
Platelet Metabolites as Candidate Biomarkers in Sepsis Diagnosis and Management Using the Proposed Explainable Artificial Intelligence Approach

 , , , and
, , , and
Abstract
:1. Introduction
Literature Review
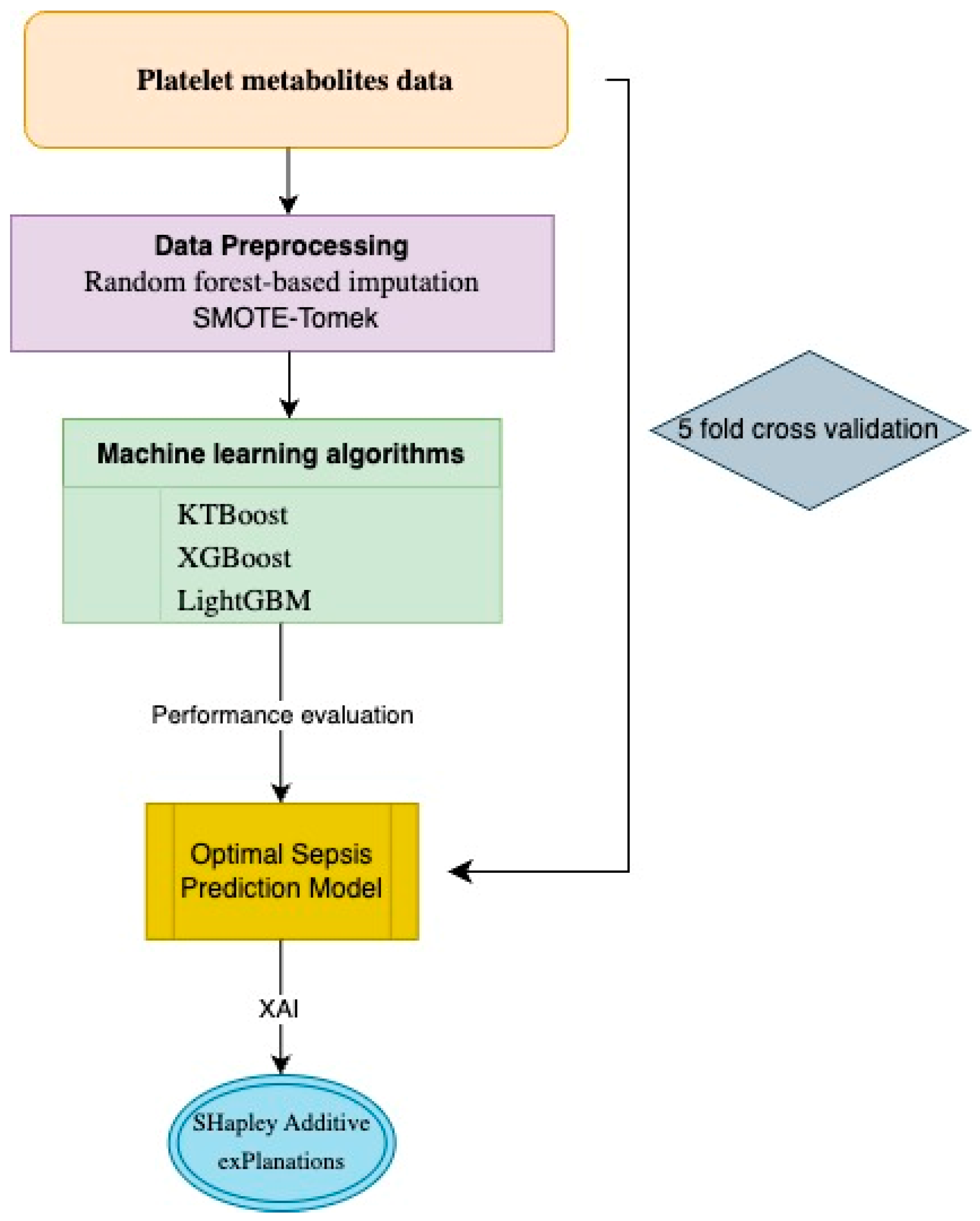
2. Materials and Methods
2.1. Study Design and Participants
Inclusion and Exclusion Criteria
- −
- Sepsis Group:
- Suspected/proven infection (Organ dysfunction was identified as an acute change SOFA score ≥ 2 points consequent to the infection.) [1].
- Any two of the four systemic inflammatory response criteria present in the ED [46].
- Must be at least eighteen years old.
- At least 2.0 mmol/L of lactate.
- Enrollment started two hours after a quantitative resuscitation protocol was started.
- −
- Control Group:
- Admitted to the ED.
- No medical problems requiring chronic treatment impacting platelet function (e.g., aspirin, P2Y12 inhibitors).
- −
- Exclusion Criteria:
- Apart from sepsis as the primary diagnosis.
- Declared the status of “Do Not Resuscitate.”
- Transfer with previous sepsis treatment from another hospital.
- Cardiopulmonary resuscitation before enrollment.
2.2. Sample Collection
- Centrifugation at 200× g for 6 min at room temperature to separate platelet-rich plasma.
- A further centrifugation at 4500× g for 5 min at 4 °C to pellet the platelets.
