
Angiopoietin-like Proteins and Lipoprotein Lipase: The Waltz Partners That Govern Triglyceride-Rich Lipoprotein Metabolism? Impact on Atherogenesis, Dietary Interventions, and Emerging Therapies
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Lipoprotein Lipase Structure and Function
3. Overview of Main LPL Regulators
3.1. Apolipoproteins (Apo)
3.2. Angiopoietin-like Proteins (ANGPTLs)
3.3. CREBH
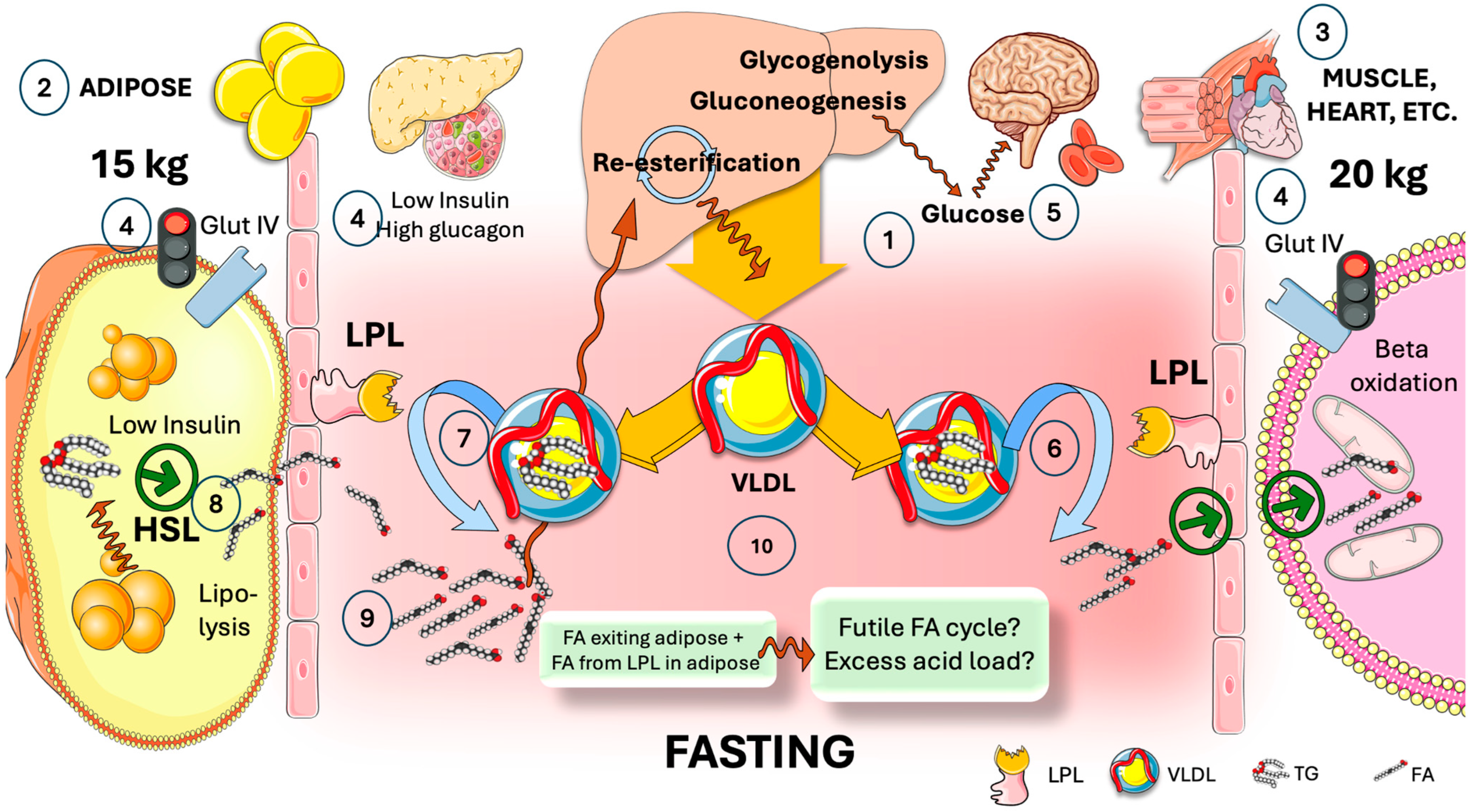
4. The Need for Tissue-Specific Regulation of LPL in the Fast-Fed Cycle
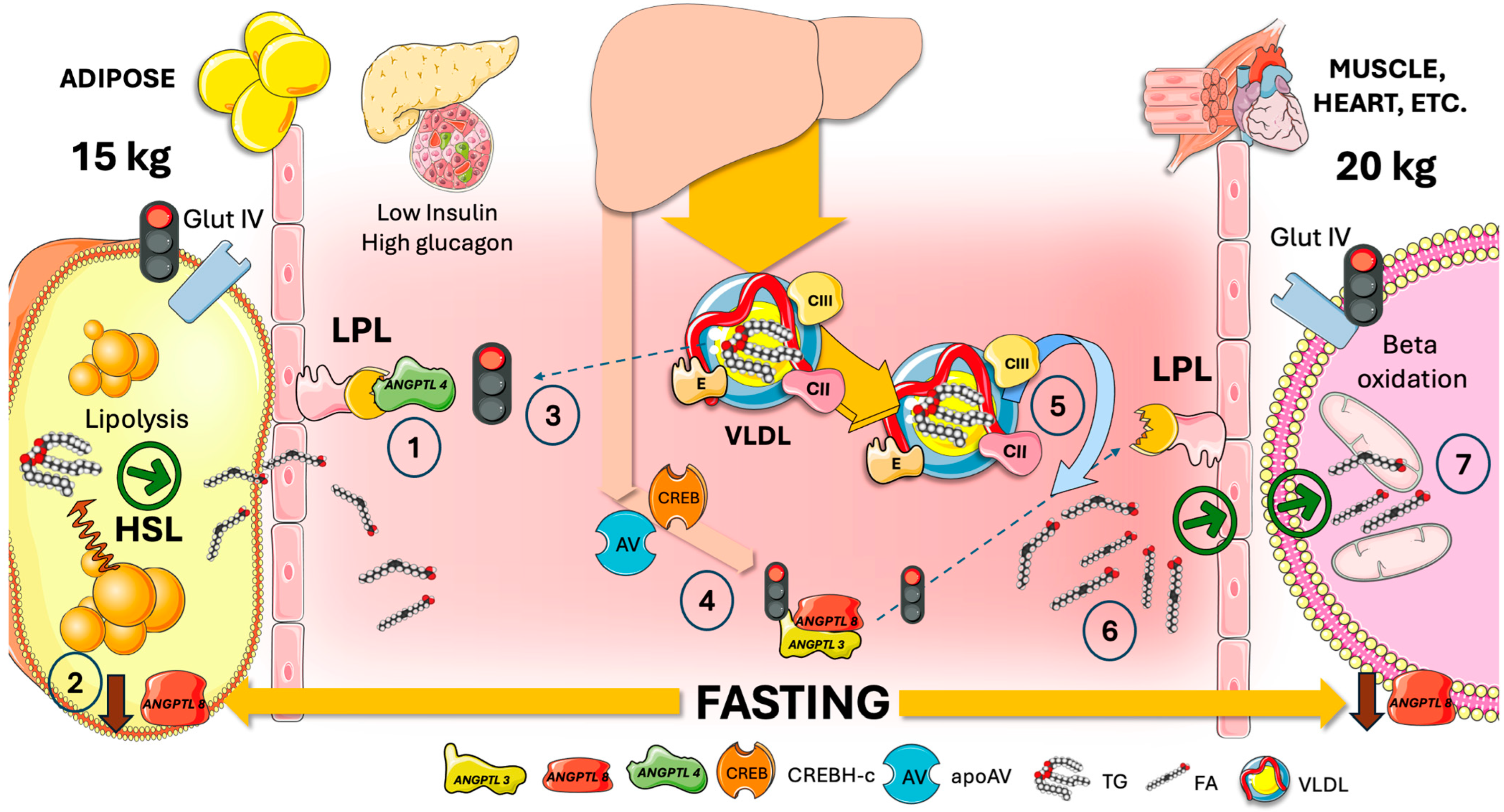
5. Adipocyte ANGPTL4 Diverts Most TRL during Fasting to Oxidative Tissues by Transiently Blocking Adipose Tissue LPL Activity
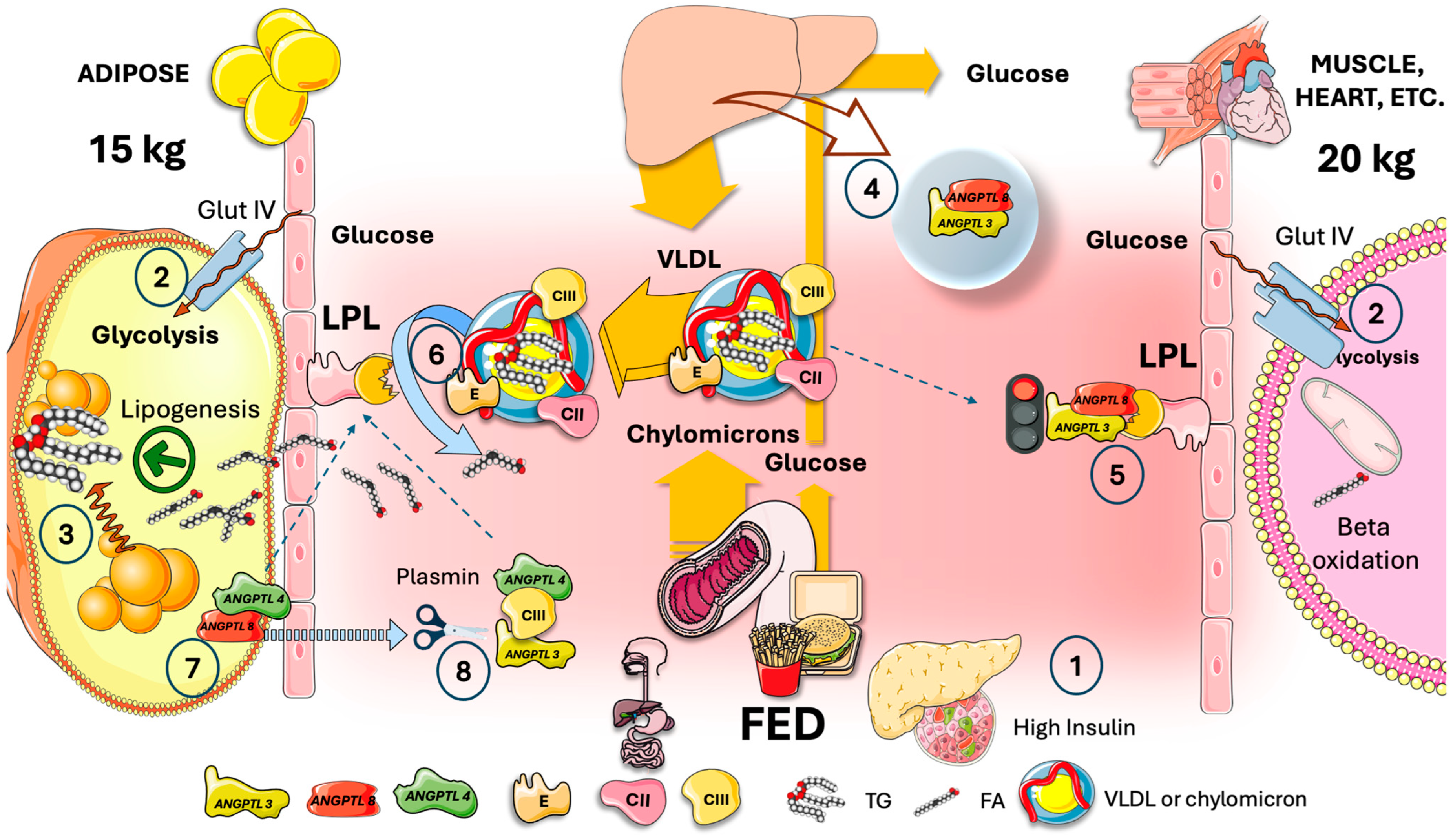
6. In the Fed State, Hepatic ANGPTL3/8 Acts on Muscle in an Endocrine Manner to Redirect VLDL and Chylomicrons to Fat Tissue
7. ANGPTL8, the Main Circadian LPL Switch?
8. Cross-Regulation of LPL in the Fast-Fed Cycle, Current Model
- Firstly, ANGPTL8 breaks down ANGPTL4 and inhibits its secretion, and it also prevents ANGPTL4 from inhibiting LPL. After forming a strong bond with LPL, ANGPTL4 /8 is translocated to the capillary lumen;
