

Arterial Hypertension: Novel Pharmacological Targets and Future Perspectives

 , ,
, , 

Abstract
:1. Introduction
2. New Drug Targets in Arterial Hypertension
2.1. Natriuretic Peptide and Neprilysin
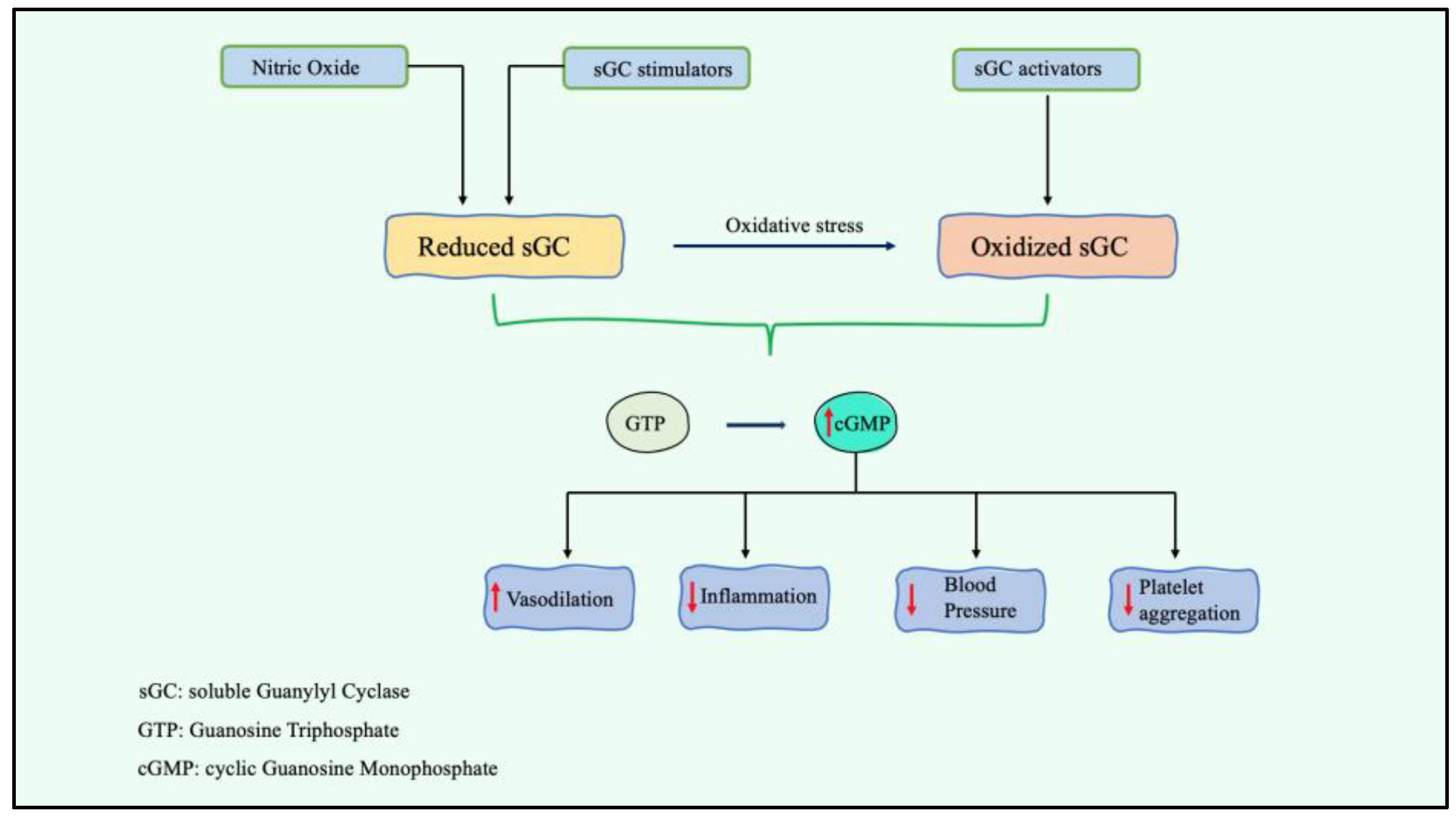
2.2. Stimulation of Soluble Guanylyl Cyclase A
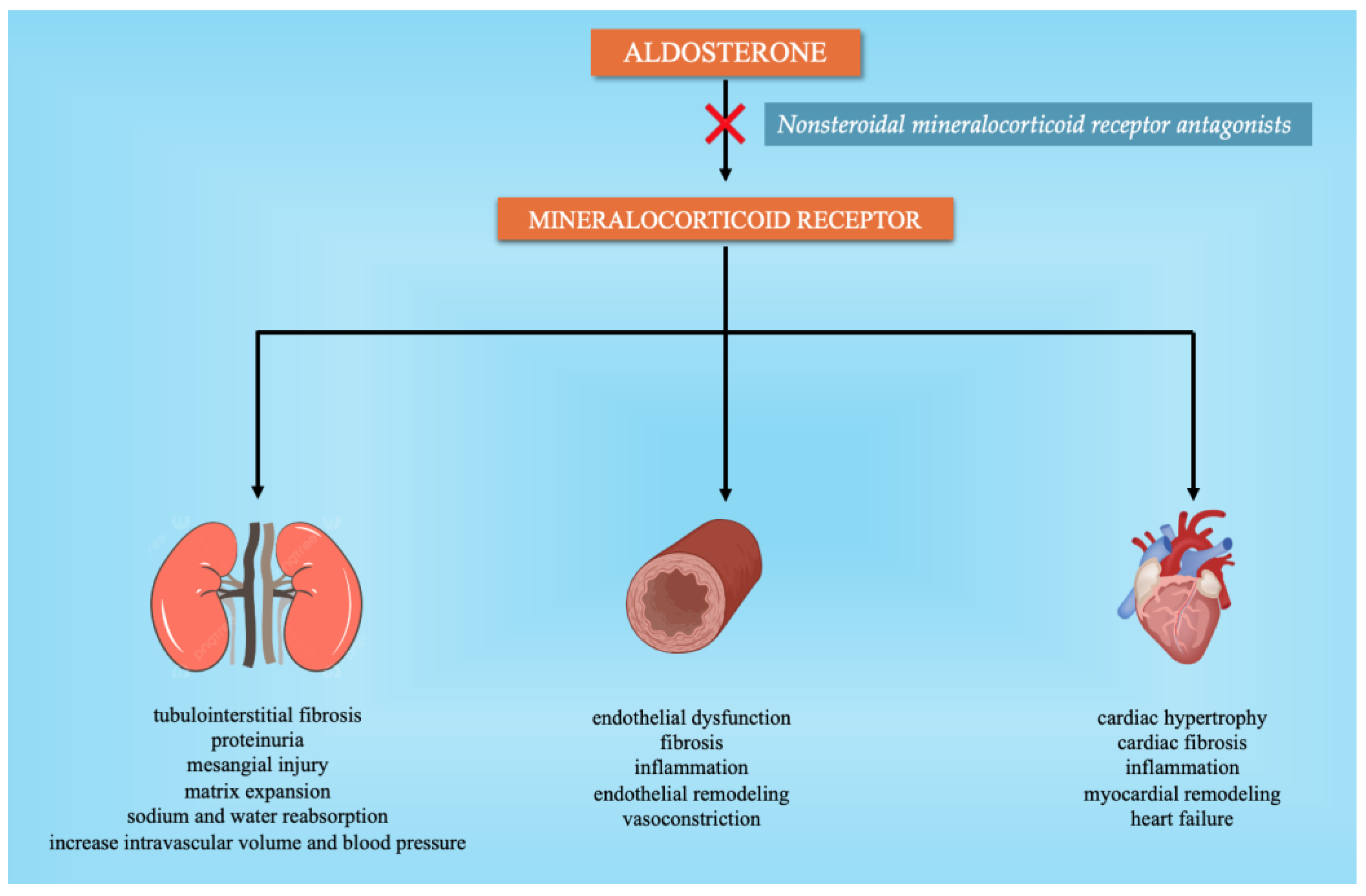
2.3. Nonsteroidal Mineralocorticoid Receptor Antagonists (MRAs)
2.4. Sodium/Glucose Cotransporter-2 Inhibitors (SGLT2i)
2.5. Aminopeptidase of the Brain Renin-Angiotensin System
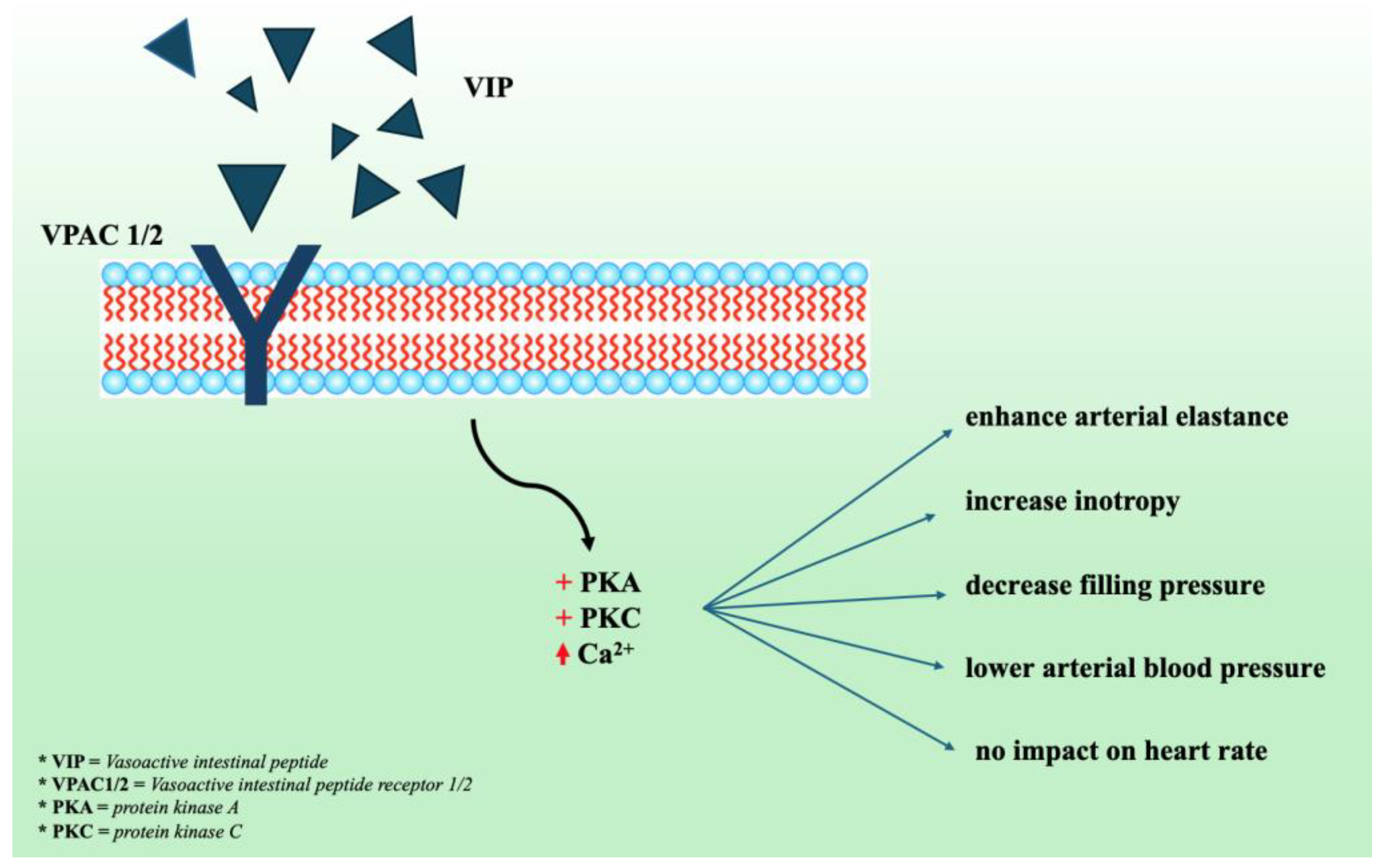
2.6. Vasoactive Intestinal Peptide (VIP) Receptor
2.7. Intestinal Sodium (Na+)/Hydrogen (H+) Exchanger 3 (NHE3)
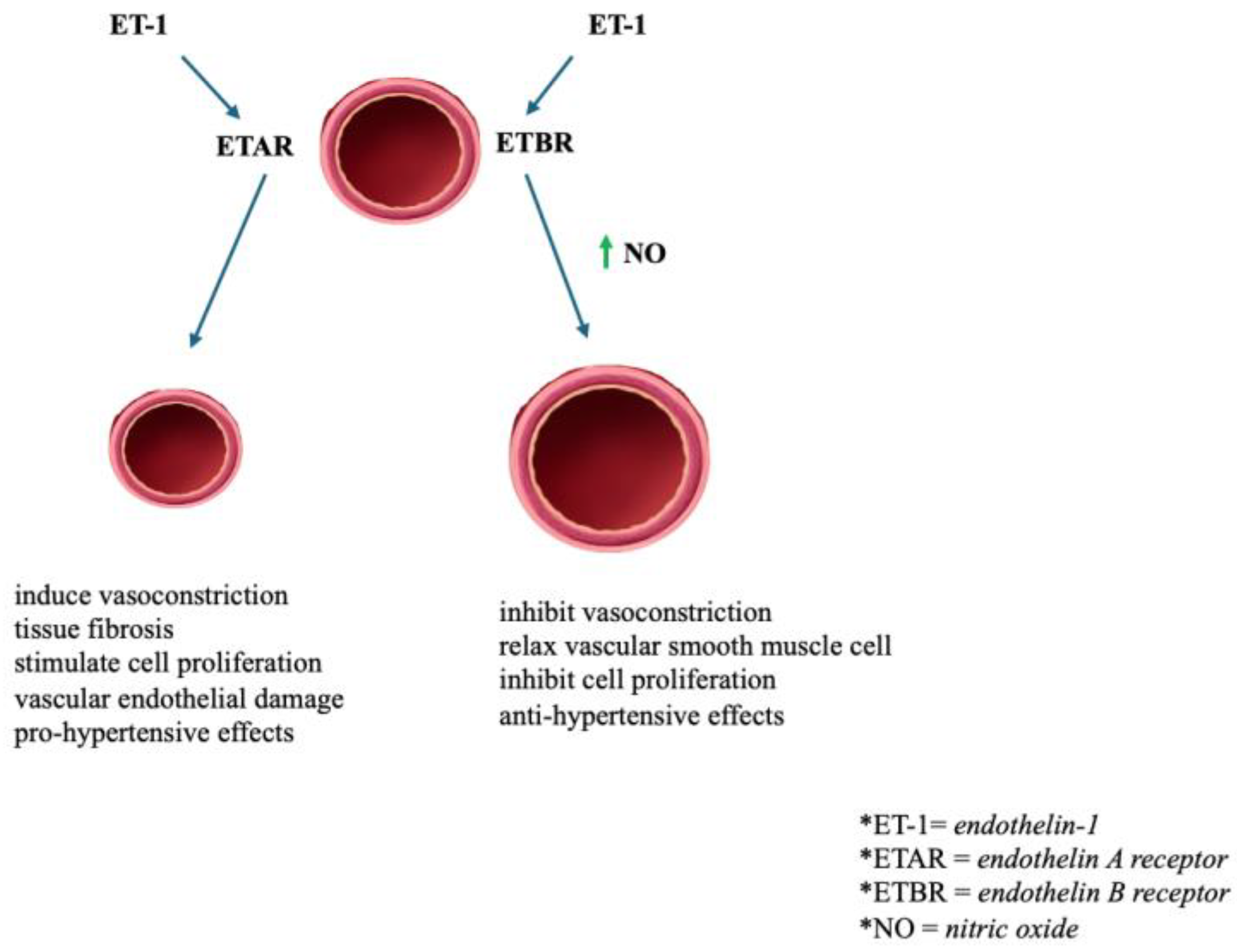
2.8. Endothelin Receptor (ETR)
2.9. Dual L-Type Calcium Channel Blocker/Endothelin A/B2 Receptor Antagonist
2.10. Drugs Targeting the NO Pathway
2.11. Dopamine β-Hydroxylase (DβH) Inhibitor
2.12. Ouabain Inhibitors
2.13. Leptin
2.14. Insulin-Resistant Aminopeptidase (IRAP)
2.15. Gastrointestinal Microbiota
2.16. Vaccines

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Drug | Mechanism | Main Findings | Clinical Trial/Study |
|---|---|---|---|---|
| NP, pGC-A | CRRL269 MANP (ZD100) ANX042 | pGC-A activator | - Vasorelaxation and antihypertensive properties in a canine model of ischemia-induced acute renal dysfunction - Effective at lowering BP, with considerable renal protective function and decreased aldosterone levels - Significant diuretic and natriuretic effects lacking vasodilatory hypotensive features - Higher decreases in office SBP and DBP and 24-h ambulatory SBP than valsartan alone | Chen et al. MANP-HTN-MS (completed 2020) Chen et al. |
| Neprilysin+ Angiotensin II Receptor Blockers | Valsartan/sacubitril (LCZ 696) | Angiotensin receptor-neprilysin inhibitors | NCT00549770 (completed 2015) | |
| Guanylyl cyclase A | Praliciguat (IW1973) | Stimulator of guanylate cyclase | - Attenuated hypertension and NO deficiency-related diseases | Shea et al. |
| Nonsteroidal mineralocorticoid receptors (MRAs) | Finerenone Esaxerenone KBP-5074 | Mineralocorticoid receptor antagonist | - Suppresses mineralocorticoid receptor-mediated fibrotic remodeling in mice cardiac fibroblasts - As monotherapy or as an addition to RAAS inhibitor treatment showed significant antihypertensive benefits - Dose-dependently decreased BP and 24-h urinary albumin excretion in the Dahl salt-sensitive hypertensive rat model - Substantially decreased SBP at the completion of the human study | Lavall et al. NCT02722265 (completed 2019) Chow et al. BLOCK-CKD (Phase 2b—completed 2024) |
| Sodium/glucose cotransporter-2 | Canagliflozin Dapagliflozin Empagliflozin | SGLT2 inhibitor | - A decrease in SBP was observed. - Considerably lowered BP and HbA1c and was comparable to placebo in terms of tolerability - Reduced SBP and DBP versus placebo | NCT00642278 (completed 2013) NCT01195662 (completed 2016) EMPA-REG BP (completed 2016) |
| Aminopeptidase A | Firibastat (RB150) NI956 | APA inhibitor | - The following was observed in experimental models of hypertension: (1) a reduction in vasopressin release from the posterior pituitary into the circulation, resulting in enhanced diuresis and lowered extracellular volume; (2) a reduction in sympathetic tone, and, thus, lower vascular resistance; and (3) an improvement in baroreflex activity. - In human trials, it failed to demonstrate efficacy in lowering unattended office systolic BP. - Ten times more potent and effective than firibastat in inhibiting brain APA enzymatic activity in vitro and in vivo in hypertensive rats | FRESH (completed 2022) Keck et al. |
| Vasoactive intestinal peptide | Vasomera (PB1046) | VIP inhibitor agonists | - Dose-dependently lowers both SBP and DBP, with no clinically relevant dose-dependent changes in HR | NCT01523067 (completed 2013) |
| Na+/H+ exchanger 3 (NHE3) | Tenapanor SAR 218034 AVE-0657 | NHE3 inhibitor | - Lowers BP, fluid volume, albuminuria, and left ventricular hypertrophy in rats with nephrectomized kidneys that have been given a salty diet - Enhances fecal sodium excretion, decreases urine sodium excretion and intestinal sodium uptake, and significantly decreases SBP in rats - Causes natriuresis and substantially lowers hypertension in Ang II-infused, high-salt-fed rats | Spencer et al. Gao et al. Li et al./Zhuo et al. |
| Endothelin receptor | Bosentan Darusentan Aprocitentan | Nonselective ETR antagonists Selective ETR antagonist Dual ETAR/ETBR antagonist | - The antihypertensive effect of bosentan was equivalent to that of enalapril. - The addition of darusentan led to a considerable reduction in BP in patients with RHTN. - Placebo and darusentan did not vary substantially after 14 weeks regarding the main endpoints, especially sitting office BP. - It was well tolerated and superior to a placebo in decreasing blood pressure at week 4, with a maintained effect at week 40 in individuals with RHTN. | Krum et al. DORADO (NCT00330369-completed 2014) DORADO-AC (NCT00389779-completed 2014) PRECISION (NCT03541174- completed 2023) |
| Dual L-Type Calcium Channel /Endothelin A/B2 Receptor | Sargachromenol-D | Dual L-type calcium channel blocker/ET A/B2 antagonist | - Lowers ET-1 and K+ depolarization-induced vasoconstriction in rabbit basilar arteries and lowers BP in rodent models of hypertension | Park et al. |
| Nitric oxide (NO) | Sphingosine-1 phosphate (FTY702) L-arginine or L-citrulline | S1PR1 antagonist NO synthase activator | - Might enhance BP and worsen HTN in an ANG II rat model - May reduce BP in hypertensive rats | Gao et al. Dumont et al. |
| Dopamineβ-hydroxylase (DβH) | Etamicastat (BIA 5–453) | Dopamine β-hydroxylase inhibitor | - In healthy males and individuals with mild to moderate HTN, 24-h ambulatory BP was reduced dose-dependently. | Almeida et al. |
| Ouabain | Rostafuroxin | Ouabain inhibitors | - Patients with the genetic profile P2a or LSS AA genotype responded more favorably (greater SBP drop) to rostafuroxin 50 μg than to losartan. | PEARL-HT |
| Insulin-resistant aminopeptidase | HFI-419 | Insulin-resistant aminopeptidase (IRAP) inhibitor | - Superiority to candesartan cilexetil in antifibrotic effectiveness and renoprotection and to the ACE inhibitor, perindopril, in mouse kidney injury produced by abnormally high salt concentrations | Gaspari et al. |
3. Other Possible Targets in Hypertension
3.1. Chemerin
3.2. Autophagy
3.3. Acetylation
3.4. Angiogenesis
3.5. Antioxidative Nutraceuticals Supplementation
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Conflicts of Interest
References
- Mills, K.T.; Stefanescu, A.; He, J. The global epidemiology of hypertension. Nat. Rev. Nephrol. 2020, 16, 223–237. [Google Scholar] [CrossRef] [PubMed]
- Tackling, G.; Borhade, M.B. Hypertensive Heart Disease. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Ettehad, D.; Emdin, C.A.; Kiran, A.; Anderson, S.G.; Callender, T.; Emberson, J.; Chalmers, J.; Rodgers, A.; Rahimi, K. Blood pressure lowering for prevention of cardiovascular disease and death: A systematic review and meta-analysis. Lancet 2016, 387, 957–967. [Google Scholar] [CrossRef] [PubMed]