2.3. Data Preprocessing and Machine Learning Modeling
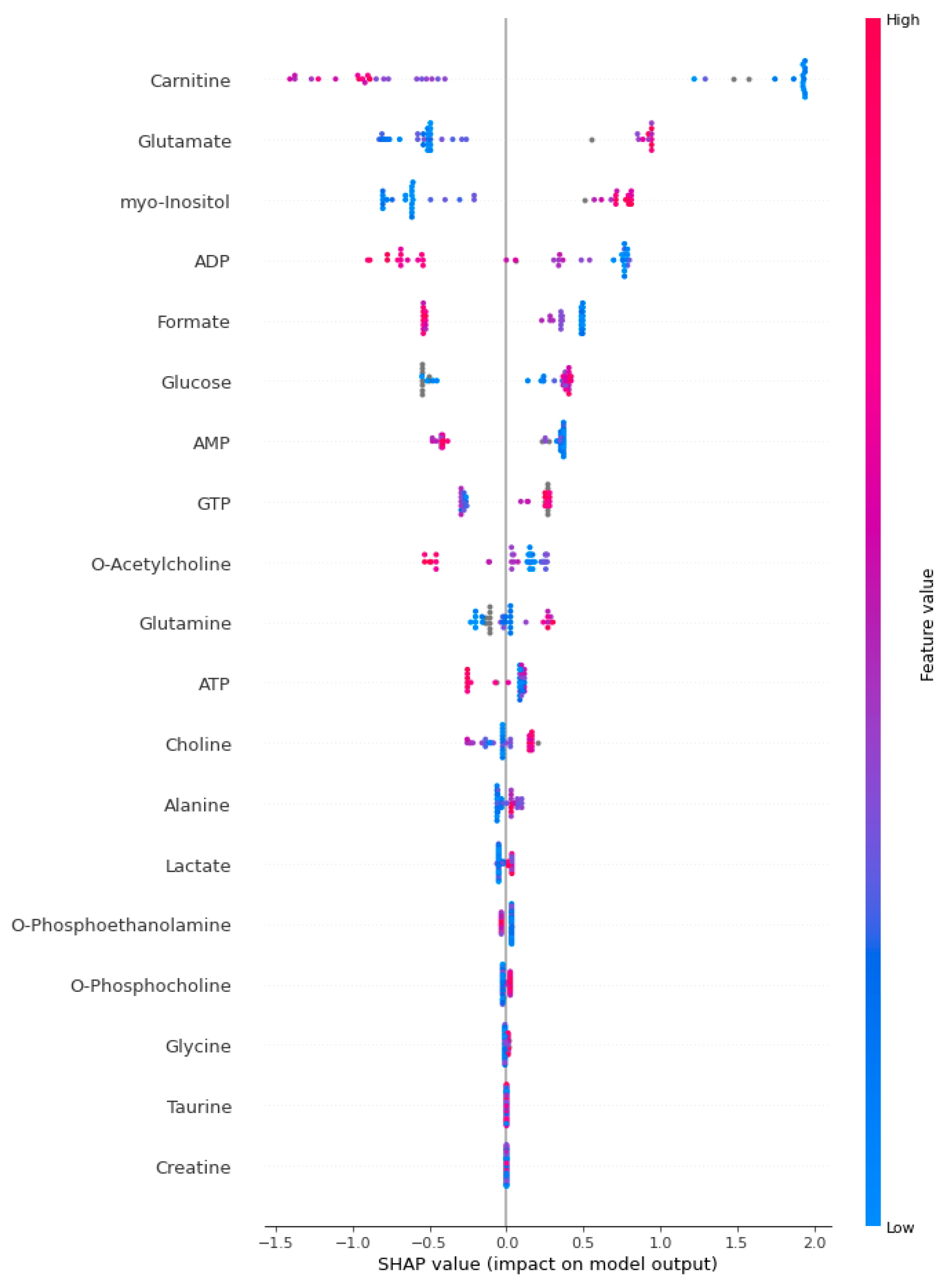
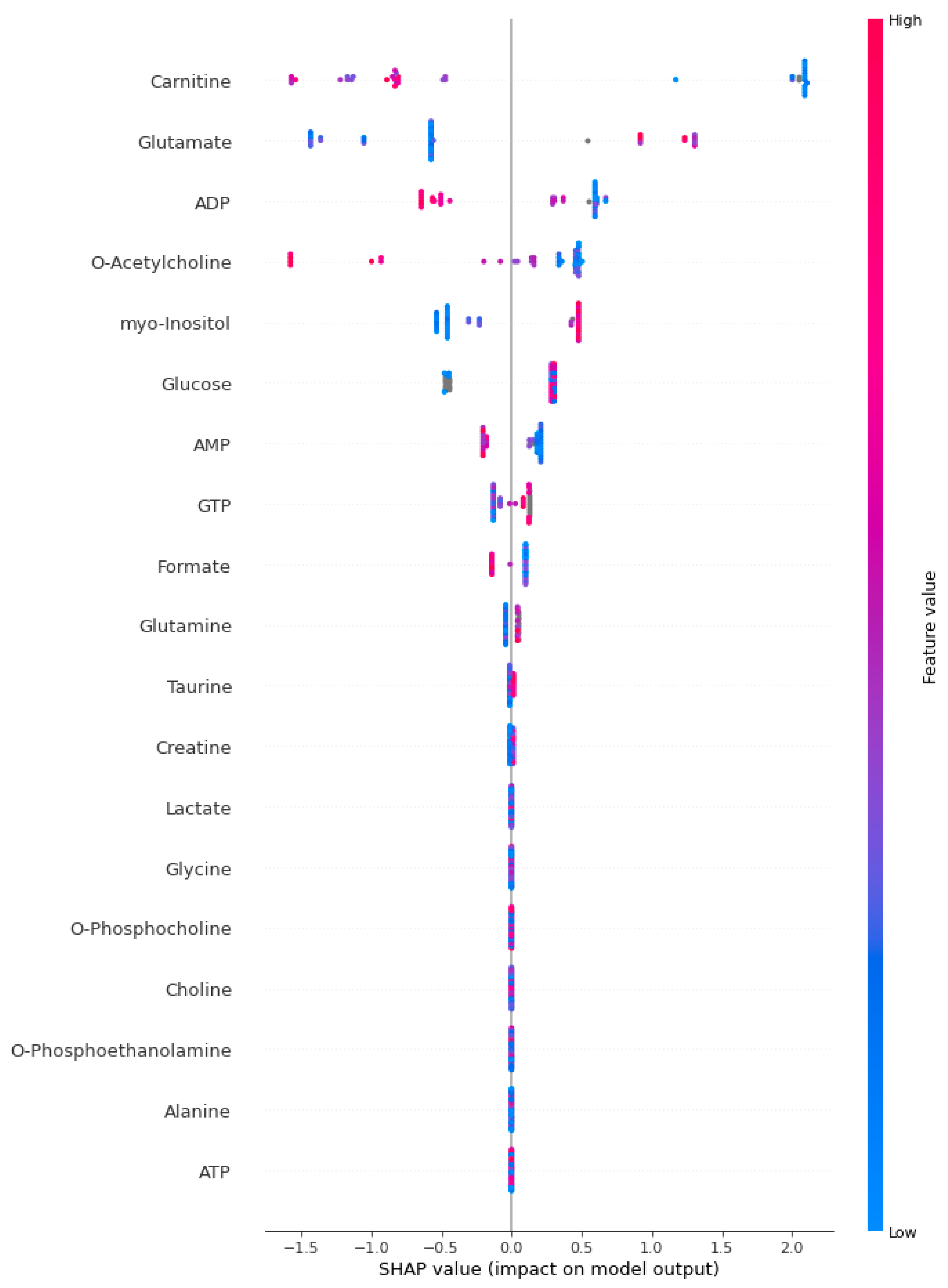
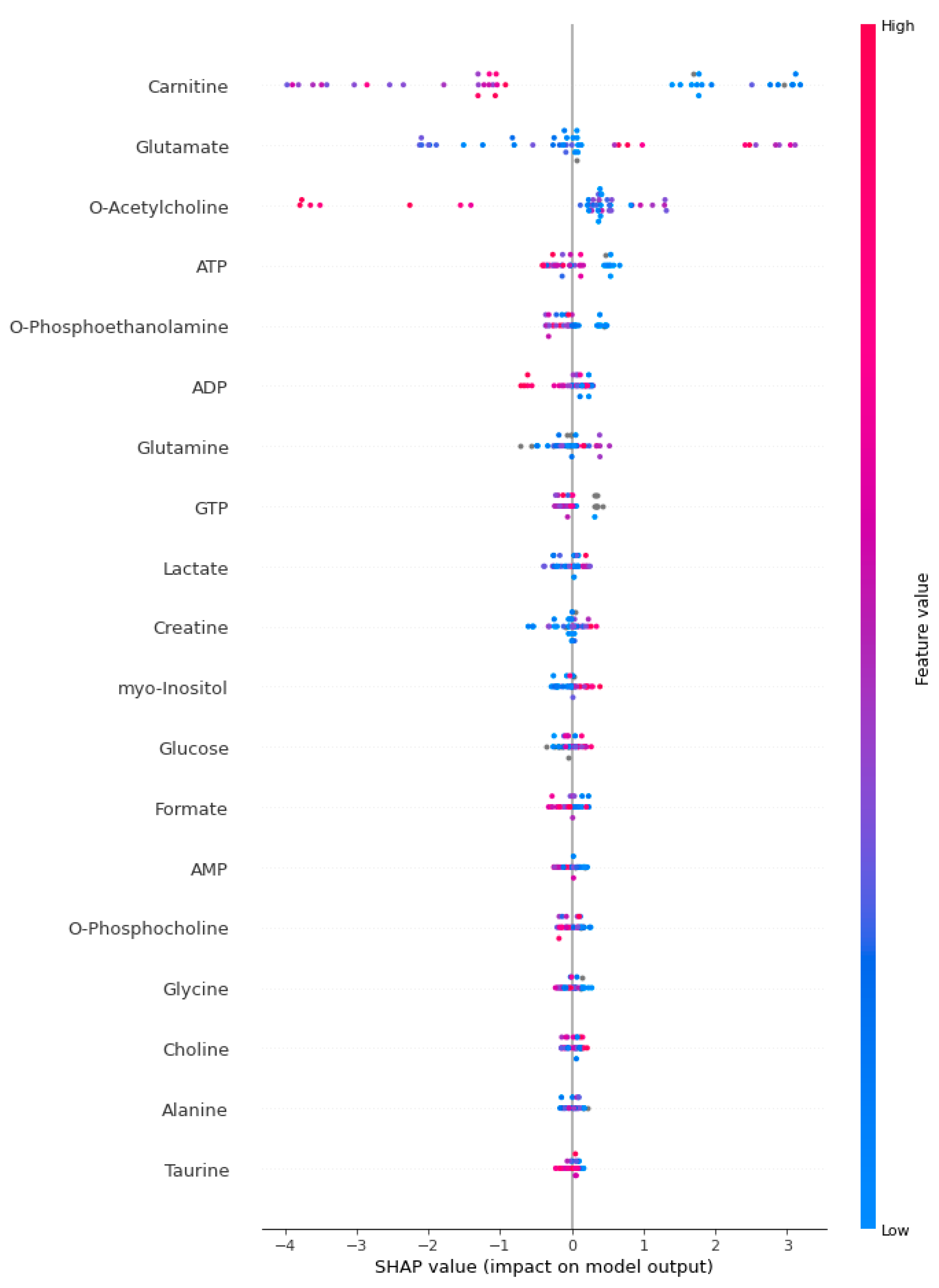
2.4. Explainable Artificial Intelligence Approach and Interpretation of Sepsis Biomarkers and Prediction
2.5. Biostatistical Analysis
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Singer, M.; Deutschman, C.S.; Seymour, C.W.; Shankar-Hari, M.; Annane, D.; Bauer, M.; Bellomo, R.; Bernard, G.R.; Chiche, J.-D.; Coopersmith, C.M. The third international consensus definitions for sepsis and septic shock (Sepsis-3). JAMA 2016, 315, 801–810. [Google Scholar] [CrossRef] [PubMed]
- Angus, D.C.; Van der Poll, T. Severe sepsis and septic shock. N. Engl. J. Med. 2013, 369, 840–851. [Google Scholar] [CrossRef] [PubMed]
- Rivers, E.; Nguyen, B.; Havstad, S.; Ressler, J.; Muzzin, A.; Knoblich, B.; Peterson, E.; Tomlanovich, M. Early goal-directed therapy in the treatment of severe sepsis and septic shock. N. Engl. J. Med. 2001, 345, 1368–1377. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, C.; Chen, M. Metabolomics-based study of potential biomarkers of sepsis. Sci. Rep. 2023, 13, 585. [Google Scholar]
- Oeser, C.; Pond, M.; Butcher, P.; Bedford Russell, A.; Henneke, P.; Laing, K.; Planche, T.; Heath, P.T.; Harris, K.J.P.O. PCR for the detection of pathogens in neonatal early onset sepsis. PLoS ONE 2020, 15, e0226817. [Google Scholar] [CrossRef]
- Tziolos, N.; Giamarellos-Bourboulis, E.J. Contemporary approaches to the rapid molecular diagnosis of sepsis. Expert Rev. Mol. Diagn. 2016, 16, 1201–1207. [Google Scholar] [CrossRef]
- Leli, C.; Cenci, E.; Cardaccia, A.; Moretti, A.; D’Alò, F.; Pagliochini, R.; Barcaccia, M.; Farinelli, S.; Vento, S.; Bistoni, F.; et al. Rapid identification of bacterial and fungal pathogens from positive blood cultures by MALDI-TOF MS. Int. J. Med. Microbiol. 2013, 303, 205–209. [Google Scholar] [CrossRef]
- Beloborodova, N.V.; Olenin, A.Y.; Pautova, A.K. Metabolomic findings in sepsis as a damage of host-microbial metabolism integration. J. Crit. Care 2018, 43, 246–255. [Google Scholar] [CrossRef]
- Gawaz, M.; Langer, H.; May, A.E. Platelets in inflammation and atherogenesis. J. Clin. Investig. 2005, 115, 3378–3384. [Google Scholar] [CrossRef]
- Russwurm, S.; Vickers, J.; Meier-Hellmann, A.; Spangenberg, P.; Bredle, D.; Reinhart, K.; Lösche, W. Platelet and leukocyte activation correlate with the severity of septic organ dysfunction. Shock 2002, 17, 263–268. [Google Scholar] [CrossRef]
- Nicholson, J.K.; Lindon, J.C.; Holmes, E. ’Metabonomics‘: Understanding the metabolic responses of living systems to pathophysiological stimuli via multivariate statistical analysis of biological NMR spectroscopic data. Xenobiotica 1999, 29, 1181–1189. [Google Scholar] [CrossRef] [PubMed]