- Secondly, plasminogen together with tPA binds to ANGPTL4 /8, which in turn produces plasmin. Plasmin then locally cleaves LPL inhibitors such as ANGPTL3 /8, ANGPTL4, and APOCIII. WAT LPL activity is thus entirely restored while the ANGPTL3 /8 is in turn capable of blocking oxidative-tissue LPL. The conversion of plasminogen to plasmin is made possible by the ANGPTL4 /8-LPL complex’s interactions with luminal plasminogen receptors and endothelial-released tPA;
- Plasma ANGPTL3/8 levels in healthy individuals are around 15 ng/mL while fasting but increase to approximately 28 ng/mL two hours after a meal. Consequently, fasting lowers the amount of ANGPTL3/8 in circulation, but considerable amounts are still present. This begs the following question: when fasting, how can the body restore oxidative-tissue LPL activity and dampen plasma ANGPTL3/8? The discovery that ApoAV is an endogenous ANGPTL3/8 inhibitor provides a first hint. By acting on the ANGPTL3/8 complex, ApoAV lifts LPL inhibition as shown earlier. It appears that the long-sought method by which ApoAV reduces serum TG is by selectively suppressing the ANGPTL3/8 complex’s LPL-inhibitory activity. The peculiar characteristics of this system contribute to the explanation of why, even after ApoAV was identified as a crucial component of TG metabolism two decades ago, the precise mechanism by which it reduces TG remained a persistent mystery [44,60,71,72].