- Banegas, J.R.; López-García, E.; Dallongeville, J.; Guallar, E.; Halcox, J.P.; Borghi, C.; Massó-González, E.L.; Jiménez, F.J.; Perk, J.; Steg, P.G.; et al. Achievement of treatment goals for primary prevention of cardiovascular disease in clinical practice across Europe: The EURIKA study. Eur. Heart J. 2011, 32, 2143–2152. [Google Scholar] [CrossRef] [PubMed]
- Chow, C.K.; Teo, K.K.; Rangarajan, S.; Islam, S.; Gupta, R.; Avezum, A.; Bahonar, A.; Chifamba, J.; Dagenais, G.; Diaz, R.; et al. Prevalence, awareness, treatment, and control of hypertension in rural and urban communities in high-, middle-, and low-income countries. JAMA 2013, 310, 959–968. [Google Scholar] [CrossRef] [PubMed]
- Falaschetti, E.; Mindell, J.; Knott, C.; Poulter, N. Hypertension management in England: A serial cross-sectional study from 1994 to 2011. Lancet 2014, 383, 1912–1919. [Google Scholar] [CrossRef]
- Tocci, G.; Rosei, E.A.; Ambrosioni, E.; Borghi, C.; Ferri, C.; Ferrucci, A.; Mancia, G.; Morganti, A.; Pontremoli, R.; Trimarco, B.; et al. Blood pressure control in Italy: Analysis of clinical data from 2005-2011 surveys on hypertension. J. Hypertens. 2012, 30, 1065–1074. [Google Scholar] [CrossRef]
- Ghatage, T.; Goyal, S.G.; Dhar, A.; Bhat, A. Novel therapeutics for the treatment of hypertension and its associated complications: Peptide- and nonpeptide-based strategies. Hypertens. Res. 2021, 44, 740–755. [Google Scholar] [CrossRef]
- Arendse, L.B.; Danser, A.H.J.; Poglitsch, M.; Touyz, R.M.; Burnett, J.C.; Llorens-Cortes, C.; Ehlers, M.R.; Sturrock, E.D. Novel Therapeutic Approaches Targeting the Renin-Angiotensin System and Associated Peptides in Hypertension and Heart Failure. Pharmacol. Rev. 2019, 71, 539–570. [Google Scholar] [CrossRef] [PubMed]
- Malek, V.; Gaikwad, A.B. Neprilysin inhibitors: A new hope to halt the diabetic cardiovascular and renal complications? Biomed. Pharmacother. 2017, 90, 752–759. [Google Scholar] [CrossRef]
- Chen, Y.; Harty, G.J.; Zheng, Y.; Iyer, S.R.; Sugihara, S.; Sangaralingham, S.J.; Ichiki, T.; Grande, J.P.; Lee, H.-C.; Wang, X.; et al. CRRL269. Circ. Res. 2019, 124, 1462–1472. [Google Scholar] [CrossRef]
- Chen, H.H.; Neutel, J.M.; Smith, D.H.; Heublein, D.; Burnett, J.C. Abstract 15143: ZD100: BP Lowering, Renal Enhancing and Aldosterone Suppressing Properties via pGC-A in Human Resistant “Like” Hypertension—A First in Human Study. Circulation 2016, 134, A15143. [Google Scholar]
- Chen, H.H.; Simari, R.D.; Youngberg, S.P.; Grogan, D.R.; Miller, J.W.; Burnett, J.C. Abstract 14201: ANX-042, a Novel Natriuretic Peptide (NP), is Safe and Stimulates Cyclic Guanosine Monophosphate (cGMP) in Healthy Volunteers. Circulation 2013, 128, A14201. [Google Scholar]
- Mills, J.; Vardeny, O. The role of neprilysin inhibitors in cardiovascular disease. Curr. Heart Fail. Rep. 2015, 12, 389–394. [Google Scholar] [CrossRef]
- Braunwald, E. The path to an angiotensin receptor antagonist-neprilysin inhibitor in the treatment of heart failure. J. Am. Coll. Cardiol. 2015, 65, 1029–1041. [Google Scholar] [CrossRef]
- Judge, P.; Haynes, R.; Landray, M.J.; Baigent, C. Neprilysin inhibition in chronic kidney disease. Nephrol. Dial. Transplant. 2015, 30, 738–743. [Google Scholar] [CrossRef] [PubMed]
- Kalk, P.; Sharkovska, Y.; Kashina, E.; von Websky, K.; Relle, K.; Pfab, T.; Alter, M.; Guillaume, P.; Provost, D.; Hoffmann, K.; et al. Endothelin-converting enzyme/neutral endopeptidase inhibitor SLV338 prevents hypertensive cardiac remodeling in a blood pressure-independent manner. Hypertension 2011, 57, 755–763. [Google Scholar] [CrossRef]
- Kario, K.; Williams, B. Angiotensin receptor-neprilysin inhibitors for hypertension-hemodynamic effects and relevance to hypertensive heart disease. Hypertens. Res. 2022, 45, 1097–1110. [Google Scholar] [CrossRef]
- Ruilope, L.M.; Dukat, A.; Böhm, M.; Lacourcière, Y.; Gong, J.; Lefkowitz, M.P. Blood-pressure reduction with LCZ696, a novel dual-acting inhibitor of the angiotensin II receptor and neprilysin: A randomised, double-blind, placebo-controlled, active comparator study. Lancet 2010, 375, 1255–1266. [Google Scholar] [CrossRef]
- Kario, K.; Tamaki, Y.; Okino, N.; Gotou, H.; Zhu, M.; Zhang, J. LCZ696, a First-in-Class Angiotensin Receptor-Neprilysin Inhibitor: The First Clinical Experience in Patients with Severe Hypertension. J. Clin. Hypertens. 2016, 18, 308–314. [Google Scholar] [CrossRef]
- Buys, E.; Sips, P. New insights into the role of soluble guanylate cyclase in blood pressure regulation. Curr. Opin. Nephrol. Hypertens. 2014, 23, 135–142. [Google Scholar] [CrossRef]
- Nagasaka, Y.; Buys, E.S.; Spagnolli, E.; Steinbicker, A.U.; Hayton, S.R.; Rauwerdink, K.M.; Brouckaert, P.; Zapol, W.M.; Bloch, K.D. Soluble guanylate cyclase-α1 is required for the cardioprotective effects of inhaled nitric oxide. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, H1477–H1483. [Google Scholar] [CrossRef] [PubMed]
- Buys, E.S.; Cauwels, A.; Raher, M.J.; Passeri, J.J.; Hobai, I.; Cawley, S.M.; Rauwerdink, K.M.; Thibault, H.; Sips, P.Y.; Thoonen, R.; et al. sGC(α)1(β)1 attenuates cardiac dysfunction and mortality in murine inflammatory shock models. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H654–H663. [Google Scholar] [CrossRef]
- Atochin, D.N.; Yuzawa, I.; Li, Q.; Rauwerdink, K.M.; Malhotra, R.; Chang, J.; Brouckaert, P.; Ayata, C.; Moskowitz, M.A.; Bloch, K.D.; et al. Soluble guanylate cyclase α1β1 limits stroke size and attenuates neurological injury. Stroke 2010, 41, 1815–1819. [Google Scholar] [CrossRef]
- Krishnan, S.M.; Kraehling, J.R.; Eitner, F.; Bénardeau, A.; Sandner, P. The impact of the nitric oxide (no)/soluble guanylyl cyclase (sgc) signaling cascade on kidney health and disease: A preclinical perspective. Int. J. Mol. Sci. 2018, 19, 1712. [Google Scholar] [CrossRef]
- Kang, Y.; Liu, R.; Wu, J.-X.; Chen, L. Structural insights into the mechanism of human soluble guanylate cyclase. Nature 2019, 574, 206–210. [Google Scholar] [CrossRef]
- Sandner, P.; Stasch, J.P. Anti-fibrotic effects of soluble guanylate cyclase stimulators and activators: A review of the preclinical evidence. Respir. Med. 2017, 122 (Suppl. S1), S1–S9. [Google Scholar] [CrossRef] [PubMed]
- Stasch, J.-P.; Schlossmann, J.; Hocher, B. Renal effects of soluble guanylate cyclase stimulators and activators: A review of the preclinical evidence. Curr. Opin. Pharmacol. 2015, 21, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Shea, C.M.; Price, G.M.; Liu, G.; Sarno, R.; Buys, E.S.; Currie, M.G.; Masferrer, J.L. Soluble guanylate cyclase stimulator praliciguat attenuates inflammation, fibrosis, and end-organ damage in the Dahl model of cardiorenal failure. Am. J. Physiol. Renal Physiol. 2020, 318, F148–F159. [Google Scholar] [CrossRef]
- Carey, R.M.; Calhoun, D.A.; Bakris, G.L.; Brook, R.D.; Daugherty, S.L.; Dennison-Himmelfarb, C.R.; Egan, B.M.; Flack, J.M.; Gidding, S.S.; Judd, E.; et al. Resistant hypertension: Detection, evaluation, and management: A scientific statement from the american heart association. Hypertension 2018, 72, e53–e90. [Google Scholar] [CrossRef]