- McCann, M.R.; McHugh, C.E.; Kirby, M.; Jennaro, T.S.; Jones, A.E.; Stringer, K.A.; Puskarich, M.A. A multivariate metabolomics method for estimating platelet mitochondrial oxygen consumption rates in patients with sepsis. Metabolites 2020, 10, 139. [Google Scholar] [CrossRef]
- Wang, J.; Sun, Y.; Teng, S.; Li, K. Prediction of sepsis mortality using metabolite biomarkers in the blood: A meta-analysis of death-related pathways and prospective validation. BMC Med. 2020, 18, 83. [Google Scholar] [CrossRef]
- Chen, Q.; Liang, X.; Wu, T.; Jiang, J.; Jiang, Y.; Zhang, S.; Ruan, Y.; Zhang, H.; Zhang, C.; Chen, P. Integrative analysis of metabolomics and proteomics reveals amino acid metabolism disorder in sepsis. J. Transl. Med. 2022, 20, 123. [Google Scholar] [CrossRef] [PubMed]
- Parthasarathy, U.; Kuang, Y.; Thakur, G.; Hogan, J.D.; Wyche, T.P.; Norton, J.E.; Killough, J.R.; Sana, T.R.; Beakes, C.; Shyong, B.J.I. Distinct subsets of neutrophils crosstalk with cytokines and metabolites in patients with sepsis. Iscience 2023, 26, 105948. [Google Scholar] [CrossRef] [PubMed]
- McBride, M.A.; Owen, A.M.; Stothers, C.L.; Hernandez, A.; Luan, L.; Burelbach, K.R.; Patil, T.K.; Bohannon, J.K.; Sherwood, E.R.; Patil, N.K. The metabolic basis of immune dysfunction following sepsis and trauma. Front. Immunol. 2020, 11, 1043. [Google Scholar] [CrossRef] [PubMed]
- Mickiewicz, B.; Vogel, H.J.; Wong, H.R.; Winston, B.W. Metabolomics as a novel approach for early diagnosis of pediatric septic shock and its mortality. Am. J. Respir. Crit. Care Med. 2013, 187, 967–976. [Google Scholar] [CrossRef]
- Schmerler, D.; Neugebauer, S.; Ludewig, K.; Bremer-Streck, S.; Brunkhorst, F.M.; Kiehntopf, M. Targeted metabolomics for discrimination of systemic inflammatory disorders in critically ill patients. J. Lipid Res. 2012, 53, 1369–1375. [Google Scholar] [CrossRef]
- Zhang, Y.; Weng, Y.; Lund, J. Applications of explainable artificial intelligence in diagnosis and surgery. Diagnostics 2022, 12, 237. [Google Scholar] [CrossRef]
- Takeyama, N.; Takagi, D.; Matsuo, N.; Kitazawa, Y.; Tanaka, T. Altered hepatic fatty acid metabolism in endotoxicosis: Effect of L-carnitine on survival. Am. J. Physiol. Endocrinol. Metab. 1989, 256, E31–E38. [Google Scholar] [CrossRef]
- Winter, B.; Fiskum, G.; Gallo, L. Effects of L-carnitine on serum triglyceride and cytokine levels in rat models of cachexia and septic shock. Br. J. Cancer 1995, 72, 1173–1179. [Google Scholar] [CrossRef]
- Gibault, J.P.; Frey, A.; Guiraud, M.; Schirardin, H.; Bouletreau, P.; Bach, A.C. Effects of L-carnitine infusion on intralipid clearance and utilization. Study carried out in septic patients of an intensive care unit. J. Parenter. Enter. Nutr. 1988, 12, 29–34. [Google Scholar] [CrossRef]