9. ANGPTL8: Master Switch Gone Awry?
10. A Special Case: Postprandial Exercise
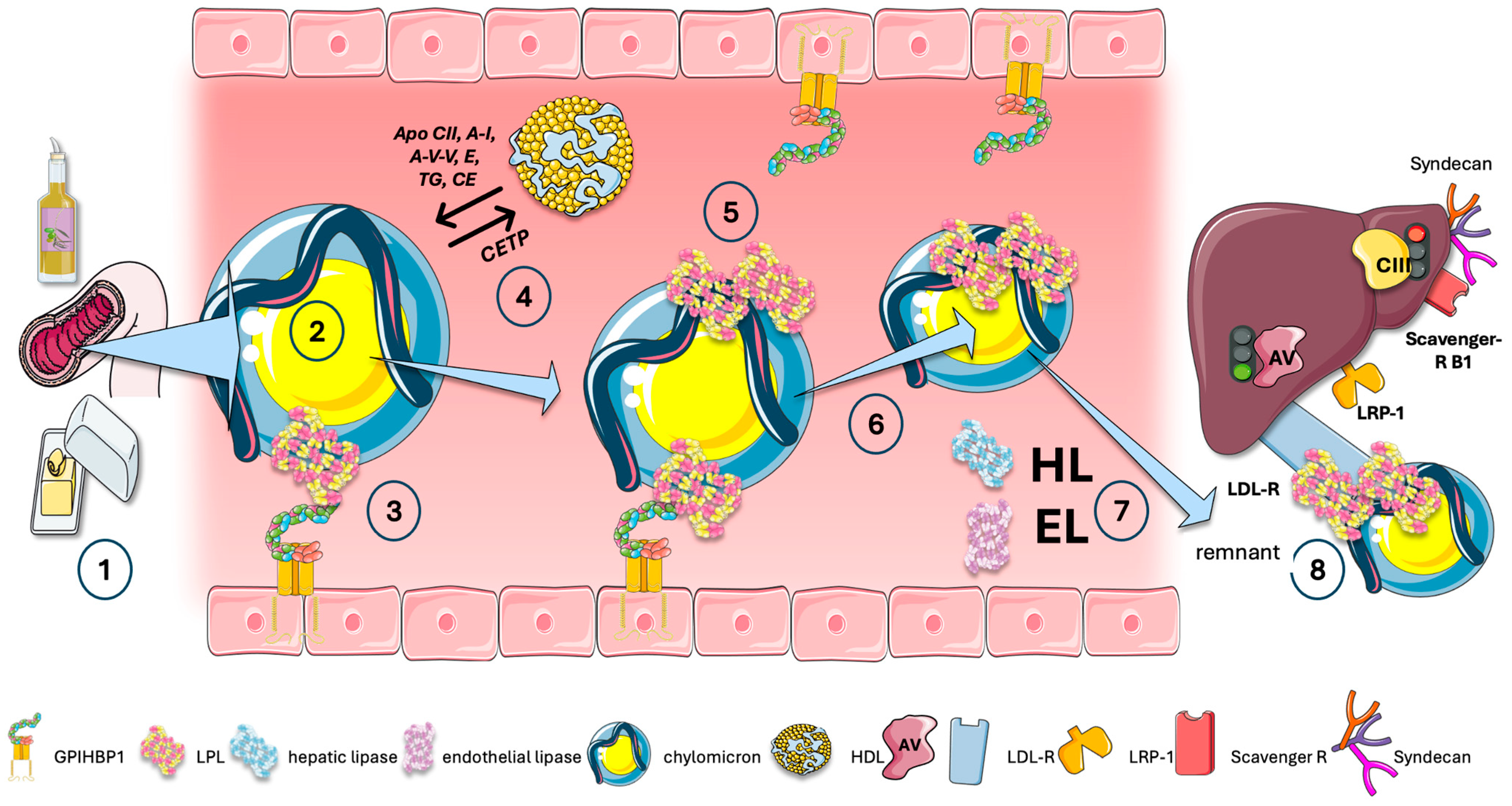
11. Exchange of Lipids and Proteins Is Also a Key Component of the Intravascular Traffic of TRL
12. Blocking ANGPTLs: The Next Frontier?
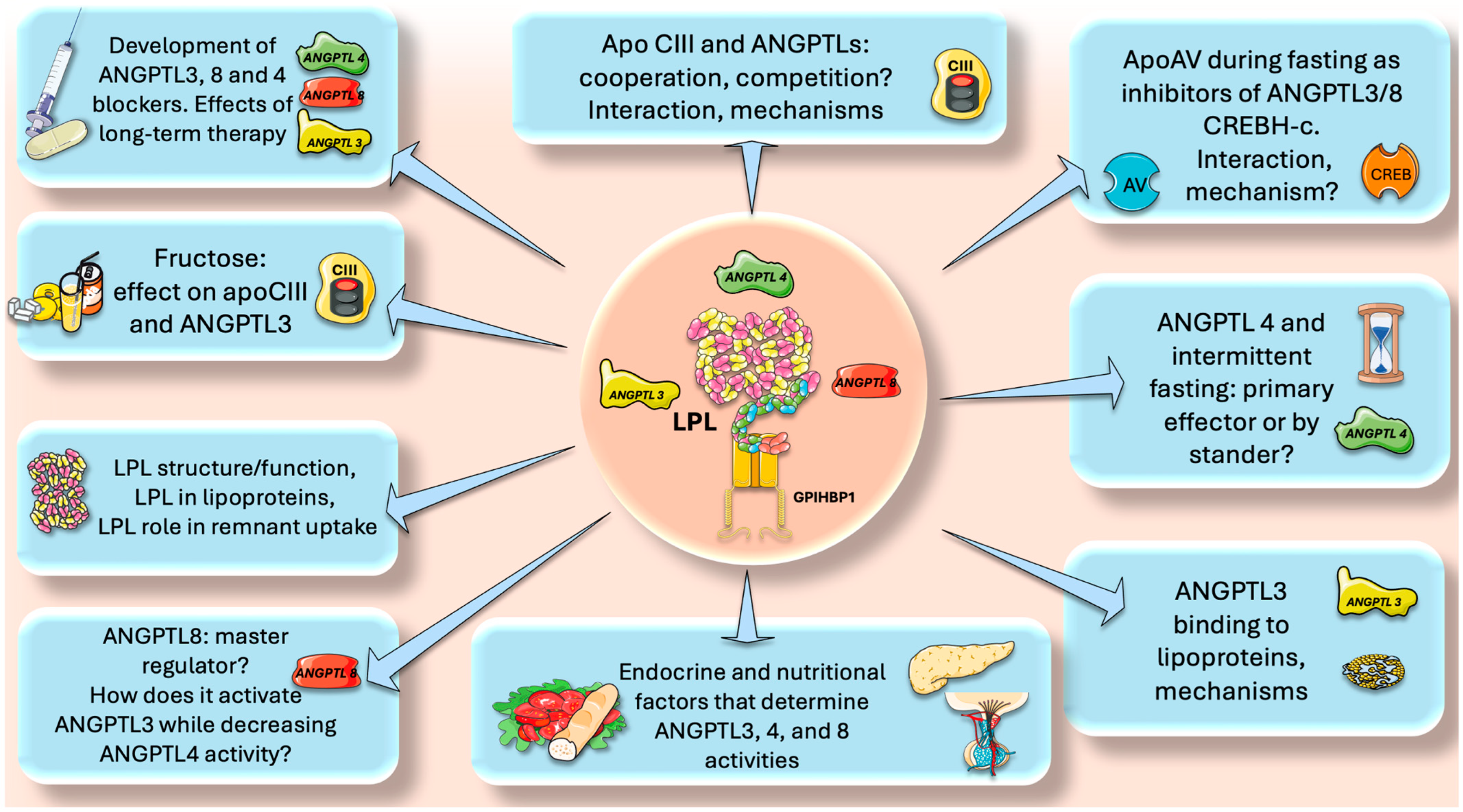
13. Perspectives and Questions to Be Addressed in Future Research
- Intermittent fasting and ANGPTL4. Multiple studies have demonstrated that intermittent fasting reduces insulin resistance, favorably shifts leptin and adiponectin levels, and is beneficial for weight loss [43,91,92,93,94,95,96]. Moreover, numerous disorders, such as obesity, type 2 diabetes, hypertension, and cardiovascular risk factors, can benefit from intermittent fasting, as shown by pre-clinical and clinical investigations [94,95,96]. As we have seen, ANGPTL4 is a master regulator of lipid traffic in the fasting state, shunting FA to muscle and reducing fat mass. How does intermittent fasting affect ANGPTL4? Is ANGPTL4 the main responsible for the lipid-lowering effects of intermittent fasting or just a bystander? Are there unknown regulatory loops ANGPTL4-adiponectin or ANGPTL4-leptin?