- Ponikowski, P.; Voors, A.A.; Anker, S.D.; Bueno, H.; Cleland, J.G.F.; Coats, A.J.S.; Falk, V.; González-Juanatey, J.R.; Harjola, V.-P.; Jankowska, E.A.; et al. The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC)Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur. Heart J. 2016, 37, 2129–2200. [Google Scholar] [CrossRef]
- Williams, B.; Mancia, G.; Spiering, W.; Agabiti Rosei, E.; Azizi, M.; Burnier, M.; Clement, D.L.; Coca, A.; de Simone, G.; Dominiczak, A.; et al. 2018 ESC/ESH Guidelines for the management of arterial hypertension. Eur. Heart J. 2018, 39, 3021–3104. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, A. Pathophysiological mechanisms of mineralocorticoid receptor-dependent cardiovascular and chronic kidney disease. Hypertens. Res. 2019, 42, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Azizi, M.; Rossignol, P.; Hulot, J.-S. Emerging drug classes and their potential use in hypertension. Hypertension 2019, 74, 1075–1083. [Google Scholar] [CrossRef] [PubMed]
- Lavall, D.; Jacobs, N.; Mahfoud, F.; Kolkhof, P.; Böhm, M.; Laufs, U. The non-steroidal mineralocorticoid receptor antagonist finerenone prevents cardiac fibrotic remodeling. Biochem. Pharmacol. 2019, 168, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Itoh, H.; Rakugi, H.; Okuda, Y.; Yamakawa, S. Efficacy and safety of esaxerenone (CS-3150) for the treatment of essential hypertension: A phase 2 randomized, placebo-controlled, double-blind study. J. Hum. Hypertens. 2019, 33, 542–551. [Google Scholar] [CrossRef]
- Ito, S.; Itoh, H.; Rakugi, H.; Okuda, Y.; Iijima, S. Antihypertensive effects and safety of esaxerenone in patients with moderate kidney dysfunction. Hypertens. Res. 2021, 44, 489–497. [Google Scholar] [CrossRef]
- Ito, S.; Itoh, H.; Rakugi, H.; Okuda, Y.; Yoshimura, M.; Yamakawa, S. Double-Blind Randomized Phase 3 Study Comparing Esaxerenone (CS-3150) and Eplerenone in Patients with Essential Hypertension (ESAX-HTN Study). Hypertension 2020, 75, 51–58. [Google Scholar] [CrossRef]
- Cp, C. Pharmacological Profile of KBP-5074, a Novel NonSteroidal Mineralocorticoid Receptor Antagonist for the Treatment of Cardiorenal Diseases. J. Drug Res. Dev. 2017, 3, 1–9. [Google Scholar] [CrossRef]
- Bakris, G.; Pergola, P.E.; Delgado, B.; Genov, D.; Doliashvili, T.; Vo, N.; Yang, Y.F.; McCabe, J.; Benn, V.; Pitt, B. Effect of KBP-5074 on Blood Pressure in Advanced Chronic Kidney Disease: Results of the BLOCK-CKD Study. Hypertension 2021, 78, 74–81. [Google Scholar] [CrossRef]
- Kalra, S. Sodium Glucose Co-Transporter-2 (SGLT2) Inhibitors: A Review of Their Basic and Clinical Pharmacology. Diabetes Ther. 2014, 5, 355–366. [Google Scholar] [CrossRef]
- Ni, L.; Yuan, C.; Chen, G.; Zhang, C.; Wu, X. SGLT2i: Beyond the glucose-lowering effect. Cardiovasc. Diabetol. 2020, 19, 98. [Google Scholar] [CrossRef] [PubMed]
- Briasoulis, A.; Al Dhaybi, O.; Bakris, G.L. SGLT2 inhibitors and mechanisms of hypertension. Curr. Cardiol. Rep. 2018, 20, 1. [Google Scholar] [CrossRef]
- Gupta, R.; Maitz, T.; Egeler, D.; Mehta, A.; Nyaeme, M.; Hajra, A.; Goel, A.; Sreenivasan, J.; Patel, N.; Aronow, W.S. SGLT2 inhibitors in hypertension: Role beyond diabetes and heart failure. Trends Cardiovasc. Med. 2022, 33, 479–486. [Google Scholar] [CrossRef] [PubMed]
- DeFronzo, R.A.; Norton, L.; Abdul-Ghani, M. Renal, metabolic and cardiovascular considerations of SGLT2 inhibition. Nat. Rev. Nephrol. 2017, 13, 11–26. [Google Scholar] [CrossRef]
- Shin, S.J.; Chung, S.; Kim, S.J.; Lee, E.-M.; Yoo, Y.-H.; Kim, J.-W.; Ahn, Y.-B.; Kim, E.-S.; Moon, S.-D.; Kim, M.-J.; et al. Effect of Sodium-Glucose Co-Transporter 2 Inhibitor, Dapagliflozin, on Renal Renin-Angiotensin System in an Animal Model of Type 2 Diabetes. PLoS ONE 2016, 11, e0165703. [Google Scholar] [CrossRef]
- Cherney, D.Z.; Perkins, B.A.; Soleymanlou, N.; Har, R.; Fagan, N.; Johansen, O.E.; Woerle, H.-J.; von Eynatten, M.; Broedl, U.C. The effect of empagliflozin on arterial stiffness and heart rate variability in subjects with uncomplicated type 1 diabetes mellitus. Cardiovasc. Diabetol. 2014, 13, 28. [Google Scholar] [CrossRef] [PubMed]
- Pfeifer, M.; Townsend, R.R.; Davies, M.J.; Vijapurkar, U.; Ren, J. Effects of canagliflozin, a sodium glucose co-transporter 2 inhibitor, on blood pressure and markers of arterial stiffness in patients with type 2 diabetes mellitus: A post hoc analysis. Cardiovasc. Diabetol. 2017, 16, 29. [Google Scholar] [CrossRef]
- Villafaña, S.; Huang, F.; Hong, E. Role of the sympathetic and renin angiotensin systems in the glucose-induced increase of blood pressure in rats. Eur. J. Pharmacol. 2004, 506, 143–150. [Google Scholar] [CrossRef]
- Yoshikawa, T.; Kishi, T.; Shinohara, K.; Takesue, K.; Shibata, R.; Sonoda, N.; Inoguchi, T.; Sunagawa, K.; Tsutsui, H.; Hirooka, Y. Arterial pressure lability is improved by sodium-glucose cotransporter 2 inhibitor in streptozotocin-induced diabetic rats. Hypertens. Res. 2017, 40, 646–651. [Google Scholar] [CrossRef]
- Tahara, A.; Kurosaki, E.; Yokono, M.; Yamajuku, D.; Kihara, R.; Hayashizaki, Y.; Takasu, T.; Imamura, M.; Li, Q.; Tomiyama, H.; et al. Effects of SGLT2 selective inhibitor ipragliflozin on hyperglycemia, hyperlipidemia, hepatic steatosis, oxidative stress, inflammation, and obesity in type 2 diabetic mice. Eur. J. Pharmacol. 2013, 715, 246–255. [Google Scholar] [CrossRef]
- Han, J.H.; Oh, T.J.; Lee, G.; Maeng, H.J.; Lee, D.H.; Kim, K.M.; Choi, S.H.; Jang, H.C.; Lee, H.S.; Park, K.S.; et al. The beneficial effects of empagliflozin, an SGLT2 inhibitor, on atherosclerosis in ApoE-/- mice fed a western diet. Diabetologia 2017, 60, 364–376. [Google Scholar] [CrossRef] [PubMed]
- Vasilakou, D.; Karagiannis, T.; Athanasiadou, E.; Mainou, M.; Liakos, A.; Bekiari, E.; Sarigianni, M.; Matthews, D.R.; Tsapas, A. Sodium-glucose cotransporter 2 inhibitors for type 2 diabetes: A systematic review and meta-analysis. Ann. Intern. Med. 2013, 159, 262–274. [Google Scholar] [CrossRef] [PubMed]
- Mazidi, M.; Rezaie, P.; Gao, H.-K.; Kengne, A.P. Effect of Sodium-Glucose Cotransport-2 Inhibitors on Blood Pressure in People with Type 2 Diabetes Mellitus: A Systematic Review and Meta-Analysis of 43 Randomized Control Trials With 22 528 Patients. J. Am. Heart Assoc. 2017, 6, e004007. [Google Scholar] [CrossRef] [PubMed]
- Baker, W.L.; Buckley, L.F.; Kelly, M.S.; Bucheit, J.D.; Parod, E.D.; Brown, R.; Carbone, S.; Abbate, A.; Dixon, D.L. Effects of Sodium-Glucose Cotransporter 2 Inhibitors on 24-Hour Ambulatory Blood Pressure: A Systematic Review and Meta-Analysis. J. Am. Heart Assoc. 2017, 6, e005686. [Google Scholar] [CrossRef]
- Mancia, G.; Cannon, C.P.; Tikkanen, I.; Zeller, C.; Ley, L.; Woerle, H.J.; Broedl, U.C.; Johansen, O.E. Impact of empagliflozin on blood pressure in patients with type 2 diabetes mellitus and hypertension by background antihypertensive medication. Hypertension 2016, 68, 1355–1364. [Google Scholar] [CrossRef] [PubMed]