- Puskarich, M.A.; Kline, J.A.; Krabill, V.; Claremont, H.; Jones, A.E. Preliminary safety and efficacy of L-carnitine infusion for the treatment of vasopressor-dependent septic shock: A randomized control trial. J. Parenter. Enter. Nutr. 2014, 38, 736–743. [Google Scholar] [CrossRef] [PubMed]
- Nanni, G.; Pittiruti, M.; Giovannini, I.; Boldrini, G.; Ronconi, P.; Castagneto, M. Plasma carnitine levels and urinary carnitine excretion during sepsis. J. Parenter. Enter. Nutr. 1985, 9, 483–490. [Google Scholar] [CrossRef] [PubMed]
- Jennaro, T.S.; Puskarich, M.A.; Flott, T.L.; McLellan, L.A.; Jones, A.E.; Pai, M.P.; Stringer, K.A. Kidney function as a key driver of the pharmacokinetic response to high-dose L-carnitine in septic shock. Pharmacother. J. Hum. Pharmacol. Drug Ther. 2023, 43, 1240–1250. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, R.; Sailasuta, N.; Hurd, R.; Nelson, S.; Pelletier, D. Evidence of elevated glutamate in multiple sclerosis using magnetic resonance spectroscopy at 3 T. Brain 2005, 128, 1016–1025. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.-W.; Kwon, J.-I.; Woo, C.-W.; Heo, H.; Kim, K.W.; Woo, D.-C.; Kim, J.K.; Lee, D.-H. In vivo measurement of neurochemical abnormalities in the hippocampus in a rat model of cuprizone-induced demyelination. Diagnostics 2020, 11, 45. [Google Scholar] [CrossRef]
- Piani, D.; Frei, K.; Do, K.Q.; Cuénod, M.; Fontana, A. Murine brain macrophages induce NMDA receptor mediated neurotoxicity in vitro by secreting glutamate. Neurosci. Lett. 1991, 133, 159–162. [Google Scholar] [CrossRef]
- Wang, R.; Reddy, P.H. Role of glutamate and NMDA receptors in Alzheimer’s disease. J. Alzheimer’s Dis. 2017, 57, 1041–1048. [Google Scholar] [CrossRef]
- Vishnoi, S.; Raisuddin, S.; Parvez, S. Glutamate excitotoxicity and oxidative stress in epilepsy: Modulatory role of melatonin. J. Environ. Pathol. Toxicol. Oncol. 2016, 35, 365–374. [Google Scholar] [CrossRef]
- Majdi, A.; Mahmoudi, J.; Sadigh-Eteghad, S.; Farhoudi, M.; Shotorbani, S.S. The interplay of microRNAs and post-ischemic glutamate excitotoxicity: An emergent research field in stroke medicine. Neurol. Sci. 2016, 37, 1765–1771. [Google Scholar] [CrossRef] [PubMed]
- Tehse, J.; Taghibiglou, C. The overlooked aspect of excitotoxicity: Glutamate-independent excitotoxicity in traumatic brain injuries. Eur. J. Neurosci. 2019, 49, 1157–1170. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Xu, M.; Xie, J.; Liu, T.; Xu, X.; Gao, W.; Li, Z.; Bai, X.; Liu, X. Inhibition of ferroptosis attenuates glutamate excitotoxicity and nuclear autophagy in a CLP septic mouse model. Shock 2022, 57, 694–702. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Yu, C.; Long, Y. Myo-inositol oxygenase (MIOX) accelerated inflammation in the model of infection-induced cardiac dysfunction by NLRP3 inflammasome. Immun. Inflamm. Dis. 2023, 11, e829. [Google Scholar] [CrossRef]