- Endocrine and nutritional factors. The role of insulin in determining the correct expression of the LPL gene is well known. What are the roles of insulin and glucagon ratios in the expression of ANGPTL3,4 and 8? What are the other endocrine factors involved? Do incretins play a role? Regarding nutritional factors, fructose has received a lot of attention in the past decade as an important dyslipidemia CVD risk factor [97,98,99,100,101]. Indeed, our studies have shown (among other beneficial effects) that isocaloric fructose restriction reduced the AUC of TG, apoCIII, ANGPTL3, and apoB48, showing a continuous benefit throughout most of the postprandial period [20,73,102,103,104,105,106]. This has been corroborated by other studies that suggest that fructose-induced delayed catabolism of TRL is even more relevant than increased production [107,108]. Does fructose directly enhance ANGPTL3 expression?
- ApoCIII and ANGPTLs, cooperation, competition? The role of apoCIII as a baseline inhibitor of LPL and its potential benefit throughout evolution has been discussed previously. One clear clue is the rarity of LOF mutations for apoCIII. What was protective in times of food uncertainty has become deleterious in times of caloric abundance. We have seen that on top of the apoCII/CIII regulation, there are the ANGPTL3,4, 8 axes. How do they interact? Do they compete? What are the mechanisms for putative additive or subtractive activities?
- ApoAV and CHREBH-C activate LPL during fasting by inhibiting ANGPTL3/8. What are the precise mechanisms?
- Plasmin clears up remaining ANGPTL3/8 complexes locally where ANGPTL4 is up (adipose capillary bed during fasting). What is the interaction between this process and the tPA-fibrinolysis pathway?
- ANGPTL8 is a master switch in the fast-fed cycle. How does it, on one hand, activate ANGPTL3 whereas, on the other, decrease ANGPTL4 activity?
- LPL structure-function. Much is known about this enzyme, but a lot remains to be uncovered, including its interaction with lipoproteins and its quantitative role in remnant uptake;
- Development of new, effective, and safe ANGPTL blockers. ANGPTL3 inhibitors are already on the market, and new ones are continuously being developed. ANGPTL8 blockers show promise and ANGPTL4 blockers need more study. For all, what would be the effect of long-term therapy?
14. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Thanassoulis, G.; Welsh, R.C.; Hegele, R.A. What Guidelines Say About Risk Reduction: Major Data on the Link Between Lipid Lowering and Outcomes. Can. J. Cardiol. 2024, 40, S13–S19. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.I.; Yu, J.; Hayashi, T.; Han, S.H.; Koh, K.K. Strategies to Overcome Residual Risk during Statins Era. Circ. J. 2019, 83, 1973–1979. [Google Scholar] [CrossRef]
- Gugliucci, A. Beyond LDL: Understanding Triglyceride-Rich Lipoproteins to Tackle Residual Risk. J. Clin. Med. 2023, 12, 3991. [Google Scholar] [CrossRef] [PubMed]
- Rikhi, R.; Shapiro, M.D. Newer and Emerging LDL-C Lowering Agents and Implications for ASCVD Residual Risk. J. Clin. Med. 2022, 11, 4611. [Google Scholar] [CrossRef] [PubMed]
- Vallejo-Vaz, A.J.; Corral, P.; Schreier, L.; Ray, K.K. Triglycerides and Residual Risk. Curr. Opin. Endocrinol. Diabetes Obes. 2020, 27, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Gugliucci, A. The Chylomicron Saga: Time to Focus on Postprandial Metabolism. Front. Endocrinol. 2023, 14, 1322869. [Google Scholar] [CrossRef]
- Gugliucci, A. Triglyceride-Rich Lipoprotein Metabolism: Key Regulators of Their Flux. J. Clin. Med. 2023, 12, 4399. [Google Scholar] [CrossRef]
- Zilversmit, D.B. Atherogenic Nature of Triglycerides, Postprandial Lipidemia, and Triglyceride-Rich Remnant, Lipoproteins. Clin. Chem. 1995, 41, 153–158. [Google Scholar] [CrossRef]
- Borén, J.; Taskinen, M.R. Metabolism of Triglyceride-Rich Lipoproteins. Handb. Exp. Pharmacol. 2022, 270, 133–156. [Google Scholar] [CrossRef]
- Ference, B.A.; Kastelein, J.J.P.; Ray, K.K.; Ginsberg, H.N.; Chapman, M.J.; Packard, C.J.; Laufs, U.; Oliver-Williams, C.; Wood, A.M.; Butterworth, A.S.; et al. Association of Triglyceride-Lowering LPL Variants and LDL-C-Lowering LDLR Variants with Risk of Coronary Heart Disease. JAMA—J. Am. Med. Assoc. 2019, 321, 364–373. [Google Scholar] [CrossRef]
- Baratta, F.; Cocomello, N.; Coronati, M.; Ferro, D.; Pastori, D.; Angelico, F.; Del Ben, M. Cholesterol Remnants, Triglyceride-Rich Lipoproteins and Cardiovascular Risk. Int. J. Mol. Sci. 2023, 24, 4268. [Google Scholar] [CrossRef] [PubMed]
- Wierzbicki, A.S.; Clarke, R.E.; Viljoen, A.; Mikhailidis, D.P. Triglycerides: A Case for Treatment? Curr. Opin. Cardiol. 2012, 27, 398–404. [Google Scholar] [CrossRef] [PubMed]
- Reith, C.; Armitage, J. Management of Residual Risk after Statin Therapy. Atherosclerosis 2016, 245, 161–170. [Google Scholar] [CrossRef] [PubMed]
- González-Juanatey, J.R.; Almendro-Delia, M.; Cosín-Sales, J.; Bellmunt-Montoya, S.; Gómez-Doblas, J.J.; Riambau, V.; García-Moll, X.; García-Alegría, J.; Hernández, J.L.; Lozano, F.S.; et al. Residual Risk Reduction Opportunities in Patients with Chronic Coronary Syndrome. Role of Dual Pathway Inhibition. Expert. Rev. Clin. Pharmacol. 2020, 13, 695–706. [Google Scholar] [CrossRef] [PubMed]
- Taskinen, M.R.; Matikainen, N.; Björnson, E.; Söderlund, S.; Inkeri, J.; Hakkarainen, A.; Parviainen, H.; Sihlbom, C.; Thorsell, A.; Andersson, L.; et al. Contribution of Intestinal Triglyceride-Rich Lipoproteins to Residual Atherosclerotic Cardiovascular Disease Risk in Individuals with Type 2 Diabetes on Statin Therapy. Diabetologia 2023, 66, 2307–2319. [Google Scholar] [CrossRef]
