CFHL3, DOWN16, FHR-3, FHR3, HLF4 | 605336 |
| Ahr ID: 11622 | aryl-hydrocarbon receptor [Mus musculus (house mouse)] | Chromosome 12, NC_000078.7 (35547978..35584988, complement) | Ah, Ahhe, In, bHLHe76, Ahr | |
| PGF ID: 5228 | placental growth factor [Homo sapiens (human)] | Chromosome 14, NC_000014.9 (74941830..74955764, complement) | D12S1900L, PIGF, PLGF, PLGF-2, SHGC-10760, PGF | 601121 |
| CD36 ID: 948 | CD36 molecule (CD36 blood group) [Homo sapiens (human)] | Chromosome 7, NC_000007.14 (80602207..80679274) | BDPLT10, CHDS7, FAT, GP3B, GP4, GPIV, PASIV, SCARB3 | 173510 |
| THBS1 ID: 7057 | thrombospondin 1 [Homo sapiens (human)] | Chromosome 15, NC_000015.10 (39581079..39599466) | THBS, THBS-1, TSP, TSP-1, TSP1 | 188060 |
| RORA ID: 6095 | RAR related orphan receptor A [Homo sapiens (human)] | Chromosome 15, NC_000015.10 (60488284..61229302, complement) | IDDECA, NR1F1, ROR1, ROR2, ROR3, RORa1, RORalpha, RZR-ALPHA, RZRA | 600825 |
| TOMM40 ID: 10452 | translocase of outer mitochondrial membrane 40 [Homo sapiens (human)] | Chromosome 19, NC_000019.10 (44891254..44903689) | C19orf1, D19S1177E, PER-EC1, PEREC1, TOM40 | 608061 |
| CFP ID: 5199 | complement factor properdin [Homo sapiens (human)] | Chromosome X, NC_000023.11 (47623282..47630305, complement) | BFD, PFC, PFD, PROPERDIN | 300383 |
| C9 ID: 735 | complement C9 [Homo sapiens (human)] | Chromosome 5, NC_000005.10 (39284140..39364495, complement) | ARMD15D, C9 | 120940 |
| TGFB1 ID: 7040 | transforming growth factor beta 1 [Homo sapiens (human)] | Chromosome 19, NC_000019.10 (41330323..41353922, complement) | CED, DPD1, IBDIMDE, LAP, TGF-beta1, TGFB | 190180 |
| CXCL12 ID: 6387 | C-X-C motif chemokine ligand 12 [Homo sapiens (human)] | Chromosome 10, NC_000010.11 (44370165..44385097, complement) | IRH, PBSF, SCYB12, SDF1, TLSF, TPAR1 | 600835 |
| TIMP3 ID: 7078 | TIMP metallopeptidase inhibitor 3 [Homo sapiens (human)] | Chromosome 22, NC_000022.11 (32801705..32863041) | HSMRK222, K222, K222TA2, SFD | 188826 |
| ELN ID: 2006 | elastin [Homo sapiens (human)] | Chromosome 7, NC_000007.14 (74028173..74069907) | ADCL1, SVAS, WBS, WS | 130160 |
| CXCR3 ID: 2833 | C-X-C motif chemokine receptor 3 [Homo sapiens (human)] | Chromosome X, NC_000023.11 (71615919..71618511, complement) | CD182, CD183, CKR-L2, CMKAR3, GPR9, IP10-R, Mig-R, MigR | 300574 |
| ACAD10 ID: 80724 | acyl-CoA dehydrogenase family member 10 [Homo sapiens (human)] | Chromosome 12, NC_000012.12 (111686053..111757099) | 611181 | |
| MIR4513 ID: 100616183 | microRNA 4513 [Homo sapiens (human)] | Chromosome 15, NC_000015.10 (74788672..74788757, complement) |
| Name of the Study | Date | Investigated Drug | Number of Participants | Results | Ocular Adverse Events |
|---|---|---|---|---|---|
| OAKS [27] | 2018–2020 (24 months) | pegcetacoplan | 637 | % GA reduction in treated vs. sham Monthly: 21% (p = 0.0528) | 1.6% |
| DERBY [27] | 2018–2020 (24 months) | pegcetacoplan | 621 | % GA reduction in treated vs. sham Monthly: 12% (p = 0.0528) | 1.3% |
| GATHER1 [28] | 2016–2019 (18 months) | avacincaptad pegol | 286 | % GA reduction in treated vs. sham 2 mg: 27.4% (p = 0.0072) 4 mg: 27.8% (p = 0.0051) | ≥2% |
| GATHER2 [29] | 2020–2021 (12 months) | avacincaptad pegol | 448 | % GA reduction in treated vs. sham 2 mg:14% (p = 0.0064) | 49% |
| BEACON [30] | 2014–2018 (30 months) | brimonidine | 310 | % GA reduction in treated vs. sham 400 μg: 10% (p = 0.033) | 62.3% |