- Wang, L.-X.; Tang, L.-M.; Pan, G.-Q.; Yan, C.-X.; Lu, L.; Liang, Y.-F. Evaluation of correlation between NF-κB mediated PAI-1 gene and sepsis. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 30–36. [Google Scholar]
- Lin, Y.; Rong, J.; Zhang, Z. Silent existence of eosinopenia in sepsis: A systematic review and meta-analysis. BMC Infect. Dis. 2021, 21, 471. [Google Scholar] [CrossRef]
- Howlett, A.; Plakkal, N. Inositol in preterm infants at risk for or having respiratory distress syndrome. Cochrane Database Syst. Rev. 2019, CD000366. [Google Scholar] [CrossRef]
- Leite, R.O.; de Souza, P.O.; Haas, C.B.; da Silveira, F.; Mohr, K.; Bertoni, A.P.S.; Soares, M.; Azambuja, J.H.; Prá, M.D.; da Cruz, L.L.P. ATPergic signaling disruption in human sepsis as a potential source of biomarkers for clinical use. Clin. Exp. Med. 2023, 23, 3651–3662. [Google Scholar] [CrossRef]
- Santos, S.S.; Brunialti, M.K.C.; Soriano, F.G.; Szabo, C.; Salomão, R. Repurposing of clinically approved poly-(ADP-ribose) polymerase inhibitors for the therapy of sepsis. Shock 2021, 56, 901–909. [Google Scholar] [CrossRef]
- Zhang, L.; Yao, J.; Wang, X.; Li, H.; Liu, T.; Zhao, W. Poly (ADP-ribose) synthetase inhibitor has a heart protective effect in a rat model of experimental sepsis. Int. J. Clin. Exp. Pathol. 2015, 8, 9824. [Google Scholar]
- Lin, Z.Y.; Xu, P.B.; Yan, S.K.; Meng, H.B.; Yang, G.J.; Dai, W.X.; Liu, X.R.; Li, J.B.; Deng, X.M.; Zhang, W.D. A metabonomic approach to early prognostic evaluation of experimental sepsis by 1H NMR and pattern recognition. NMR Biomed. Int. J. Devoted Dev. Appl. Magn. Reson. Vivo 2009, 22, 601–608. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Qi, Y.; Wang, F.; Zhang, B.; Jianguo, T. Circulating sepsis-related metabolite sphinganine could protect against intestinal damage during sepsis. Front. Immunol. 2023, 14, 1151728. [Google Scholar] [CrossRef] [PubMed]
- Basoglu, A.; Baspinar, N.; Tenori, L.; Hu, X.; Yildiz, R. NMR based metabolomics evaluation in neonatal calves with acute diarrhea and suspected sepsis: A new approach for biomarker/S. Metabolomics 2014, 4, 1. [Google Scholar]
- Nyamundanda, G.; Gormley, I.C.; Fan, Y.; Gallagher, W.M.; Brennan, L. MetSizeR: Selecting the optimal sample size for metabolomic studies using an analysis based approach. BMC Bioinform. 2013, 14, 338. [Google Scholar] [CrossRef]
- McHugh, C. Estimating Platelet Mitochondrial Function in Patients with Sepsis. Metabolomics Workbench. 2024. Available online: https://www.metabolomicsworkbench.org/data/DRCCMetadata.php?Mode=Study&StudyID=ST001294 (accessed on 14 July 2024).