- Goldberg, I.J.; Sharma, G.; Fisher, E.A. Atherosclerosis: Making a U Turn. Annu. Rev. Med. 2020, 71, 191–201. [Google Scholar] [CrossRef]
- Getz, G.S.; Reardon, C.A. Atherosclerosis: Cell Biology and Lipoproteins. Curr. Opin. Lipidol. 2020, 31, 286–290. [Google Scholar] [CrossRef]
- Qiao, Y.-N.; Zou, Y.-L.; Guo, S.-D. Low-Density Lipoprotein Particles in Atherosclerosis. Front. Physiol. 2022, 13, 931931. [Google Scholar] [CrossRef]
- Neels, J.G.; Leftheriotis, G.; Chinetti, G. Atherosclerosis Calcification: Focus on Lipoproteins. Metabolites 2023, 13, 457. [Google Scholar] [CrossRef]
- Jones, G.M.; Caccavello, R.; Palii, S.P.; Pullinger, C.R.; Kane, J.P.; Mulligan, K.; Gugliucci, A.; Schwarz, J.M. Separation of Postprandial Lipoproteins: Improved Purification of Chylomicrons Using an ApoB100 Immunoaffinity Method. J. Lipid Res. 2020, 61, 455–463. [Google Scholar] [CrossRef]
- Adiels, M.; Matikainen, N.; Westerbacka, J.; Söderlund, S.; Larsson, T.; Olofsson, S.O.; Borén, J.; Taskinen, M.R. Postprandial Accumulation of Chylomicrons and Chylomicron Remnants Is Determined by the Clearance Capacity. Atherosclerosis 2012, 222, 222–228. [Google Scholar] [CrossRef]
- Masuda, D.; Yamashita, S. Postprandial Hyperlipidemia and Remnant Lipoproteins. J. Atheroscler. Thromb. 2017, 24, 95–109. [Google Scholar] [CrossRef] [PubMed]
- Wheless, A.; Gunn, K.H.; Neher, S.B. Macromolecular Interactions of Lipoprotein Lipase (LPL). Subcell. Biochem. 2024, 104, 139–179. [Google Scholar] [CrossRef]
- Wu, S.A.; Kersten, S.; Qi, L. Lipoprotein Lipase and Its Regulators: An Unfolding Story. Trends Endocrinol. Metab. 2021, 32, 48–61. [Google Scholar] [CrossRef] [PubMed]
- Tramontano, D.; Bini, S.; D’Erasmo, L.; Arca, M. Recent Apolipoprotein CIII Trials. Curr. Opin. Lipidol. 2022, 33, 309–318. [Google Scholar] [CrossRef]
- Zhang, R.; Zhang, K. An Updated ANGPTL3-4-8 Model as a Mechanism of Triglyceride Partitioning between Fat and Oxidative Tissues. Prog. Lipid Res. 2022, 85, 101140. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Beigneux, A.P.; Weston, T.A.; Chen, K.; Yang, Y.; Nguyen, L.P.; Guagliardo, P.; Jung, H.; Tran, A.P.; Tu, Y.; et al. The Lipoprotein Lipase That Is Shuttled into Capillaries by GPIHBP1 Enters the Glycocalyx Where It Mediates Lipoprotein Processing. Proc. Natl. Acad. Sci. USA 2023, 120, e2313825120. [Google Scholar] [CrossRef] [PubMed]
- Moon, J.H.; Kim, K.; Choi, S.H. Lipoprotein Lipase: Is It a Magic Target for the Treatment of Hypertriglyceridemia. Endocrinol. Metab. 2022, 37, 575–586. [Google Scholar] [CrossRef]
- He, P.P.; Jiang, T.; OuYang, X.P.; Liang, Y.Q.; Zou, J.Q.; Wang, Y.; Shen, Q.Q.; Liao, L.; Zheng, X.L. Lipoprotein Lipase: Biosynthesis, Regulatory Factors, and Its Role in Atherosclerosis and Other Diseases. Clin. Chim. Acta 2018, 480, 126–137. [Google Scholar] [CrossRef]
- Kumari, A.; Kristensen, K.K.; Ploug, M.; Winther, A.-M.L. The Importance of Lipoprotein Lipase Regulation in Atherosclerosis. Biomedicines 2021, 9, 782. [Google Scholar] [CrossRef]
- Wen, Y.; Chen, Y.Q.; Konrad, R.J. The Regulation of Triacylglycerol Metabolism and Lipoprotein Lipase Activity. Adv. Biol. 2022, 6, e2200093. [Google Scholar] [CrossRef] [PubMed]
- Gunn, K.H.; Neher, S.B. Structure of Dimeric Lipoprotein Lipase Reveals a Pore Adjacent to the Active Site. Nat. Commun. 2023, 14, 2569. [Google Scholar] [CrossRef]
- Chen, Y.Q.; Pottanat, T.G.; Siegel, R.W.; Ehsani, M.; Qian, Y.W.; Zhen, E.Y.; Regmi, A.; Roell, W.C.; Guo, H.; Jane Luo, M.; et al. Angiopoietin-like Protein 8 Differentially Regulates ANGPTL3 and ANGPTL4 during Postprandial Partitioning of Fatty Acids. J. Lipid Res. 2020, 61, 1203–1220. [Google Scholar] [CrossRef] [PubMed]
- Ploug, M. ANGPTL4: A New Mode in the Regulation of Intravascular Lipolysis. Curr. Opin. Lipidol. 2022, 33, 112–119. [Google Scholar] [CrossRef]
- Guo, C.; Wang, C.; Deng, X.; He, J.; Yang, L.; Yuan, G. ANGPTL8 in Metabolic Homeostasis: More Friend than Foe? Open Biol. 2021, 11, 210106. [Google Scholar] [CrossRef]
- Sylvers-Davie, K.L.; Davies, B.S.J. Regulation of Lipoprotein Metabolism by ANGPTL3, ANGPTL4, and ANGPTL8. Am. J. Physiol. Endocrinol. Metab. 2021, 321, E493–E508. [Google Scholar] [CrossRef]
- Borén, J.; Packard, C.J.; Taskinen, M.-R. The Roles of ApoC-III on the Metabolism of Triglyceride-Rich Lipoproteins in Humans. Front. Endocrinol. 2020, 11, 474. [Google Scholar] [CrossRef]
- Borén, J.; Taskinen, M.R.; Björnson, E.; Packard, C.J. Metabolism of Triglyceride-Rich Lipoproteins in Health and Dyslipidaemia. Nat. Rev. Cardiol. 2022, 19, 577–592. [Google Scholar] [CrossRef]
- Shah, N.P.; Pajidipati, N.J.; McGarrah, R.W.; Navar, A.M.; Vemulapalli, S.; Blazing, M.A.; Shah, S.H.; Hernandez, A.F.; Patel, M.R. Lipoprotein (a): An Update on a Marker of Residual Risk and Associated Clinical Manifestations. Am. J. Cardiol. 2020, 126, 94–102. [Google Scholar] [CrossRef] [PubMed]
- De la Parra Soto, L.G.; Gutiérrez-Uribe, J.A.; Sharma, A.; Ramírez-Jiménez, A.K. Is Apo-CIII the New Cardiovascular Target? An Analysis of Its Current Clinical and Dietetic Therapies. Nutr. Metab. Cardiovasc. Dis. 2022, 32, 295–308. [Google Scholar] [CrossRef]
- Jakel, H.; Nowak, M.; Helleboid-Chapman, A.; Fruchart-Najib, J.; Fruchart, J.C. Is Apolipoprotein A5 a Novel Regulator of Triglyceride-Rich Lipoproteins? Ann. Med. 2006, 38, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Su, X.; Kong, Y.; Peng, D.Q. New Insights into Apolipoprotein A5 in Controlling Lipoprotein Metabolism in Obesity and the Metabolic Syndrome Patients. Lipids Health Dis. 2018, 17, 174. [Google Scholar] [CrossRef] [PubMed]