- Rosenstock, J.; Aggarwal, N.; Polidori, D.; Zhao, Y.; Arbit, D.; Usiskin, K.; Capuano, G.; Canovatchel, W.; Canagliflozin DIA 2001 Study Group. Dose-ranging effects of canagliflozin, a sodium-glucose cotransporter 2 inhibitor, as add-on to metformin in subjects with type 2 diabetes. Diabetes Care 2012, 35, 1232–1238. [Google Scholar] [CrossRef]
- Weber, M.A.; Mansfield, T.A.; Cain, V.A.; Iqbal, N.; Parikh, S.; Ptaszynska, A. Blood pressure and glycaemic effects of dapagliflozin versus placebo in patients with type 2 diabetes on combination antihypertensive therapy: A randomised, double-blind, placebo-controlled, phase 3 study. Lancet Diabetes Endocrinol. 2016, 4, 211–220. [Google Scholar] [CrossRef]
- Tran, K.C.; Hiremath, S. SGLT2 inhibitors in resistant hypertension: A sweet solution. Am. J. Hypertens. 2020, 33, 1071–1074. [Google Scholar] [CrossRef]
- Benjamin, E.J.; Muntner, P.; Alonso, A.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Das, S.R.; et al. Disease and Stroke Statistics—2019 Update: A Report from the American Heart Association. Circulation 2019, 139, e56–e528. [Google Scholar] [CrossRef]
- Nguyen, Q.; Dominguez, J.; Nguyen, L.; Gullapalli, N. Hypertension management: An update. Am. Health Drug Benefits 2010, 3, 47–56. [Google Scholar]
- Ganten, D.; Hermann, K.; Bayer, C.; Unger, T.; Lang, R.E. Angiotensin synthesis in the brain and increased turnover in hypertensive rats. Science 1983, 221, 869–871. [Google Scholar] [CrossRef] [PubMed]
- Basso, N.; Ruiz, P.; Mangiarua, E.; Taquini, A.C. Renin-like activity in the rat brain during the development of DOC-salt hypertension. Hypertension 1981, 3, II-14. [Google Scholar] [CrossRef] [PubMed]
- Davisson, R.L.; Yang, G.; Beltz, T.G.; Cassell, M.D.; Johnson, A.K.; Sigmund, C.D. The brain renin-angiotensin system contributes to the hypertension in mice containing both the human renin and human angiotensinogen transgenes. Circ. Res. 1998, 83, 1047–1058. [Google Scholar] [CrossRef]
- Marc, Y.; Llorens-Cortes, C. The role of the brain renin-angiotensin system in hypertension: Implications for new treatment. Prog. Neurobiol. 2011, 95, 89–103. [Google Scholar] [CrossRef]
- Bodineau, L.; Frugière, A.; Marc, Y.; Claperon, C.; Llorens-Cortes, C. Aminopeptidase A inhibitors as centrally acting antihypertensive agents. Heart Fail. Rev. 2008, 13, 311–319. [Google Scholar] [CrossRef]
- Nagatsu, I.; Nagatsu, T.; Yamamoto, T.; Glenner, G.G.; Mehl, J.W. Purification of aminopeptidase a in human serum and degradation of angiotensin II by the purified enzyme. Biochim. Biophys. Acta (BBA)—Enzymol. 1970, 198, 255–270. [Google Scholar] [CrossRef]
- Keck, M.; Hmazzou, R.; Llorens-Cortes, C. Orally active aminopeptidase A inhibitor prodrugs: Current state and future directions. Curr. Hypertens. Rep. 2019, 21, 50. [Google Scholar] [CrossRef]
- Gao, J.; Marc, Y.; Iturrioz, X.; Leroux, V.; Balavoine, F.; Llorens-Cortes, C. A new strategy for treating hypertension by blocking the activity of the brain renin-angiotensin system with aminopeptidase A inhibitors. Clin. Sci. 2014, 127, 135–148. [Google Scholar] [CrossRef]
- Alomar, S.A.; Alghabban, S.A.; Alharbi, H.A.; Almoqati, M.F.; Alduraibi, Y.; Abu-Zaid, A. Firibastat, the first-in-class brain aminopeptidase a inhibitor, in the management of hypertension: A review of clinical trials. Avicenna J. Med. 2021, 11, 1–7. [Google Scholar] [CrossRef]
- Reaux, A.; Fournie-Zaluski, M.C.; David, C.; Zini, S.; Roques, B.P.; Corvol, P.; Llorens-Cortes, C. Aminopeptidase A inhibitors as potential central antihypertensive agents. Proc. Natl. Acad. Sci. USA 1999, 96, 13415–13420. [Google Scholar] [CrossRef]
- Fournie-Zaluski, M.-C.; Fassot, C.; Valentin, B.; Djordjijevic, D.; Reaux-Le Goazigo, A.; Corvol, P.; Roques, B.P.; Llorens-Cortes, C. Brain renin-angiotensin system blockade by systemically active aminopeptidase A inhibitors: A potential treatment of salt-dependent hypertension. Proc. Natl. Acad. Sci. USA 2004, 101, 7775–7780. [Google Scholar] [CrossRef] [PubMed]
- Bodineau, L.; Frugière, A.; Marc, Y.; Inguimbert, N.; Fassot, C.; Balavoine, F.; Roques, B.; Llorens-Cortes, C. Orally active aminopeptidase A inhibitors reduce blood pressure: A new strategy for treating hypertension. Hypertension 2008, 51, 1318–1325. [Google Scholar] [CrossRef] [PubMed]
- Keck, M.; De Almeida, H.; Compère, D.; Inguimbert, N.; Flahault, A.; Balavoine, F.; Roques, B.; Llorens-Cortes, C. NI956/QGC006, a Potent Orally Active, Brain-Penetrating Aminopeptidase A Inhibitor for Treating Hypertension. Hypertension 2019, 73, 1300–1307. [Google Scholar] [CrossRef] [PubMed]
- Marc, Y.; Gao, J.; Balavoine, F.; Michaud, A.; Roques, B.P.; Llorens-Cortes, C. Central antihypertensive effects of orally active aminopeptidase A inhibitors in spontaneously hypertensive rats. Hypertension 2012, 60, 411–418. [Google Scholar] [CrossRef] [PubMed]
- Marc, Y.; Hmazzou, R.; Balavoine, F.; Flahault, A.; Llorens-Cortes, C. Central antihypertensive effects of chronic treatment with RB150: An orally active aminopeptidase A inhibitor in deoxycorticosterone acetate-salt rats. J. Hypertens. 2018, 36, 641–650. [Google Scholar] [CrossRef]
- Wright, J.W.; Tamura-Myers, E.; Wilson, W.L.; Roques, B.P.; Llorens-Cortes, C.; Speth, R.C.; Harding, J.W. Conversion of brain angiotensin II to angiotensin III is critical for pressor response in rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2003, 284, R725–R733. [Google Scholar] [CrossRef]
- Marc, Y.; Boitard, S.E.; Balavoine, F.; Azizi, M.; Llorens-Cortes, C. Targeting brain aminopeptidase A: A new strategy for the treatment of hypertension and heart failure. Can. J. Cardiol. 2020, 36, 721–731. [Google Scholar] [CrossRef]
- Huang, B.S.; Ahmad, M.; White, R.A.; Marc, Y.; Llorens-Cortes, C.; Leenen, F.H.H. Inhibition of brain angiotensin III attenuates sympathetic hyperactivity and cardiac dysfunction in rats post-myocardial infarction. Cardiovasc. Res. 2013, 97, 424–431. [Google Scholar] [CrossRef]
- Balavoine, F.; Azizi, M.; Bergerot, D.; De Mota, N.; Patouret, R.; Roques, B.P.; Llorens-Cortes, C. Randomised, double-blind, placebo-controlled, dose-escalating phase I study of QGC001, a centrally acting aminopeptidase a inhibitor prodrug. Clin. Pharmacokinet. 2014, 53, 385–395. [Google Scholar] [CrossRef]
- Azizi, M.; Courand, P.-Y.; Denolle, T.; Delsart, P.; Zhygalina, V.; Amar, L.; Lantelme, P.; Mounier-Vehier, C.; De Mota, N.; Balavoine, F.; et al. A pilot double-blind randomized placebo-controlled crossover pharmacodynamic study of the centrally active aminopeptidase A inhibitor, firibastat, in hypertension. J. Hypertens. 2019, 37, 1722–1728. [Google Scholar] [CrossRef]
- Ferdinand, K.C.; Balavoine, F.; Besse, B.; Black, H.R.; Desbrandes, S.; Dittrich, H.C.; Nesbitt, S.D.; NEW HOPE Investigators. Efficacy and Safety of Firibastat, A First-in-Class Brain Aminopeptidase A Inhibitor, in Hypertensive Overweight Patients of Multiple Ethnic Origins. Circulation 2019, 140, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Firibastat in Treatment-Resistant Hypertension—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT04277884 (accessed on 19 July 2022).