- Bone, R.C.; Balk, R.A.; Cerra, F.B.; Dellinger, R.P.; Fein, A.M.; Knaus, W.A.; Schein, R.M.; Sibbald, W.J.J.C. Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. Chest 1992, 101, 1644–1655. [Google Scholar] [CrossRef]
- Puskarich, M.A.; Kline, J.A.; Watts, J.A.; Shirey, K.; Hosler, J.; Jones, A.E. Early alterations in platelet mitochondrial function are associated with survival and organ failure in patients with septic shock. J. Crit. Care 2016, 31, 63–67. [Google Scholar] [CrossRef] [PubMed]
- Paglia, G.; Magnúsdóttir, M.; Thorlacius, S.; Sigurjónsson, Ó.E.; Guðmundsson, S.; Palsson, B.Ø.; Thiele, I. Intracellular metabolite profiling of platelets: Evaluation of extraction processes and chromatographic strategies. J. Chromatogr. B 2012, 898, 111–120. [Google Scholar] [CrossRef] [PubMed]
- McHugh, C.E.; Flott, T.L.; Schooff, C.R.; Smiley, Z.; Puskarich, M.A.; Myers, D.D.; Younger, J.G.; Jones, A.E.; Stringer, K.A. Rapid, reproducible, quantifiable NMR metabolomics: Methanol and methanol: Chloroform precipitation for removal of macromolecules in serum and whole blood. Metabolites 2018, 8, 93. [Google Scholar] [CrossRef]
- Blondel, C.; Khelalfa, F.; Reynaud, S.; Fauvelle, F.; Raveton, M. Effect of organochlorine pesticides exposure on the maize root metabolome assessed using high-resolution magic-angle spinning 1H NMR spectroscopy. Environ. Pollut. 2016, 214, 539–548. [Google Scholar] [CrossRef]
- Diserens, G.; Vermathen, M.; Precht, C.; Broskey, N.; Boesch, C.; Amati, F.; Dufour, J.-F.; Vermathen, P. Separation of small metabolites and lipids in spectra from biopsies by diffusion-weighted HR-MAS NMR: A feasibility study. Analyst 2015, 140, 272–279. [Google Scholar] [CrossRef]
- Hong, Y.S.; Coen, M.; Rhode, C.M.; Reily, M.D.; Robertson, D.G.; Holmes, E.; Lindon, J.C.; Nicholson, J.K. Chemical shift calibration of 1H MAS NMR liver tissue spectra exemplified using a study of glycine protection of galactosamine toxicity. Magn. Reson. Chem. 2009, 47, S47–S53. [Google Scholar] [CrossRef]
- Jang, W.G.; Park, J.Y.; Lee, J.; Bang, E.; Kim, S.R.; Lee, E.K.; Yun, H.J.; Kang, C.M.; Hwang, G.S. Investigation of relative metabolic changes in the organs and plasma of rats exposed to X-ray radiation using HR-MAS 1H NMR and solution 1H NMR. NMR Biomed. 2016, 29, 507–518. [Google Scholar] [CrossRef] [PubMed]
- Emwas, A.-H.; Roy, R.; McKay, R.T.; Tenori, L.; Saccenti, E.; Gowda, G.N.; Raftery, D.; Alahmari, F.; Jaremko, L.; Jaremko, M.; et al. NMR spectroscopy for metabolomics research. Metabolites 2019, 9, 123. [Google Scholar] [CrossRef]
- Yang, F.; Chen, Y. Urinary phytoestrogens and the risk of uterine leiomyomata in US women. BMC Women’s Health 2023, 23, 261. [Google Scholar] [CrossRef] [PubMed]
- Swana, E.F.; Doorsamy, W.; Bokoro, P. Tomek link and SMOTE approaches for machine fault classification with an imbalanced dataset. Sensors 2022, 22, 3246. [Google Scholar] [CrossRef]
- Chandra, W.; Suprihatin, B.; Resti, Y. Median-KNN Regressor-SMOTE-Tomek links for handling missing and imbalanced data in air quality prediction. Symmetry 2023, 15, 887. [Google Scholar] [CrossRef]
- Sain, H.; Purnami, S.W. Combine sampling support vector machine for imbalanced data classification. Procedia Comput. Sci. 2015, 72, 59–66. [Google Scholar] [CrossRef]
- Fushiki, T. Estimation of prediction error by using K-fold cross-validation. Stat. Comput. 2011, 21, 137–146. [Google Scholar] [CrossRef]
- Zhou, W.; Yan, Z.; Zhang, L. A comparative study of 11 non-linear regression models highlighting autoencoder, DBN, and SVR, enhanced by SHAP importance analysis in soybean branching prediction. Sci. Rep. 2024, 14, 5905. [Google Scholar] [CrossRef]
- Abbas, Z.; ur Rehman, M.; Tayara, H.; Zou, Q.; Chong, K.T. XGBoost framework with feature selection for the prediction of RNA N5-methylcytosine sites. Mol. Ther. 2023, 31, 2543–2551. [Google Scholar] [CrossRef]
- Yagin, F.H.; Al-Hashem, F.; Ahmad, I.; Ahmad, F.; Alkhateeb, A. Pilot-Study to Explore Metabolic Signature of Type 2 Diabetes: A Pipeline of Tree-Based Machine Learning and Bioinformatics Techniques for Biomarkers Discovery. Nutrients 2024, 16, 1537. [Google Scholar] [CrossRef] [PubMed]