- May-Zhang, L.; Liu, M.; Black, D.; Tso, P. Apolipoprotein A5, a Unique Modulator of Fasting and Postprandial Triglycerides. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2022, 1867, 159185. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Konrad, R.J.; Ploug, M.; Young, S.G. APOA5 Deficiency Causes Hypertriglyceridemia by Reducing Amounts of Lipoprotein Lipase in Capillaries. J. Lipid Res. 2024, 65, 100578. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Chen, Y.Q.; Konrad, R.J. Angiopoietin-like Protein 8: A Multifaceted Protein Instrumental in Regulating Triglyceride Metabolism. Curr. Opin. Lipidol. 2024, 35, 58–65. [Google Scholar] [CrossRef]
- Kim, H.; Song, Z.; Zhang, R.; Davies, B.S.J.; Zhang, K. A Hepatokine Derived from the ER Protein CREBH Promotes Triglyceride Metabolism by Stimulating Lipoprotein Lipase Activity. Sci. Signal 2023, 16, eadd6702. [Google Scholar] [CrossRef]
- Zhang, R.; Zhang, K. A Unified Model for Regulating Lipoprotein Lipase Activity. Trends Endocrinol. Metab. 2024, 35, 490–504. [Google Scholar] [CrossRef]
- Hoffmann, W.G.; Chen, Y.Q.; Schwartz, C.S.; Barber, J.L.; Dev, P.K.; Reasons, R.J.; Miranda Maravi, J.S.; Armstrong, B.; Gerszten, R.E.; Silbernagel, G.; et al. Effects of Exercise Training on ANGPTL3/8 and ANGPTL4/8 and Their Associations with Cardiometabolic Traits. J. Lipid Res. 2024, 65, 100495. [Google Scholar] [CrossRef]
- Dewey, F.E.; Gusarova, V.; O’Dushlaine, C.; Gottesman, O.; Trejos, J.; Hunt, C.; Van Hout, C.V.; Habegger, L.; Buckler, D.; Lai, K.-M.V.; et al. Inactivating Variants in ANGPTL4 and Risk of Coronary Artery Disease. N. Engl. J. Med. 2016, 374, 1123–1133. [Google Scholar] [CrossRef]
- Stitziel, N.O. Variants in ANGPTL4 and the Risk of Coronary Artery Disease. N. Engl. J. Med. 2016, 375, 2303–2306. [Google Scholar] [CrossRef]
- Gusarova, V.; O’Dushlaine, C.; Teslovich, T.M.; Benotti, P.N.; Mirshahi, T.; Gottesman, O.; Van Hout, C.V.; Murray, M.F.; Mahajan, A.; Nielsen, J.B.; et al. Genetic Inactivation of ANGPTL4 Improves Glucose Homeostasis and Is Associated with Reduced Risk of Diabetes. Nat. Commun. 2018, 9, 2252. [Google Scholar] [CrossRef] [PubMed]
- Alex, S.; Lichtenstein, L.; Dijk, W.; Mensink, R.P.; Tan, N.S.; Kersten, S. ANGPTL4 Is Produced by Entero-Endocrine Cells in the Human Intestinal Tract. Histochem. Cell Biol. 2014, 141, 383–391. [Google Scholar] [CrossRef] [PubMed]
- Kersten, S.; Lichtenstein, L.; Steenbergen, E.; Mudde, K.; Hendriks, H.F.J.; Hesselink, M.K.; Schrauwen, P.; Müller, M. Caloric Restriction and Exercise Increase Plasma ANGPTL4 Levels in Humans via Elevated Free Fatty Acids. Arter. Thromb. Vasc. Biol. 2009, 29, 969–974. [Google Scholar] [CrossRef] [PubMed]
- Lichtenstein, L.; Mattijssen, F.; De Wit, N.J.; Georgiadi, A.; Hooiveld, G.J.; Van Der Meer, R.; He, Y.; Qi, L.; Köster, A.; Tamsma, J.T.; et al. Angptl4 Protects against Severe Proinflammatory Effects of Saturated Fat by Inhibiting Fatty Acid Uptake into Mesenteric Lymph Node Macrophages. Cell Metab. 2010, 12, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Mysling, S.; Kristensen, K.K.; Larsson, M.; Kovrov, O.; Bensadouen, A.; Jørgensen, T.J.D.; Olivecrona, G.; Young, S.G.; Ploug, M. The Angiopoietin-like Protein Angptl4 Catalyzes Unfolding of the Hydrolase Domain in Lipoprotein Lipase and the Endothelial Membrane Protein Gpihbp1 Counteracts This Unfolding. Elife 2016, 5, 18. [Google Scholar] [CrossRef]
- Balasubramaniam, D.; Schroeder, O.; Russell, A.M.; Fitchett, J.R.; Austin, A.K.; Beyer, T.P.; Chen, Y.Q.; Day, J.W.; Ehsani, M.; Heng, A.R.; et al. An Anti-ANGPTL3/8 Antibody Decreases Circulating Triglycerides by Binding to a LPL-Inhibitory Leucine Zipper-like Motif. J. Lipid Res. 2022, 63, 100198. [Google Scholar] [CrossRef]
- Arefanian, H.; Al-Khairi, I.; Khalaf, N.A.; Cherian, P.; Kavalakatt, S.; Madhu, D.; Mathur, A.; Qaddoumi, M.G.; Al-Mulla, F.; Abubaker, J.; et al. Increased Expression Level of ANGPTL8 in White Adipose Tissue under Acute and Chronic Cold Treatment. Lipids Health Dis. 2021, 20, 117. [Google Scholar] [CrossRef]
- Kovrov, O.; Kristensen, K.K.; Larsson, E.; Ploug, M.; Olivecrona, G. On the Mechanism of Angiopoietin-like Protein 8 for Control of Lipoprotein Lipase Activity. J. Lipid Res. 2019, 60, 783–793. [Google Scholar] [CrossRef]
- Zhen, E.Y.; Chen, Y.Q.; Russell, A.M.; Ehsani, M.; Siegel, R.W.; Qian, Y.; Konrad, R.J. Angiopoietin-like Protein 4/8 Complex-Mediated Plasmin Generation Leads to Cleavage of the Complex and Restoration of LPL Activity. Proc. Natl. Acad. Sci. USA 2023, 120, e2214081120. [Google Scholar] [CrossRef]
- Chen, Y.Q.; Pottanat, T.G.; Zhen, E.Y.; Siegel, R.W.; Ehsani, M.; Qian, Y.W.; Konrad, R.J. ApoA5 Lowers Triglyceride Levels via Suppression of ANGPTL3/8-Mediated LPL Inhibition. J. Lipid Res. 2021, 62, 100068. [Google Scholar] [CrossRef]
- Gallo, A.; Béliard, S.; D’Erasmo, L.; Bruckert, E. Familial Chylomicronemia Syndrome (FCS): Recent Data on Diagnosis and Treatment. Curr. Atheroscler. Rep. 2020, 22, 63. [Google Scholar] [CrossRef]
- Björnson, E.; Packard, C.J.; Adiels, M.; Andersson, L.; Matikainen, N.; Söderlund, S.; Kahri, J.; Hakkarainen, A.; Lundbom, N.; Lundbom, J.; et al. Apolipoprotein B48 Metabolism in Chylomicrons and Very Low-Density Lipoproteins and Its Role in Triglyceride Transport in Normo- and Hypertriglyceridemic Human Subjects. J. Intern. Med. 2020, 288, 422–438. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R. The ANGPTL3-4-8 Model, a Molecular Mechanism for Triglyceride Trafficking. Open Biol. 2016, 6, 150272. [Google Scholar] [CrossRef] [PubMed]