- Henning, R.J.; Sawmiller, D.R. Vasoactive intestinal peptide: Cardiovascular effects. Cardiovasc. Res. 2001, 49, 27–37. [Google Scholar] [CrossRef]
- Kasacka, I.; Piotrowska, Ż.; Janiuk, I. Influence of renovascular hypertension on the distribution of vasoactive intestinal peptide in the stomach and heart of rats. Exp. Biol. Med. 2015, 240, 1402–1407. [Google Scholar] [CrossRef] [PubMed]
- Couvineau, A.; Laburthe, M. VPAC receptors: Structure, molecular pharmacology and interaction with accessory proteins. Br. J. Pharmacol. 2012, 166, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.; Xu, L.; Cai, J. New drug targets for hypertension: A literature review. Biochim. Biophys. Acta Mol. Basis Dis. 2021, 1867, 166037. [Google Scholar] [CrossRef]
- Davis, J.; Oparil, S. Novel medical treatments for hypertension and related comorbidities. Curr. Hypertens. Rep. 2018, 20, 90. [Google Scholar] [CrossRef]
- Free, A.; Brazg, R.; Matson, M.; Smith, W.; Chuck, L.; Georgopoulos, L.; Malatesta, J.; Arnold, S.; Kramer, W.; Strange, P.; et al. Abstract 19112: A Phase 1, Multi-center, Randomized, Double-blind, Placebo Controlled Study to Evaluate the Safety/Tolerability, Pharmacokinetic and Hemodynamic Response Following Single Ascending Subcutaneous Doses of PB1046 (VasomeraTM) in Subjects with Essential Hypertension. Circulation 2014, 130, A19112. [Google Scholar]
- Linz, B.; Saljic, A.; Hohl, M.; Gawałko, M.; Jespersen, T.; Sanders, P.; Böhm, M.; Linz, D. Inhibition of sodium-proton-exchanger subtype 3-mediated sodium absorption in the gut: A new antihypertensive concept. Int. J. Cardiol. Heart Vasc. 2020, 29, 100591. [Google Scholar] [CrossRef]
- Lambers Heerspink, H.J.; Holtkamp, F.A.; Parving, H.-H.; Navis, G.J.; Lewis, J.B.; Ritz, E.; de Graeff, P.A.; de Zeeuw, D. Moderation of dietary sodium potentiates the renal and cardiovascular protective effects of angiotensin receptor blockers. Kidney Int. 2012, 82, 330–337. [Google Scholar] [CrossRef]
- Felker, G.M.; O’Connor, C.M.; Braunwald, E. Loop diuretics in acute decompensated heart failure: Necessary? Evil? A necessary evil? Circ. Heart Fail. 2009, 2, 56–62. [Google Scholar] [CrossRef]
- Nwia, S.M.; Li, X.C.; Leite, A.P.D.O.; Hassan, R.; Zhuo, J.L. The Na+/H+ Exchanger 3 in the Intestines and the Proximal Tubule of the Kidney: Localization, Physiological Function, and Key Roles in Angiotensin II-Induced Hypertension. Front. Physiol. 2022, 13, 861659. [Google Scholar] [CrossRef] [PubMed]
- Linz, D.; Wirth, K.; Linz, W.; Heuer, H.O.O.; Frick, W.; Hofmeister, A.; Heinelt, U.; Arndt, P.; Schwahn, U.; Böhm, M.; et al. Antihypertensive and laxative effects by pharmacological inhibition of sodium-proton-exchanger subtype 3-mediated sodium absorption in the gut. Hypertension 2012, 60, 1560–1567. [Google Scholar] [CrossRef] [PubMed]
- Linz, B.; Hohl, M.; Reil, J.C.; Böhm, M.; Linz, D. Inhibition of NHE3-mediated Sodium Absorption in the Gut Reduced Cardiac End-organ Damage Without Deteriorating Renal Function in Obese Spontaneously Hypertensive Rats. J. Cardiovasc. Pharmacol. 2016, 67, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Onishi, A.; Fu, Y.; Patel, R.; Darshi, M.; Crespo-Masip, M.; Huang, W.; Song, P.; Freeman, B.; Kim, Y.C.; Soleimani, M.; et al. A role for tubular Na+/H+ exchanger NHE3 in the natriuretic effect of the SGLT2 inhibitor empagliflozin. Am. J. Physiol. Renal Physiol. 2020, 319, F712–F728. [Google Scholar] [CrossRef]
- Spencer, A.G.; Labonte, E.D.; Rosenbaum, D.P.; Plato, C.F.; Carreras, C.W.; Leadbetter, M.R.; Kozuka, K.; Kohler, J.; Koo-McCoy, S.; He, L.; et al. Intestinal inhibition of the Na+/H+ exchanger 3 prevents cardiorenal damage in rats and inhibits Na+ uptake in humans. Sci. Transl. Med. 2014, 6, 227ra36. [Google Scholar] [CrossRef]
- Wang, Q.; Zhou, R.; Zhang, C.; Dong, H.; Ma, J.; Wang, G. Inhibition of central Na(+)/H(+) exchanger type 3 can alleviate sleep apnea in Sprague-Dawley rats. Chin. Med. J. 2014, 127, 48–53. [Google Scholar] [CrossRef]
- Li, X.C.; Zhu, D.; Chen, X.; Zheng, X.; Zhao, C.; Zhang, J.; Soleimani, M.; Rubera, I.; Tauc, M.; Zhou, X.; et al. Proximal Tubule-Specific Deletion of the NHE3 (Na+/H+ Exchanger 3) in the Kidney Attenuates Ang II (Angiotensin II)-Induced Hypertension in Mice. Hypertension 2019, 74, 526–535. [Google Scholar] [CrossRef]
- Zhuo, J.L.; Soleimani, M.; Li, X.C. New Insights into the Critical Importance of Intratubular Na+/H+ Exchanger 3 and Its Potential Therapeutic Implications in Hypertension. Curr. Hypertens. Rep. 2021, 23, 34. [Google Scholar] [CrossRef]
- Yanagisawa, M.; Kurihara, H.; Kimura, S.; Tomobe, Y.; Kobayashi, M.; Mitsui, Y.; Yazaki, Y.; Goto, K.; Masaki, T. A novel potent vasoconstrictor peptide produced by vascular endothelial cells. Nature 1988, 332, 411–415. [Google Scholar] [CrossRef]
- Davenport, A.P. International Union of Pharmacology. XXIX. Update on endothelin receptor nomenclature. Pharmacol. Rev. 2002, 54, 219–226. [Google Scholar] [CrossRef]
- Davenport, A.P.; Hyndman, K.A.; Dhaun, N.; Southan, C.; Kohan, D.E.; Pollock, J.S.; Pollock, D.M.; Webb, D.J.; Maguire, J.J. Endothelin. Pharmacol. Rev. 2016, 68, 357–418. [Google Scholar] [CrossRef] [PubMed]
- Burnier, M. Update on endothelin receptor antagonists in hypertension. Curr. Hypertens. Rep. 2018, 20, 51. [Google Scholar] [CrossRef] [PubMed]
- Kiowski, W.; Lüscher, T.F.; Linder, L.; Bühler, F.R. Endothelin-1-induced vasoconstriction in humans. Reversal by calcium channel blockade but not by nitrovasodilators or endothelium-derived relaxing factor. Circulation 1991, 83, 469–475. [Google Scholar] [CrossRef] [PubMed]
- Iglarz, M.; Steiner, P.; Wanner, D.; Rey, M.; Hess, P.; Clozel, M. Vascular effects of endothelin receptor antagonists depends on their selectivity for ETA versus ETB receptors and on the functionality of endothelial ETB receptors. J. Cardiovasc. Pharmacol. 2015, 66, 332–337. [Google Scholar] [CrossRef]
- Boesen, E.I. Endothelin receptors, renal effects and blood pressure. Curr. Opin. Pharmacol. 2015, 21, 25–34. [Google Scholar] [CrossRef]
- Herrera, M.; Garvin, J.L. Endothelin stimulates endothelial nitric oxide synthase expression in the thick ascending limb. Am. J. Physiol. Renal Physiol. 2004, 287, F231–F235. [Google Scholar] [CrossRef]
- Krum, H.; Viskoper, R.J.; Lacourciere, Y.; Budde, M.; Charlon, V. The effect of an endothelin-receptor antagonist, bosentan, on blood pressure in patients with essential hypertension. Bosentan Hypertension Investigators. N. Engl. J. Med. 1998, 338, 784–790. [Google Scholar] [CrossRef]
- Nakov, R.; Pfarr, E.; Eberle, S.; HEAT Investigators. Darusentan: An effective endothelin A receptor antagonist for treatment of hypertension. Am. J. Hypertens. 2002, 15, 583–589. [Google Scholar] [CrossRef]
- Yuan, W.; Cheng, G.; Li, B.; Li, Y.; Lu, S.; Liu, D.; Xiao, J.; Zhao, Z. Endothelin-receptor antagonist can reduce blood pressure in patients with hypertension: A meta-analysis. Blood Press. 2017, 26, 139–149. [Google Scholar] [CrossRef]
- DORADO—Fixed Doses of Darusentan as Compared to Placebo in Resistant Hypertension—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT00330369 (accessed on 22 July 2022).