- Sigrist, F. KTBoost: Combined kernel and tree boosting. Neural Process. Lett. 2021, 53, 1147–1160. [Google Scholar] [CrossRef]
- Lai, Y.; Lin, P.; Lin, F.; Chen, M.; Lin, C.; Lin, X.; Wu, L.; Zheng, M.; Chen, J. Identification of immune microenvironment subtypes and signature genes for Alzheimer’s disease diagnosis and risk prediction based on explainable machine learning. Front. Immunol. 2022, 13, 1046410. [Google Scholar] [CrossRef]
- Yagin, B.; Yagin, F.H.; Colak, C.; Inceoglu, F.; Kadry, S.; Kim, J. Cancer metastasis prediction and genomic biomarker identification through machine learning and eXplainable artificial intelligence in breast cancer research. Diagnostics 2023, 13, 3314. [Google Scholar] [CrossRef] [PubMed]
- Gozukara Bag, H.G.; Yagin, F.H.; Gormez, Y.; González, P.P.; Colak, C.; Gülü, M.; Badicu, G.; Ardigò, L.P. Estimation of obesity levels through the proposed predictive approach based on physical activity and nutritional habits. Diagnostics 2023, 13, 2949. [Google Scholar] [CrossRef] [PubMed]
- Angelov, P.P.; Soares, E.A.; Jiang, R.; Arnold, N.I.; Atkinson, P.M. Explainable artificial intelligence: An analytical review. Wiley Interdiscip. Rev. Data Min. Knowl. Discov. 2021, 11, e1424. [Google Scholar] [CrossRef]
- Minh, D.; Wang, H.X.; Li, Y.F.; Nguyen, T.N. Explainable artificial intelligence: A comprehensive review. Artif. Intell. Rev. 2022, 55, 1–66. [Google Scholar]
- Li, Z. Extracting spatial effects from machine learning model using local interpretation method: An example of SHAP and XGBoost. Comput. Environ. Urban Syst. 2022, 96, 101845. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, Z.; Luo, X.; Zhao, H. Diagnosis of Parkinson’s disease based on SHAP value feature selection. Biocybern. Biomed. Eng. 2022, 42, 856–869. [Google Scholar] [CrossRef]
- Wang, Q.; Wang, C.; Zhang, W.; Tao, Y.; Guo, J.; Liu, Y.; Liu, Z.; Liu, D.; Mei, J.; Chen, F. Identification of biomarkers related to sepsis diagnosis based on bioinformatics and machine learning and experimental verification. Front. Immunol. 2023, 14, 1087691. [Google Scholar] [CrossRef]
- Fleuren, L.M.; Klausch, T.L.; Zwager, C.L.; Schoonmade, L.J.; Guo, T.; Roggeveen, L.F.; Swart, E.L.; Girbes, A.R.; Thoral, P.; Ercole, A. Machine learning for the prediction of sepsis: A systematic review and meta-analysis of diagnostic test accuracy. Intensive Care Med. 2020, 46, 383–400. [Google Scholar] [CrossRef] [PubMed]
- Adams, R.; Henry, K.E.; Sridharan, A.; Soleimani, H.; Zhan, A.; Rawat, N.; Johnson, L.; Hager, D.N.; Cosgrove, S.E.; Markowski, A. Prospective, multi-site study of patient outcomes after implementation of the TREWS machine learning-based early warning system for sepsis. Nat. Med. 2022, 28, 1455–1460. [Google Scholar] [CrossRef]
- Antcliffe, D.B.; Burnham, K.L.; Al-Beidh, F.; Santhakumaran, S.; Brett, S.J.; Hinds, C.J.; Ashby, D.; Knight, J.C.; Gordon, A.C. Transcriptomic signatures in sepsis and a differential response to steroids. From the VANISH randomized trial. Am. J. Respir. Crit. Care Med. 2019, 199, 980–986. [Google Scholar]
- Koenig, R.; Kolte, A.; Ahlers, O.; Oswald, M.; Krauss, V.; Roell, D.; Sommerfeld, O.; Dimopoulos, G.; Tsangaris, I.; Antoniadou, E. Use of IFNγ/IL10 ratio for stratification of hydrocortisone therapy in patients with septic shock. Front. Immunol. 2021, 12, 607217. [Google Scholar] [CrossRef] [PubMed]
- Wong, H.R.; Cvijanovich, N.Z.; Anas, N.; Allen, G.L.; Thomas, N.J.; Bigham, M.T.; Weiss, S.L.; Fitzgerald, J.; Checchia, P.A.; Meyer, K. Developing a clinically feasible personalized medicine approach to pediatric septic shock. Am. J. Respir. Crit. Care Med. 2015, 191, 309–315. [Google Scholar] [CrossRef]
- Sinha, P.; Furfaro, D.; Cummings, M.J.; Abrams, D.; Delucchi, K.; Maddali, M.V.; He, J.; Thompson, A.; Murn, M.; Fountain, J. Latent class analysis reveals COVID-19–related acute respiratory distress syndrome subgroups with differential responses to corticosteroids. Am. J. Respir. Crit. Care Med. 2021, 204, 1274–1285. [Google Scholar]