| Name of the Study | Date | Investigated Drug | Number of Participants | Results | Ocular Adverse Events |
|---|---|---|---|---|---|
| VISION-1 [45] | 2001–2002 54 weeks | pegaptanib | 1190 | In the pegaptanib 0.3 mg group, 80% achieved the primary endpoint of <15 ETDRS charts (Early Treatment Diabetic Retinopathy Study) letters lost, 47% maintained VA, and 20% gained ≥15 letters of vision | Serious ocular adverse events: 0.16% endophthalmitis and 0.08% retinal detachment |
| ABC [46] | 2006–2007 | bevacizumab | 131 | In the bevacizumab group, 21 (32%) patients achieved 15 or more letters above baseline VA compared with two (3%) in the standard treatment group (p < 0.001). | Serious ocular adverse events associated with bevacizumab were uncommon. Rates of adverse events of intraocular inflammation graded as ≥1 |
| CATT [47] | 2008–2012 | bevacizumab ranibizumab | 1208 | VA was similar for both drugs (bevacizumab-ranibizumab difference, −1.4 letters; 95% confidence interval–CI, −3.7 to 0.8; p = 0.21); the mean gain was greater for monthly than for as needed treatment (difference, −2.4 letters; 95% CI, −4.8 to −0.1; p = 0.046) | 24.1% bevacizumab 19.0% ranibizumab |
| MARINA [48] | 2003 12 months | ranibizumab | 716 | VA improved by 15 or more letters in 24.8% of the 0.3-mg group and 33.8% of the 0.5-mg group, as compared with 5.0% of the sham-injection group (p < 0.001 for both doses) | 1.3% |
| ANCHOR [49] | 2003–2006 12 months | ranibizumab | 423 | VA improved 15 letters more at 12 months: 40% ranibizumab 0.5 mg, 36% Ranibizumab 0.3 mg, and 6% PDT (p < 0.0001) | 0.3 mg ranibizumab 1.3% 0.5 mg ranibizumab 2.9% |
| PIER [50] | 2004–2005 12 months | ranibizumab | 184 | Gaining at least 15 letters: 9.5% in the sham group, 11.7% ranibizumab 0.3 mg, and 13.1% ranibizumab 0.5 mg | The incidence of ocular adverse events was low. Ocular hemorrhage: sham 1.6% 0.3 mg ranibizumab 3.4% 0.5 mg ranibizumab 0% Macular edema Sham 1.6% 0.3 mg ranibizumab 1.7% 0.5 mg ranibizumab 0% |
| PrONTO [51] | 2004–2005 24 months | ranibizumab | 40 | VA improved by 11.1 letters (p < 0.001); the OCT-CRT decreased by 212 microm (p < 0.001) | There were no ocular adverse events attributable to the injection of ranibizumab |
| SUSTAIN [52] | 2006–2008 12 months | ranibizumab | 531 | Mean best-corrected VA increased from baseline to month 3 to reach +5.8 letters, decreased slightly from month 3 to 6, and remained stable from month 6 to 12, reaching +3.6 at month 12 | Serious ocular AE: 1.2% both 0.3 mg and 0.5 mg of ranibizmab |
| VIEW1 [53] | 2007–2011 12 months | alibercept | 1217 | Mean improvements from baseline in the ETDRS letter score for 0.5 mg ranibizmab 8.1 letters, 0.5 aflibercept 6.9 letters, 2 mg aflibercept every month 10.9 letters, 2 mg aflibercept every 2 months 7.9 letters, and 2 mg afibercept every month was significantly better (p < 0.01) than 0.5 mg ranibizumab | Serious AE: 3.3% ranibizumab and 1.0% alibercept. The most frequent ocular AEs were conjunctival hemorrhage, macular degeneration, eye pain, vitreous detachment, and vitreous floaters |
| VIEW2 [54] | 2008–2011 | aflibercept | 1240 | Mean improvements from baseline in ETDRS letter score for 0.5 mg ranibizmab 9.4 letters, 2 mg aflibercept every month 7.6 letters, and 2 mg aflibercept every 2 months 8.9 letters | Serious AE: 3.1% ranibizumab and 2.9% alibercept. The most frequent ocular AEs were conjunctival hemorrhage, macular degeneration, eye pain, vitreous detachment, and vitreous floaters |