- DORADO-AC—Optimized Doses of Darusentan as Compared to an Active Control in Resistant Hypertension—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT00389779 (accessed on 22 July 2022).
- Weber, M.A.; Black, H.; Bakris, G.; Krum, H.; Linas, S.; Weiss, R.; Linseman, J.V.; Wiens, B.L.; Warren, M.S.; Lindholm, L.H. A selective endothelin-receptor antagonist to reduce blood pressure in patients with treatment-resistant hypertension: A randomised, double-blind, placebo-controlled trial. Lancet 2009, 374, 1423–1431. [Google Scholar] [CrossRef]
- Bakris, G.L.; Lindholm, L.H.; Black, H.R.; Krum, H.; Linas, S.; Linseman, J.V.; Arterburn, S.; Sager, P.; Weber, M. Divergent results using clinic and ambulatory blood pressures: Report of a darusentan-resistant hypertension trial. Hypertension 2010, 56, 824–830. [Google Scholar] [CrossRef]
- Iglarz, M.; Binkert, C.; Morrison, K.; Fischli, W.; Gatfield, J.; Treiber, A.; Weller, T.; Bolli, M.H.; Boss, C.; Buchmann, S.; et al. Pharmacology of macitentan, an orally active tissue-targeting dual endothelin receptor antagonist. J. Pharmacol. Exp. Ther. 2008, 327, 736–745. [Google Scholar] [CrossRef]
- Sidharta, P.N.; Dingemanse, J. Effect of Multiple-Dose Aprocitentan Administration on the Pharmacokinetics of Midazolam in Healthy Male Subjects. Eur. J. Drug Metab. Pharmacokinet. 2020, 45, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Sidharta, P.N.; Melchior, M.; Kankam, M.K.; Dingemanse, J. Single- and multiple-dose tolerability, safety, pharmacokinetics, and pharmacodynamics of the dual endothelin receptor antagonist aprocitentan in healthy adult and elderly subjects. Drug Des. Devel. Ther. 2019, 13, 949–964. [Google Scholar] [CrossRef] [PubMed]
- Trensz, F.; Bortolamiol, C.; Kramberg, M.; Wanner, D.; Hadana, H.; Rey, M.; Strasser, D.S.; Delahaye, S.; Hess, P.; Vezzali, E.; et al. Pharmacological Characterization of Aprocitentan, a Dual Endothelin Receptor Antagonist, Alone and in Combination with Blockers of the Renin Angiotensin System, in Two Models of Experimental Hypertension. J. Pharmacol. Exp. Ther. 2019, 368, 462–473. [Google Scholar] [CrossRef] [PubMed]
- Angeli, F.; Verdecchia, P.; Reboldi, G. Aprocitentan, A dual endothelin receptor antagonist under development for the treatment of resistant hypertension. Cardiol. Ther. 2021, 10, 397–406. [Google Scholar] [CrossRef]
- Verweij, P.; Danaietash, P.; Flamion, B.; Ménard, J.; Bellet, M. Randomized Dose-Response Study of the New Dual Endothelin Receptor Antagonist Aprocitentan in Hypertension. Hypertension 2020, 75, 956–965. [Google Scholar] [CrossRef]
- McCoy, E.K.; Lisenby, K.M. Aprocitentan (a Dual Endothelin-Receptor Antagonist) for Treatment-Resistant Hypertension. J. Cardiovasc. Pharmacol. 2021, 77, 699–706. [Google Scholar] [CrossRef]
- Schlaich, M.P.; Bellet, M.; Weber, M.A.; Danaietash, P.; Bakris, G.L.; Flack, J.M.; Dreier, R.F.; Sassi-Sayadi, M.; Haskell, L.P.; Narkiewicz, K.; et al. Dual endothelin antagonist aprocitentan for resistant hypertension (PRECISION): A multicentre, blinded, randomised, parallel-group, phase 3 trial. Lancet 2022, 400, 1927–1937. [Google Scholar] [CrossRef]
- K-Laflamme, A.; Oster, L.; Cardinal, R.; de Champlain, J. Effects of renin-angiotensin blockade on sympathetic reactivity and beta-adrenergic pathway in the spontaneously hypertensive rat. Hypertension 1997, 30, 278–287. [Google Scholar] [CrossRef]
- Khalil, R.A. Modulators of the vascular endothelin receptor in blood pressure regulation and hypertension. Curr. Mol. Pharmacol. 2011, 4, 176–186. [Google Scholar] [CrossRef] [PubMed]
- Napoli, C.; Ignarro, L.J. Nitric oxide and pathogenic mechanisms involved in the development of vascular diseases. Arch. Pharm. Res. 2009, 32, 1103–1108. [Google Scholar] [CrossRef]
- Fleming, I.; Busse, R. NO: The primary EDRF. J. Mol. Cell. Cardiol. 1999, 31, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, J.A.; Ali, F.; Bailey, L.; Moreno, L.; Harrington, L.S. Role of nitric oxide and prostacyclin as vasoactive hormones released by the endothelium. Exp. Physiol. 2008, 93, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Félétou, M.; Vanhoutte, P.M. Endothelium-derived hyperpolarizing factor: Where are we now? Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1215–1225. [Google Scholar] [CrossRef]
- Pintérová, M.; Kuneš, J.; Zicha, J. Altered neural and vascular mechanisms in hypertension. Physiol. Res. 2011, 60, 381–402. [Google Scholar] [CrossRef]
- Park, B.-G.; Shin, W.-S.; Oh, S.; Park, G.-M.; Kim, N.I.; Lee, S. A novel antihypertension agent, sargachromenol D from marine brown algae, Sargassum siliquastrum, exerts dual action as an L-type Ca2+ channel blocker and endothelin A/B2 receptor antagonist. Bioorg. Med. Chem. 2017, 25, 4649–4655. [Google Scholar] [CrossRef]
- Schiffrin, E.L. Endothelin: Potential role in hypertension and vascular hypertrophy. Hypertension 1995, 25, 1135–1143. [Google Scholar] [CrossRef]
- Schiffrin, E.L. Endothelin and endothelin antagonists in hypertension. J. Hypertens. 1998, 16, 1891–1895. [Google Scholar] [CrossRef]
- Hermann, M.; Flammer, A.; Lüscher, T.F. Nitric oxide in hypertension. J. Clin. Hypertens. 2006, 8, 17–29. [Google Scholar] [CrossRef]
- Brunner, H.; Cockcroft, J.R.; Deanfield, J.; Donald, A.; Ferrannini, E.; Halcox, J.; Kiowski, W.; Lüscher, T.F.; Mancia, G.; Natali, A.; et al. Part II: Association with cardiovascular risk factors and diseases. A statement by the Working Group on Endothelins and Endothelial Factors of the European Society of Hypertension. J. Hypertens. 2005, 23, 233–246. [Google Scholar] [CrossRef] [PubMed]
- Lüscher, T.F.; Vanhoutte, P.M. Endothelium-dependent contractions to acetylcholine in the aorta of the spontaneously hypertensive rat. Hypertension 1986, 8, 344–348. [Google Scholar] [CrossRef] [PubMed]
- Panza, J.A.; Quyyumi, A.A.; Brush, J.E.; Epstein, S.E. Abnormal endothelium-dependent vascular relaxation in patients with essential hypertension. N. Engl. J. Med. 1990, 323, 22–27. [Google Scholar] [CrossRef]
- Pinheiro, L.C.; Tanus-Santos, J.E.; Castro, M.M. The potential of stimulating nitric oxide formation in the treatment of hypertension. Expert Opin. Ther. Targets 2017, 21, 543–556. [Google Scholar] [CrossRef]
- Cantalupo, A.; Gargiulo, A.; Dautaj, E.; Liu, C.; Zhang, Y.; Hla, T.; Di Lorenzo, A. S1PR1 (Sphingosine-1-Phosphate Receptor 1) Signaling Regulates Blood Flow and Pressure. Hypertension 2017, 70, 426–434. [Google Scholar] [CrossRef]
- Dumont, Y.; D’Amours, M.; Lebel, M.; Larivière, R. Supplementation with a low dose of L-arginine reduces blood pressure and endothelin-1 production in hypertensive uraemic rats. Nephrol. Dial. Transplant. 2001, 16, 746–754. [Google Scholar] [CrossRef]
- Ast, J.; Jablecka, A.; Bogdanski, P.; Smolarek, I.; Krauss, H.; Chmara, E. Evaluation of the antihypertensive effect of L-arginine supplementation in patients with mild hypertension assessed with ambulatory blood pressure monitoring. Med. Sci. Monit. 2010, 16, CR266-71. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.-L.; Leu, S.; Lee, W.-C.; Wu, K.L.H.; Chan, J.Y.H. Maternal Melatonin Therapy Attenuated Maternal High-Fructose Combined with Post-Weaning High-Salt Diets-Induced Hypertension in Adult Male Rat Offspring. Molecules 2018, 23, 886. [Google Scholar] [CrossRef]
- Tai, I.-H.; Sheen, J.-M.; Lin, Y.-J.; Yu, H.-R.; Tiao, M.-M.; Chen, C.-C.; Huang, L.-T.; Tain, Y.-L. Maternal N-acetylcysteine therapy regulates hydrogen sulfide-generating pathway and prevents programmed hypertension in male offspring exposed to prenatal dexamethasone and postnatal high-fat diet. Nitric Oxide 2016, 53, 6–12. [Google Scholar] [CrossRef]
- Hsu, C.-N.; Lin, Y.-J.; Lu, P.-C.; Tain, Y.-L. Maternal Resveratrol Therapy Protects Male Rat Offspring against Programmed Hypertension Induced by TCDD and Dexamethasone Exposures: Is It Relevant to Aryl Hydrocarbon Receptor? Int. J. Mol. Sci. 2018, 19, 2459. [Google Scholar] [CrossRef]
- Beliaev, A.; Learmonth, D.A.; Soares-da-Silva, P. Synthesis and biological evaluation of novel, peripherally selective chromanyl imidazolethione-based inhibitors of dopamine beta-hydroxylase. J. Med. Chem. 2006, 49, 1191–1197. [Google Scholar] [CrossRef]
- Almeida, L.; Nunes, T.; Costa, R.; Rocha, J.F.; Vaz-da-Silva, M.; Soares-da-Silva, P. Etamicastat, a novel dopamine β-hydroxylase inhibitor: Tolerability, pharmacokinetics, and pharmacodynamics in patients with hypertension. Clin. Ther. 2013, 35, 1983–1996. [Google Scholar] [CrossRef] [PubMed]
- Palatini, P.; Dabbeni-Sala, F.; Finotti, P. Kinetic studies with competitive inhibitors indicate a sequential mechanism for dopamine beta-hydroxylase. Biochem. Int. 1984, 9, 675–682. [Google Scholar]
- Dharmasena, S.P.; Wimalasena, D.S.; Wimalasena, K. A slow-tight binding inhibitor of dopamine beta-monooxygenase: A transition state analogue for the product release step. Biochemistry 2002, 41, 12414–12420. [Google Scholar] [CrossRef] [PubMed]
- Dopamine β-Monooxygenase: Mechanism, Substrates and Inhibito...: Ingenta Connect. Available online: https://www.ingentaconnect.com/content/ben/cei/2009/00000005/00000001/art00002 (accessed on 2 August 2022).
- Dey, S.K.; Saini, M.; Prabhakar, P.; Kundu, S. Dopamine β hydroxylase as a potential drug target to combat hypertension. Expert Opin. Investig. Drugs 2020, 29, 1043–1057. [Google Scholar] [CrossRef]
- Oparil, S.; Schmieder, R.E. New approaches in the treatment of hypertension. Circ. Res. 2015, 116, 1074–1095. [Google Scholar] [CrossRef] [PubMed]
- Igreja, B.; Pires, N.M.; Bonifácio, M.J.; Loureiro, A.I.; Fernandes-Lopes, C.; Wright, L.C.; Soares-da-Silva, P. Blood pressure-decreasing effect of etamicastat alone and in combination with antihypertensive drugs in the spontaneously hypertensive rat. Hypertens. Res. 2015, 38, 30–38. [Google Scholar] [CrossRef]
- Safety, Tolerability, Pharmacokinetic, Including Food Interaction, and Pharmacodynamic Profile of BIA 5-1058.—Study Results—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/results/NCT02151994?term=ZAMICASTAT&draw=2&rank=9 (accessed on 2 August 2022).