- Aryal, B.; Price, N.L.; Suarez, Y.; Fernández-Hernando, C. ANGPTL4 in Metabolic and Cardiovascular Disease. Trends Mol. Med. 2019, 25, 723–734. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Quagliarini, F.; Gusaroèa, È.; Gromada, J.; Èalenzuela, D.M.; Cohen, J.C.; Hobbs, H.H. Mice Lacking ANGPTL8 (Betatrophin) Manifest Disrupted Triglyceride Metabolism without Impaired Glucose Homeostasis. Proc. Natl. Acad. Sci. USA 2013, 110, 16109–16114. [Google Scholar] [CrossRef] [PubMed]
- Haller, J.F.; Mintah, I.J.; Shihanian, L.M.; Stevis, P.; Buckler, D.; Alexa-Braun, C.A.; Kleiner, S.; Banfi, S.; Cohen, J.C.; Hobbs, H.H.; et al. ANGPTL8 Requires ANGPTL3 to Inhibit Lipoprotein Lipase and Plasma Triglyceride Clearance. J. Lipid Res. 2017, 58, 1166–1173. [Google Scholar] [CrossRef]
- Oldoni, F.; Cheng, H.; Banfi, S.; Gusarova, V.; Cohen, J.C.; Hobbs, H.H. ANGPTL8 Has Both Endocrine and Autocrine Effects on Substrate Utilization. JCI Insight 2020, 5, e138777. [Google Scholar] [CrossRef]
- Abu-Farha, M.; Ghosh, A.; Al-Khairi, I.; Madiraju, S.R.M.; Abubaker, J.; Prentki, M. The Multi-Faces of Angptl8 in Health and Disease: Novel Functions beyond Lipoprotein Lipase Modulation. Prog. Lipid Res. 2020, 80, 101067. [Google Scholar] [CrossRef]
- Quagliarini, F.; Wang, Y.; Kozlitina, J.; Grishin, N.V.; Hyde, R.; Boerwinkle, E.; Valenzuela, D.M.; Murphy, A.J.; Cohen, J.C.; Hobbs, H.H. Atypical Angiopoietin-like Protein That Regulates ANGPTL3. Proc. Natl. Acad. Sci. USA 2012, 109, 19751–19756. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, Y.; Han, B.; Li, L.; Li, Y.; Ma, Y.; Kang, S.; Li, Q.; Kong, L.; Huang, K.; et al. Postprandial Exercise Regulates Tissue-Specific Triglyceride Uptake through Angiopoietin-like Proteins. JCI Insight 2024, 9, 1–16. [Google Scholar] [CrossRef]
- Chen, Y.Q.; Yang, Y.; Zhen, E.Y.; Beyer, T.P.; Li, H.; Wen, Y.; Ehsani, M.; Jackson, N.; Xie, K.; Jung, H.; et al. Carboxyl-Terminal Sequences in APOA5 Are Important for Suppressing ANGPTL3/8 Activity. Proc. Natl. Acad. Sci. USA 2024, 121, e2322332121. [Google Scholar] [CrossRef]
- Calandra, S.; Oliva, C.P.; Tarugi, P.; Bertolini, S. APOA5 and Triglyceride Metabolism, Lesson from Human APOA5 Deficiency. Curr. Opin. Lipidol. 2006, 17, 122–127. [Google Scholar] [CrossRef]
- Gugliucci, A. Sugar and Dyslipidemia: A Double-Hit, Perfect Storm. J. Clin. Med. 2023, 12, 5660. [Google Scholar] [CrossRef]
- Rhainds, D.; Tardif, J.C. From HDL-Cholesterol to HDL-Function: Cholesterol Efflux Capacity Determinants. Curr. Opin. Lipidol. 2019, 30, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Zakiev, E.; Feng, M.; Sukhorukov, V.; Kontush, A. HDL-Targeting Therapeutics: Past, Present and Future. Curr. Pharm. Des. 2016, 23, 1207–1215. [Google Scholar] [CrossRef]
- Nicholls, S.J. CETP-Inhibition and HDL-Cholesterol: A Story of CV Risk or CV Benefit, or Both. Clin. Pharmacol. Ther. 2018, 104, 297–300. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.H. The Current Status of Research on High-Density Lipoproteins (HDL): A Paradigm Shift from HDL Quantity to HDL Quality and HDL Functionality. Int. J. Mol. Sci. 2022, 23, 3967. [Google Scholar] [CrossRef]
- Vergès, B.; Adiels, M.; Boren, J.; Barrett, P.H.; Watts, G.F.; Chan, D.; Duvillard, L.; Söderlund, S.; Matikainen, N.; Kahri, J.; et al. Interrelationships between the Kinetics of VLDL Subspecies and Hdl Catabolism in Abdominal Obesity: A Multicenter Tracer Kinetic Study. J. Clin. Endocrinol. Metab. 2014, 99, 4281–4290. [Google Scholar] [CrossRef] [PubMed]
- Stankov, S.; Cuchel, M. Gene Editing for Dyslipidemias: New Tools to “Cut” Lipids. Atherosclerosis 2023, 368, 14–24. [Google Scholar] [CrossRef]
- Mohamed, F.; Botha, T.C.; Raal, F.J. Inhibition of Angiopoietin-like 3 for the Management of Severe Hypercholesterolemia. Curr. Opin. Lipidol. 2021, 32, 213–218. [Google Scholar] [CrossRef]
- Michaeli, D.T.; Michaeli, J.C.; Albers, S.; Boch, T.; Michaeli, T. Established and Emerging Lipid-Lowering Drugs for Primary and Secondary Cardiovascular Prevention. Am. J. Cardiovasc. Drugs 2023, 23, 477–495. [Google Scholar] [CrossRef] [PubMed]
- Gill, P.K.; Hegele, R.A. New Biological Therapies for Low-Density Lipoprotein Cholesterol. Can. J. Cardiol. 2023, 39, 1913–1930. [Google Scholar] [CrossRef]
- Makhmudova, U.; Steinhagen-Thiessen, E.; Volpe, M.; Landmesser, U. Advances in Nucleic Acid-Targeted Therapies for Cardiovascular Disease Prevention. Cardiovasc. Res. 2024, cvae136. [Google Scholar] [CrossRef]
- Mehta, N.; Gilbert, R.; Chahal, P.S.; Moreno, M.J.; Nassoury, N.; Coulombe, N.; Lytvyn, V.; Mercier, M.; Fatehi, D.; Lin, W.; et al. Preclinical Development and Characterization of Novel Adeno-Associated Viral Vectors for the Treatment of Lipoprotein Lipase Deficiency. Hum. Gene Ther. 2023, 34, 927–946. [Google Scholar] [CrossRef] [PubMed]
- Thorin, E.; Labbé, P.; Lambert, M.; Mury, P.; Dagher, O.; Miquel, G.; Thorin-Trescases, N. Angiopoietin-Like Proteins: Cardiovascular Biology and Therapeutic Targeting for the Prevention of Cardiovascular Diseases. Can. J. Cardiol. 2023, 39, 1736–1756. [Google Scholar] [CrossRef] [PubMed]
- Shang, R.; Rodrigues, B. Lipoprotein Lipase as a Target for Obesity/Diabetes Related Cardiovascular Disease. J. Pharm. Pharm. Sci. 2024, 27, 13199. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Kim, N.H. New Therapeutic Approaches to the Treatment of Dyslipidemia 1: ApoC-III and ANGPTL3. J. Lipid Atheroscler. 2023, 12, 23–36. [Google Scholar] [CrossRef] [PubMed]
- Geladari, E.; Tsamadia, P.; Vallianou, N.G. ANGPTL3 Inhibitors: Their Role in Cardiovascular Disease through Regulation of Lipid Metabolism. Circ. J. 2019, 83, 267–273. [Google Scholar] [CrossRef]