- Shi, S.; Pan, X.; Zhang, L.; Wang, X.; Zhuang, Y.; Lin, X.; Shi, S.; Zheng, J.; Lin, W. An application based on bioinformatics and machine learning for risk prediction of sepsis at first clinical presentation using transcriptomic data. Front. Genet. 2022, 13, 979529. [Google Scholar]
- Rashid, A.; Anwary, A.R.; Al-Obeidat, F.; Brierley, J.; Uddin, M.; Alkhzaimi, H.; Sarpal, A.; Toufiq, M.; Malik, Z.A.; Kadwa, R. Application of a gene modular approach for clinical phenotype genotype association and sepsis prediction using machine learning in meningococcal sepsis. Inform. Med. Unlocked 2023, 41, 101293. [Google Scholar] [CrossRef]
- Biolo, G.; Toigo, G.; Ciocchi, B.; Situlin, R.; Iscra, F.; Gullo, A.; Guarnieri, G. Metabolic response to injury and sepsis: Changes in protein metabolism. Nutrition 1997, 13, 52–57. [Google Scholar] [CrossRef]
- Tang, B.; Almansa, R.; Bermejo-Martin, J.F.; Burnham, K.; Choi, A.M.; Davenport, E.E.; Ginsburg, G.; Henao, R.; Hinds, C.; Howrylak, J.A. A community approach to mortality prediction in sepsis via gene expression analysis. Nat. Commun. 2018, 9, 64. [Google Scholar]
- Cano-Gamez, E.; Burnham, K.L.; Goh, C.; Allcock, A.; Malick, Z.H.; Overend, L.; Kwok, A.; Smith, D.A.; Peters-Sengers, H.; Antcliffe, D. An immune dysfunction score for stratification of patients with acute infection based on whole-blood gene expression. Sci. Transl. Med. 2022, 14, eabq4433. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | Group | p-Value | ||
|---|---|---|---|---|
| Control | Sepsis | |||
| Gender * | Male | 8 (57.1) | 15 (60) | 0.862 |
| Female | 6 (42.9) | 10 (40) | ||
| Age ** | 44 (14) | 55 (17) | 0.056 | |
| Metabolite Name * | Group | p-Value | |
|---|---|---|---|
| Control | Sepsis | ||
| ADP | 41.95 (16.875) | 27.8 (25.8) | 0.058 |
| AMP | 8.75 (3.825) | 7 (3.8) | 0.429 |
| ATP | 57.2 (29.15) | 47.7 (36) | 0.183 |
| Alanine | 11.9 (5.5) | 12.2 (5.1) | 0.781 |
| Carnitine | 3.33 (1.075) | 1.9 (2) | 0.067 |
| Choline | 4.725 (1.9) | 4.1 (3.6) | 0.446 |
| Creatine | 3.25 (2.225) | 4.1 (2.82) | 0.379 |
| Formate | 19 (6.6) | 18 (5.7) | 0.693 |
| GTP | 16.05 (2.347) | 16.93 (5.3) | 0.507 |
| Glucose | 56.47 (17.65) | 56.47 (15.6) | 0.426 |
| Glutamate | 83.2 (19.225) | 85.7 (61.6) | 0.988 |
| Glutamine | 34.95 (8.68) | 36.83 (16.6) | 0.669 |
| Glycine | 12 (5.35) | 8.6 (6.9) | 0.183 |
| Lactate | 64.5 (19.85) | 65.6 (48.4) | 0.965 |
| O-acetylcholine | 3.95 (2.65) | 3.1 (1.9) | 0.027 |
| O-phosphocholine | 9.55 (4.85) | 6.6 (6.1) | 0.224 |
| O-phosphoethanolamine | 54.435 (17.9) | 38.2 (32.3) | 0.047 |
| Taurine | 578.3 (239.225) | 443.2 (344.5) | 0.141 |
| myo-Inositol | 22.9 (9.05) | 32.21 (29.4) | 0.377 |
| Metric/Model | KTBoost | XGBoost | LightGBM |
|---|---|---|---|
| Accuracy | 0.900 (0.817–0.983) | 0.860 (0.764–0.956) | 0.800 (0.689–0.911) |
| F1-score | 0.894 (0.808–0.979) | 0.851 (0.752–0.950) | 0.792 (0.679–0.904) |
| Sensitivity | 0.840 (0.639–0.955) | 0.800 (0.593–0.932) | 0.760 (0.549–0.906) |
| Specificity | 0.960 (0.796–0.999) | 0.920 (0.740–0.990) | 0.840 (0.639–0.955) |
| AUC | 0.943 (0.900–0.987) | 0.914 (0.860–0.968) | 0.861 (0.800–0.923) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yagin, F.H.; Aygun, U.; Algarni, A.; Colak, C.; Al-Hashem, F.; Ardigò, L.P. Platelet Metabolites as Candidate Biomarkers in Sepsis Diagnosis and Management Using the Proposed Explainable Artificial Intelligence Approach. J. Clin. Med. 2024, 13, 5002. https://doi.org/10.3390/jcm13175002
Yagin FH, Aygun U, Algarni A, Colak C, Al-Hashem F, Ardigò LP. Platelet Metabolites as Candidate Biomarkers in Sepsis Diagnosis and Management Using the Proposed Explainable Artificial Intelligence Approach. Journal of Clinical Medicine. 2024; 13(17):5002. https://doi.org/10.3390/jcm13175002
Chicago/Turabian StyleYagin, Fatma Hilal, Umran Aygun, Abdulmohsen Algarni, Cemil Colak, Fahaid Al-Hashem, and Luca Paolo Ardigò. 2024. "Platelet Metabolites as Candidate Biomarkers in Sepsis Diagnosis and Management Using the Proposed Explainable Artificial Intelligence Approach" Journal of Clinical Medicine 13, no. 17: 5002. https://doi.org/10.3390/jcm13175002