- Ferrari, P. Rostafuroxin: An ouabain-inhibitor counteracting specific forms of hypertension. Biochim. Biophys. Acta 2010, 1802, 1254–1258. [Google Scholar] [CrossRef]
- Ferrandi, M.; Minotti, E.; Salardi, S.; Florio, M.; Bianchi, G.; Ferrari, P. Ouabainlike factor in Milan hypertensive rats. Am. J. Physiol. 1992, 263, F739–F748. [Google Scholar] [CrossRef]
- Ferrandi, M.; Manunta, P.; Balzan, S.; Hamlyn, J.M.; Bianchi, G.; Ferrari, P. Ouabain-like factor quantification in mammalian tissues and plasma: Comparison of two independent assays. Hypertension 1997, 30, 886–896. [Google Scholar] [CrossRef]
- Manunta, P.; Messaggio, E.; Ballabeni, C.; Sciarrone, M.T.; Lanzani, C.; Ferrandi, M.; Hamlyn, J.M.; Cusi, D.; Galletti, F.; Bianchi, G.; et al. Plasma ouabain-like factor during acute and chronic changes in sodium balance in essential hypertension. Hypertension 2001, 38, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, G.; Tripodi, G.; Casari, G.; Salardi, S.; Barber, B.R.; Garcia, R.; Leoni, P.; Torielli, L.; Cusi, D.; Ferrandi, M. Two point mutations within the adducin genes are involved in blood pressure variation. Proc. Natl. Acad. Sci. USA 1994, 91, 3999–4003. [Google Scholar] [CrossRef] [PubMed]
- Casari, G.; Barlassina, C.; Cusi, D.; Zagato, L.; Muirhead, R.; Righetti, M.; Nembri, P.; Amar, K.; Gatti, M.; Macciardi, F. Association of the alpha-adducin locus with essential hypertension. Hypertension 1995, 25, 320–326. [Google Scholar] [CrossRef]
- Cusi, D.; Barlassina, C.; Azzani, T.; Casari, G.; Citterio, L.; Devoto, M.; Glorioso, N.; Lanzani, C.; Manunta, P.; Righetti, M.; et al. Polymorphisms of alpha-adducin and salt sensitivity in patients with essential hypertension. Lancet 1997, 349, 1353–1357. [Google Scholar] [CrossRef] [PubMed]
- Lingrel, J.B. The physiological significance of the cardiotonic steroid/ouabain-binding site of the Na,K-ATPase. Annu. Rev. Physiol. 2010, 72, 395–412. [Google Scholar] [CrossRef]
- Blanco, G.; Wallace, D.P. Novel role of ouabain as a cystogenic factor in autosomal dominant polycystic kidney disease. Am. J. Physiol. Renal Physiol. 2013, 305, F797–F812. [Google Scholar] [CrossRef]
- Wenceslau, C.F.; Rossoni, L.V. Rostafuroxin ameliorates endothelial dysfunction and oxidative stress in resistance arteries from deoxycorticosterone acetate-salt hypertensive rats: The role of Na+K+-ATPase/ cSRC pathway. J. Hypertens. 2014, 32, 542–554. [Google Scholar] [CrossRef]
- Citterio, L.; Bianchi, G.; Scioli, G.A.; Glorioso, N.; Bigazzi, R.; Cusi, D.; Staessen, J.A.; Cavuto, S.; Ferrandi, M.; Lanzani, C.; et al. Antihypertensive treatment guided by genetics: PEARL-HT, the randomized proof-of-concept trial comparing rostafuroxin with losartan. Pharmacogenomics J. 2021, 21, 346–358. [Google Scholar] [CrossRef]
- Mendoza, M.F.; Kachur, S.M.; Lavie, C.J. Hypertension in obesity. Curr. Opin. Cardiol. 2020, 35, 389–396. [Google Scholar] [CrossRef]
- Khokhar, K.K.; Sidhu, S.; Kaur, G. Correlation between leptin level and hypertension in normal and obese pre- and postmenopausal women. Eur. J. Endocrinol. 2010, 163, 873–878. [Google Scholar] [CrossRef]
- Ma, D.; Feitosa, M.F.; Wilk, J.B.; Laramie, J.M.; Yu, K.; Leiendecker-Foster, C.; Myers, R.H.; Province, M.A.; Borecki, I.B. Leptin is associated with blood pressure and hypertension in women from the National Heart, Lung, and Blood Institute Family Heart Study. Hypertension 2009, 53, 473–479. [Google Scholar] [CrossRef] [PubMed]
- Patel, J.V.; Lim, H.S.; Dubb, K.; Hughes, E.A.; Lip, G.Y.H. Circulating levels of adiponectin, leptin, and tumour necrosis factor alpha in hypertension. Ann. Med. 2009, 41, 291–300. [Google Scholar] [CrossRef]
- Sabatier, M.J.; McCully, K.K.; Marinik, E.L.; Schwark, E.H.; Haddow, S.; Cortez-Cooper, M.; Bergeron, M.F.; Sloan, G.J.; Cannon, J.G. Leptin, blood pressure, and aerobic capacity in women. Am. J. Hypertens. 2008, 21, 1245–1250. [Google Scholar] [CrossRef]
- Kramer, C.K.; von Mühlen, D.; Barrett-Connor, E. Does leptin predict incident hypertension in older adults? Clin. Endocrinol. 2010, 73, 201–205. [Google Scholar] [CrossRef]
- Bell, B.B.; Rahmouni, K. Leptin as a Mediator of Obesity-Induced Hypertension. Curr. Obes. Rep. 2016, 5, 397–404. [Google Scholar] [CrossRef]
- Simonds, S.E.; Pryor, J.T.; Ravussin, E.; Greenway, F.L.; Dileone, R.; Allen, A.M.; Bassi, J.; Elmquist, J.K.; Keogh, J.M.; Henning, E.; et al. Leptin mediates the increase in blood pressure associated with obesity. Cell 2014, 159, 1404–1416. [Google Scholar] [CrossRef] [PubMed]
- Aizawa-Abe, M.; Ogawa, Y.; Masuzaki, H.; Ebihara, K.; Satoh, N.; Iwai, H.; Matsuoka, N.; Hayashi, T.; Hosoda, K.; Inoue, G.; et al. Pathophysiological role of leptin in obesity-related hypertension. J. Clin. Investig. 2000, 105, 1243–1252. [Google Scholar] [CrossRef] [PubMed]
- Wold, L.E.; Relling, D.P.; Duan, J.; Norby, F.L.; Ren, J. Abrogated leptin-induced cardiac contractile response in ventricular myocytes under spontaneous hypertension: Role of Jak/STAT pathway. Hypertension 2002, 39, 69–74. [Google Scholar] [CrossRef]
- Mark, A.L. Selective leptin resistance revisited. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2013, 305, R566-81. [Google Scholar] [CrossRef]
- Lim, K.; Burke, S.L.; Head, G.A. Obesity-related hypertension and the role of insulin and leptin in high-fat-fed rabbits. Hypertension 2013, 61, 628–634. [Google Scholar] [CrossRef]
- Ren, J. Leptin and hyperleptinemia—From friend to foe for cardiovascular function. J. Endocrinol. 2004, 181, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Albiston, A.L.; McDowall, S.G.; Matsacos, D.; Sim, P.; Clune, E.; Mustafa, T.; Lee, J.; Mendelsohn, F.A.; Simpson, R.J.; Connolly, L.M.; et al. Evidence that the angiotensin IV (AT(4)) receptor is the enzyme insulin-regulated aminopeptidase. J. Biol. Chem. 2001, 276, 48623–48626. [Google Scholar] [CrossRef] [PubMed]
- Faure, S.; Javellaud, J.; Achard, J.-M.; Oudart, N. Vasoconstrictive effect of angiotensin IV in isolated rat basilar artery independent of AT1 and AT2 receptors. J. Vasc. Res. 2006, 43, 19–26. [Google Scholar] [CrossRef]
- Yang, R.; Walther, T.; Gembardt, F.; Smolders, I.; Vanderheyden, P.; Albiston, A.L.; Chai, S.Y.; Dupont, A.G. Renal vasoconstrictor and pressor responses to angiotensin IV in mice are AT1a-receptor mediated. J. Hypertens. 2010, 28, 487–494. [Google Scholar] [CrossRef]
- Gaspari, T.A.; Pong, W.Y.; Lee, H.W.; Welungoda, I.; Chai, S.Y.; Widdop, R.E. 444 at4 receptor/insulin regulated aminopeptidase deficiency is both vaso- and cardio-protective under condition of cardiovascular stress in mice. J. Hypertens. 2012, 30, e131–e132. [Google Scholar] [CrossRef]
- Gaspari, T.; Shen, M.; Wang, Y.; Shastry, A.; Chai, S.Y.; Samuel, C.; Widdop, R. A9684 Comparing anti-fibrotic effects of the irap inhibitor, hfi-419 to an angiotensin receptor blocker and ace inhibitor in a high salt-induced mouse model of kidney disease. J. Hypertens. 2018, 36, e56–e57. [Google Scholar] [CrossRef]
- Numaguchi, Y.; Ishii, M.; Niwa, M.; Kuwahata, T.; Chen, X.W.; Kuzuya, M.; Okumura, K.; Murohara, T. Abstract 423: Ablation of Insulin-Regulated Aminopeptidase (IRAP/AT4R) Attenuates Lipid Accumulation and Inhibits Plaque Rupture in the Carotid Artery of ApoE Deficient Mice. Circulation 2008, 118, S_302. [Google Scholar]
- Guarner, F.; Malagelada, J.-R. Gut flora in health and disease. Lancet 2003, 361, 512–519. [Google Scholar] [CrossRef] [PubMed]
- Salminen, S.; Bouley, C.; Boutron-Ruault, M.C.; Cummings, J.H.; Franck, A.; Gibson, G.R.; Isolauri, E.; Moreau, M.C.; Roberfroid, M.; Rowland, I. Functional food science and gastrointestinal physiology and function. Br. J. Nutr. 1998, 80 (Suppl. S1), S147–S171. [Google Scholar] [CrossRef]
- Tanaka, M.; Itoh, H. Hypertension as a metabolic disorder and the novel role of the gut. Curr. Hypertens. Rep. 2019, 21, 63. [Google Scholar] [CrossRef]
- Dan, X.; Mushi, Z.; Baili, W.; Han, L.; Enqi, W.; Huanhu, Z.; Shuchun, L. Differential Analysis of Hypertension-Associated Intestinal Microbiota. Int. J. Med. Sci. 2019, 16, 872–881. [Google Scholar] [CrossRef] [PubMed]
- de la Cuesta-Zuluaga, J.; Mueller, N.T.; Álvarez-Quintero, R.; Velásquez-Mejía, E.P.; Sierra, J.A.; Corrales-Agudelo, V.; Carmona, J.A.; Abad, J.M.; Escobar, J.S. Higher Fecal Short-Chain Fatty Acid Levels Are Associated with Gut Microbiome Dysbiosis, Obesity, Hypertension and Cardiometabolic Disease Risk Factors. Nutrients 2018, 11, 51. [Google Scholar] [CrossRef]
- Li, J.; Zhao, F.; Wang, Y.; Chen, J.; Tao, J.; Tian, G.; Wu, S.; Liu, W.; Cui, Q.; Geng, B.; et al. Gut microbiota dysbiosis contributes to the development of hypertension. Microbiome 2017, 5, 14. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Lulla, A.; Sioda, M.; Winglee, K.; Wu, M.C.; Jacobs, D.R.; Shikany, J.M.; Lloyd-Jones, D.M.; Launer, L.J.; Fodor, A.A.; et al. Gut microbiota composition and blood pressure. Hypertension 2019, 73, 998–1006. [Google Scholar] [CrossRef] [PubMed]