- Ginsberg, H.N.; Goldberg, I.J. Broadening the Scope of Dyslipidemia Therapy by Targeting APOC3 (Apolipoprotein C3) and ANGPTL3 (Angiopoietin-Like Protein 3). Arter. Thromb. Vasc. Biol. 2023, 43, 388–398. [Google Scholar] [CrossRef]
- Abu-Farha, M.; Cherian, P.; Qaddoumi, M.G.; AlKhairi, I.; Sriraman, D.; Alanbaei, M.; Abubaker, J. Increased Plasma and Adipose Tissue Levels of ANGPTL8/Betatrophin and ANGPTL4 in People with Hypertension. Lipids Health Dis. 2018, 17, 35. [Google Scholar] [CrossRef]
- Santos, H.O.; Genario, R.; Tinsley, G.M.; Ribeiro, P.; Carteri, R.B.; Coelho-Ravagnani, C.D.F.; Mota, J.F. A Scoping Review of Intermittent Fasting, Chronobiology, and Metabolism. Am. J. Clin. Nutr. 2022, 115, 991–1004. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Verma, N.; Thakkar, N.; Yeung, C.; Sung, H.K. Intermittent Fasting: Physiological Implications on Outcomes in Mice and Men. Physiology 2020, 35, 185–195. [Google Scholar] [CrossRef]
- Hu, D.; Xie, Z.; Ye, Y.; Bahijri, S.; Chen, M. The Beneficial Effects of Intermittent Fasting: An Update on Mechanism, and the Role of Circadian Rhythm and Gut Microbiota. Hepatobiliary Surg. Nutr. 2020, 9, 597–602. [Google Scholar] [CrossRef] [PubMed]
- Langsted, A.; Nordestgaard, B.G. Nonfasting versus Fasting Lipid Profile for Cardiovascular Risk Prediction. Pathology 2019, 51, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Antoni, R.; Johnston, K.L.; Collins, A.L.; Robertson, M.D. Effects of Intermittent Fasting on Glucose and Lipid Metabolism. Proc. Nutr. Soc. 2017, 76, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Dote-Montero, M.; Sanchez-Delgado, G.; Ravussin, E. Effects of Intermittent Fasting on Cardiometabolic Health: An Energy Metabolism Perspective. Nutrients 2022, 14, 489. [Google Scholar] [CrossRef]
- Bray, G.A. Fructose: Pure, White, and Deadly? Fructose, by Any Other Name, Is a Health Hazard. J. Diabetes Sci. Technol. 2010, 4, 1003–1007. [Google Scholar] [CrossRef]
- Bray, G.A. How Bad Is Fructose? Am. J. Clin. Nutr. 2007, 86, 895–896. [Google Scholar] [CrossRef]
- Gugliucci, A. Formation of Fructose-Mediated Advanced Glycation End Products and Their Roles in Metabolic and Inflammatory Diseases. Adv. Nutr. 2017, 8, 54–62. [Google Scholar] [CrossRef]
- Cox, C.L.; Stanhope, K.L.; Schwarz, J.M.; Graham, J.L.; Hatcher, B.; Griffen, S.C.; Bremer, A.A.; Berglund, L.; McGahan, J.P.; Havel, P.J.; et al. Consumption of Fructose-Sweetened Beverages for 10 Weeks Reduces Net Fat Oxidation and Energy Expenditure in Overweight/Obese Men and Women. Eur. J. Clin. Nutr. 2012, 66, 201–208. [Google Scholar] [CrossRef]
- Lustig, R.H. Fructose: It’s “Alcohol without the Buzz”. Adv. Nutr. 2013, 4, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Mortera, R.R.; Bains, Y.; Gugliucci, A. Fructose at the Crossroads of the Metabolic Syndrome and Obesity Epidemics. Front. Biosci. 2019, 24, 186–211. [Google Scholar] [CrossRef]
- Erkin-Cakmak, A.; Bains, Y.; Caccavello, R.; Noworolski, S.M.; Schwarz, J.M.; Mulligan, K.; Lustig, R.H.; Gugliucci, A. Isocaloric Fructose Restriction Reduces Serum D-Lactate Concentration in Children with Obesity and Metabolic Syndrome. J. Clin. Endocrinol. Metab. 2019, 104, 3003–3011. [Google Scholar] [CrossRef]
- Lustig, R.H.; Mulligan, K.; Noworolski, S.M.; Tai, V.W.; Wen, M.J.; Erkin-Cakmak, A.; Gugliucci, A.; Schwarz, J.M. Isocaloric Fructose Restriction and Metabolic Improvement in Children with Obesity and Metabolic Syndrome. Obesity 2016, 24, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Bains, Y.; Erkin-Cakmak, A.; Caccavello, R.; Mulligan, K.; Noworolski, S.; Schwarz, J.M.; Lustig, R.; Gugliucci, A. Isocaloric Fructose Restriction Improves Postprandial Chylomicron and VLDL Excursions in Adolescents With Obesity by Reducing Angiopoietin-Like Protein 3 and Apolipoprotein CIII. Circulation 2020, 142, A16511. [Google Scholar] [CrossRef]
- Gugliucci, A.; Lustig, R.H.; Caccavello, R.; Erkin-Cakmak, A.; Noworolski, S.M.; Tai, V.W.; Wen, M.J.; Mulligan, K.; Schwarz, J.M. Short-Term Isocaloric Fructose Restriction Lowers ApoC-III Levels and Yields Less Atherogenic Lipoprotein Profiles in Children with Obesity and Metabolic Syndrome. Atherosclerosis 2016, 253, 171–177. [Google Scholar] [CrossRef]
- Butler, A.A.; Graham, J.L.; Stanhope, K.L.; Wong, S.; King, S.; Bremer, A.A.; Krauss, R.M.; Hamilton, J.; Havel, P.J. Role of Angiopoietin-like Protein 3 in Sugar-Induced Dyslipidemia in Rhesus Macaques: Suppression by Fish Oil or RNAi. J. Lipid Res. 2020, 61, 376–386. [Google Scholar] [CrossRef]
- Hieronimus, B.; Griffen, S.C.; Keim, N.L.; Bremer, A.A.; Berglund, L.; Nakajima, K.; Havel, P.J.; Stanhope, K.L. Effects of Fructose or Glucose on Circulating ApoCIII and Triglyceride and Cholesterol Content of Lipoprotein Subfractions in Humans. J. Clin. Med. 2019, 8, 913. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gugliucci, A. Angiopoietin-like Proteins and Lipoprotein Lipase: The Waltz Partners That Govern Triglyceride-Rich Lipoprotein Metabolism? Impact on Atherogenesis, Dietary Interventions, and Emerging Therapies. J. Clin. Med. 2024, 13, 5229. https://doi.org/10.3390/jcm13175229
Gugliucci A. Angiopoietin-like Proteins and Lipoprotein Lipase: The Waltz Partners That Govern Triglyceride-Rich Lipoprotein Metabolism? Impact on Atherogenesis, Dietary Interventions, and Emerging Therapies. Journal of Clinical Medicine. 2024; 13(17):5229. https://doi.org/10.3390/jcm13175229
Chicago/Turabian StyleGugliucci, Alejandro. 2024. "Angiopoietin-like Proteins and Lipoprotein Lipase: The Waltz Partners That Govern Triglyceride-Rich Lipoprotein Metabolism? Impact on Atherogenesis, Dietary Interventions, and Emerging Therapies" Journal of Clinical Medicine 13, no. 17: 5229. https://doi.org/10.3390/jcm13175229