| PULSAR [55] | 24 months 2020–2021 | aflibercept | 1011 | Aflibercept 8q12 and 8q16 showed non-inferior best-corrected visual acuity (BCVA) gains versus aflibercept 2q8. Mean BCVA change from baseline: 8q12 +6·7 (standard deviation SD 12.6 letters) 8q16 +6·2 (SD 11.7 letters 2q8 +7·6 (SD 12.2 letters). | Ocular adverse events in the study eye was similar across groups: 39% aflibercept 8q12, 38% aflibercept 8q16, and 39% aflibercept 2q8 |
| HAWK [56,57] | 24 months 2014–2017 | brolucizumab | 1078 | Each brolucizumab arm demonstrated noninferiority to aflibercept in BCVA change from baseline, least squares LS mean: 6 mg brolucizumab +6.6 letters 3 mg broucizumab +6.1 letters 2 mg aflibercept +6.8 letters | Ocular adverse events: 2.2% brolucizumab 6 mg 0.3% brolucizumab 3 mg 0% aflibercept 2 mg Thromboembolic events: 1.1% brolucizumab 3 mg, 1.4% brolucizumab 6 mg, 0.3% aflibercept 2 mg |
| HARRIER [56,57] | 2015–2017 24months | brolucizumab | 739 | Brolucizumab arm demonstrated noninferiority to aflibercept in BCVA: 6 mg brolucizumab +6.4 letters 2 mg aflibercept +3.7 letters | Ocular adverse events occurring in ≥3%, thromboembolic events: 1.6% brolucizumab 6 mg 0.5% aflibercept 2 mg |
| TENAYA [58,59] | 2019–2022 112 weeks | faricimab | 671 | BCVA change from baseline with faricimab was non-inferior to aflibercept, adjusted mean change: 6 mg faricimab +3.7 letters 2 mg aflibercept +3.3 letters | 36.3% faricmabvs 38.1% aflibercept |
| LUCERNE [58,59] | 2019–2022 112 weeks | faricimab | 658 | BCVA change from baseline with faricimab was non-inferior to aflibercept: 6 mg faricimab 5.0 letters 2 mg aflibercept 5.2 letters | 40.2% faricimab 36.2 alibercept |
| TOFU [60] | 2019–2021 4 months | umedaptanib pegol (anti-FGF2) | 86 | Umedaptanib pegol alone or in combination with aflibercept did not improve BCVA, which suggests that umedaptanib pegol is effective in preventing the disease progression | Subjects with at least one Ocular a treatment-emergent adverse event (TEAE): Arm 1: 57.1% Arm 2: 65.5% Arm 3: 34.5% |
| RAMEN Extension TOFU Trial [60] | 2020–2021 4 months | umedaptanib pegol (anti-FGF2) | 22 | The RAMEN study confirmed the cessation of disease progression | No drug-related adverse events were reported Ocular adverse events were related to the intravitreal injection procedure |
| TEMPURA [60] | 2021–2022 4 months | umedaptanib pegol (anti-FGF2) | 5 | In the TEMPURA study, naïve nAMD patients showed improvement and no further macular degeneration | No drug-related adverse events were reported; 1 ocular adverse event was reported—1 subretinal hemorrhage—20% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Śpiewak, D.; Drzyzga, Ł.; Dorecka, M.; Wyględowska-Promieńska, D. Summary of the Therapeutic Options for Patients with Dry and Neovascular AMD. J. Clin. Med. 2024, 13, 4227. https://doi.org/10.3390/jcm13144227
Śpiewak D, Drzyzga Ł, Dorecka M, Wyględowska-Promieńska D. Summary of the Therapeutic Options for Patients with Dry and Neovascular AMD. Journal of Clinical Medicine. 2024; 13(14):4227. https://doi.org/10.3390/jcm13144227
Chicago/Turabian StyleŚpiewak, Dorota, Łukasz Drzyzga, Mariola Dorecka, and Dorota Wyględowska-Promieńska. 2024. "Summary of the Therapeutic Options for Patients with Dry and Neovascular AMD" Journal of Clinical Medicine 13, no. 14: 4227. https://doi.org/10.3390/jcm13144227