- Verhaar, B.J.H.; Collard, D.; Prodan, A.; Levels, J.H.M.; Zwinderman, A.H.; Bäckhed, F.; Vogt, L.; Peters, M.J.L.; Muller, M.; Nieuwdorp, M.; et al. Associations between gut microbiota, faecal short-chain fatty acids, and blood pressure across ethnic groups: The HELIUS study. Eur. Heart J. 2020, 41, 4259–4267. [Google Scholar] [CrossRef] [PubMed]
- Yan, Q.; Gu, Y.; Li, X.; Yang, W.; Jia, L.; Chen, C.; Han, X.; Huang, Y.; Zhao, L.; Li, P.; et al. Alterations of the gut microbiome in hypertension. Front. Cell. Infect. Microbiol. 2017, 7, 381. [Google Scholar] [CrossRef]
- Yang, T.; Santisteban, M.M.; Rodriguez, V.; Li, E.; Ahmari, N.; Carvajal, J.M.; Zadeh, M.; Gong, M.; Qi, Y.; Zubcevic, J.; et al. Gut dysbiosis is linked to hypertension. Hypertension 2015, 65, 1331–1340. [Google Scholar] [CrossRef]
- Kim, S.; Goel, R.; Kumar, A.; Qi, Y.; Lobaton, G.; Hosaka, K.; Mohammed, M.; Handberg, E.M.; Richards, E.M.; Pepine, C.J.; et al. Imbalance of gut microbiome and intestinal epithelial barrier dysfunction in patients with high blood pressure. Clin. Sci. 2018, 132, 701–718. [Google Scholar] [CrossRef]
- Huart, J.; Leenders, J.; Taminiau, B.; Descy, J.; Saint-Remy, A.; Daube, G.; Krzesinski, J.-M.; Melin, P.; de Tullio, P.; Jouret, F. Gut Microbiota and Fecal Levels of Short-Chain Fatty Acids Differ Upon 24-Hour Blood Pressure Levels in Men. Hypertension 2019, 74, 1005–1013. [Google Scholar] [CrossRef]
- Jackson, M.A.; Verdi, S.; Maxan, M.-E.; Shin, C.M.; Zierer, J.; Bowyer, R.C.E.; Martin, T.; Williams, F.M.K.; Menni, C.; Bell, J.T.; et al. Gut microbiota associations with common diseases and prescription medications in a population-based cohort. Nat. Commun. 2018, 9, 2655. [Google Scholar] [CrossRef]
- Flint, H.J.; Scott, K.P.; Duncan, S.H.; Louis, P.; Forano, E. Microbial degradation of complex carbohydrates in the gut. Gut Microbes 2012, 3, 289–306. [Google Scholar] [CrossRef] [PubMed]
- Pluznick, J.L.; Protzko, R.J.; Gevorgyan, H.; Peterlin, Z.; Sipos, A.; Han, J.; Brunet, I.; Wan, L.-X.; Rey, F.; Wang, T.; et al. Olfactory receptor responding to gut microbiota-derived signals plays a role in renin secretion and blood pressure regulation. Proc. Natl. Acad. Sci. USA 2013, 110, 4410–4415. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, N.; Hori, D.; Flavahan, S.; Steppan, J.; Flavahan, N.A.; Berkowitz, D.E.; Pluznick, J.L. Microbial short chain fatty acid metabolites lower blood pressure via endothelial G protein-coupled receptor 41. Physiol. Genom. 2016, 48, 826–834. [Google Scholar] [CrossRef]
- Seppo, L.; Jauhiainen, T.; Poussa, T.; Korpela, R. A fermented milk high in bioactive peptides has a blood pressure-lowering effect in hypertensive subjects. Am. J. Clin. Nutr. 2003, 77, 326–330. [Google Scholar] [CrossRef] [PubMed]
- Wakerlin, G.E. Antibodies to renin as proof of the pathogenesis of sustained renal hypertension. Circulation 1958, 17, 653–657. [Google Scholar] [CrossRef]
- Nakagami, H.; Morishita, R. Recent advances in therapeutic vaccines to treat hypertension. Hypertension 2018, 72, 1031–1036. [Google Scholar] [CrossRef] [PubMed]
- Ambühl, P.M.; Tissot, A.C.; Fulurija, A.; Maurer, P.; Nussberger, J.; Sabat, R.; Nief, V.; Schellekens, C.; Sladko, K.; Roubicek, K.; et al. A vaccine for hypertension based on virus-like particles: Preclinical efficacy and phase I safety and immunogenicity. J. Hypertens. 2007, 25, 63–72. [Google Scholar] [CrossRef]
- Ding, D.; Du, Y.; Qiu, Z.; Yan, S.; Chen, F.; Wang, M.; Yang, S.; Zhou, Y.; Hu, X.; Deng, Y.; et al. Vaccination against type 1 angiotensin receptor prevents streptozotocin-induced diabetic nephropathy. J. Mol. Med. 2016, 94, 207–218. [Google Scholar] [CrossRef]
- Downham, M.R.; Auton, T.R.; Rosul, A.; Sharp, H.L.; Sjöström, L.; Rushton, A.; Richards, J.P.; Mant, T.G.K.; Gardiner, S.M.; Bennett, T.; et al. Evaluation of two carrier protein-angiotensin I conjugate vaccines to assess their future potential to control high blood pressure (hypertension) in man. Br. J. Clin. Pharmacol. 2003, 56, 505–512. [Google Scholar] [CrossRef]
- Brown, M.J.; Coltart, J.; Gunewardena, K.; Ritter, J.M.; Auton, T.R.; Glover, J.F. Randomized double-blind placebo-controlled study of an angiotensin immunotherapeutic vaccine (PMD3117) in hypertensive subjects. Clin. Sci. 2004, 107, 167–173. [Google Scholar] [CrossRef]
- Tissot, A.C.; Maurer, P.; Nussberger, J.; Sabat, R.; Pfister, T.; Ignatenko, S.; Volk, H.-D.; Stocker, H.; Müller, P.; Jennings, G.T.; et al. Effect of immunisation against angiotensin II with CYT006-AngQb on ambulatory blood pressure: A double-blind, randomised, placebo-controlled phase IIa study. Lancet 2008, 371, 821–827. [Google Scholar] [CrossRef] [PubMed]
- Ferland, D.J.; Mullick, A.E.; Watts, S.W. Chemerin as a driver of hypertension: A consideration. Am. J. Hypertens. 2020, 33, 975–986. [Google Scholar] [CrossRef] [PubMed]
- Bozaoglu, K.; Bolton, K.; McMillan, J.; Zimmet, P.; Jowett, J.; Collier, G.; Walder, K.; Segal, D. Chemerin is a novel adipokine associated with obesity and metabolic syndrome. Endocrinology 2007, 148, 4687–4694. [Google Scholar] [CrossRef] [PubMed]
- Goralski, K.B.; McCarthy, T.C.; Hanniman, E.A.; Zabel, B.A.; Butcher, E.C.; Parlee, S.D.; Muruganandan, S.; Sinal, C.J. Chemerin, a novel adipokine that regulates adipogenesis and adipocyte metabolism. J. Biol. Chem. 2007, 282, 28175–28188. [Google Scholar] [CrossRef]
- Roh, S.; Song, S.-H.; Choi, K.-C.; Katoh, K.; Wittamer, V.; Parmentier, M.; Sasaki, S. Chemerin--A new adipokine that modulates adipogenesis via its own receptor. Biochem. Biophys. Res. Commun. 2007, 362, 1013–1018. [Google Scholar] [CrossRef]
- Ferland, D.J.; Flood, E.D.; Garver, H.; Yeh, S.T.; Riney, S.; Mullick, A.E.; Fink, G.D.; Watts, S.W. Different blood pressure responses in hypertensive rats following chemerin mRNA inhibition in dietary high fat compared to dietary high-salt conditions. Physiol. Genomics 2019, 51, 553–561. [Google Scholar] [CrossRef]
- Lobato, N.S.; Neves, K.B.; Filgueira, F.P.; Fortes, Z.B.; Carvalho, M.H.C.; Webb, R.C.; Oliveira, A.M.; Tostes, R.C. The adipokine chemerin augments vascular reactivity to contractile stimuli via activation of the MEK-ERK1/2 pathway. Life Sci. 2012, 91, 600–606. [Google Scholar] [CrossRef]
- Neves, K.B.; Lobato, N.S.; Lopes, R.A.M.; Filgueira, F.P.; Zanotto, C.Z.; Oliveira, A.M.; Tostes, R.C. Chemerin reduces vascular nitric oxide/cGMP signalling in rat aorta: A link to vascular dysfunction in obesity? Clin. Sci. 2014, 127, 111–122. [Google Scholar] [CrossRef]
- Ferland, D.J.; Darios, E.S.; Neubig, R.R.; Sjögren, B.; Truong, N.; Torres, R.; Dexheimer, T.S.; Thompson, J.M.; Watts, S.W. Chemerin-induced arterial contraction is Gi- and calcium-dependent. Vascul. Pharmacol. 2017, 88, 30–41. [Google Scholar] [CrossRef]
- Kennedy, A.J.; Yang, P.; Read, C.; Kuc, R.E.; Yang, L.; Taylor, E.J.A.; Taylor, C.W.; Maguire, J.J.; Davenport, A.P. Chemerin Elicits Potent Constrictor Actions via Chemokine-Like Receptor 1 (CMKLR1), not G-Protein-Coupled Receptor 1 (GPR1), in Human and Rat Vasculature. J. Am. Heart Assoc. 2016, 5, e004421. [Google Scholar] [CrossRef]
- Darios, E.S.; Winner, B.M.; Charvat, T.; Krasinksi, A.; Punna, S.; Watts, S.W. The adipokine chemerin amplifies electrical field-stimulated contraction in the isolated rat superior mesenteric artery. Am. J. Physiol. Heart Circ. Physiol. 2016, 311, H498–H507. [Google Scholar] [CrossRef] [PubMed]
- Hanthazi, A.; Jespers, P.; Vegh, G.; Degroot, G.-N.; Springael, J.-Y.; Lybaert, P.; Dewachter, L.; Mc Entee, K. Chemerin influences endothelin- and serotonin-induced pulmonary artery vasoconstriction in rats. Life Sci. 2019, 231, 116580. [Google Scholar] [CrossRef] [PubMed]
- Neves, K.B.; Nguyen Dinh Cat, A.; Lopes, R.A.M.; Rios, F.J.; Anagnostopoulou, A.; Lobato, N.S.; de Oliveira, A.M.; Tostes, R.C.; Montezano, A.C.; Touyz, R.M. Chemerin regulates crosstalk between adipocytes and vascular cells through nox. Hypertension 2015, 66, 657–666. [Google Scholar] [CrossRef] [PubMed]
- Kunimoto, H.; Kazama, K.; Takai, M.; Oda, M.; Okada, M.; Yamawaki, H. Chemerin promotes the proliferation and migration of vascular smooth muscle and increases mouse blood pressure. Am. J. Physiol. Heart Circ. Physiol. 2015, 309, H1017–H1028. [Google Scholar] [CrossRef]
- Bozaoglu, K.; Curran, J.E.; Stocker, C.J.; Zaibi, M.S.; Segal, D.; Konstantopoulos, N.; Morrison, S.; Carless, M.; Dyer, T.D.; Cole, S.A.; et al. Chemerin, a novel adipokine in the regulation of angiogenesis. J. Clin. Endocrinol. Metab. 2010, 95, 2476–2485. [Google Scholar] [CrossRef]
- Gu, P.; Wang, W.; Yao, Y.; Xu, Y.; Wang, L.; Zang, P.; Ma, J.; Yang, C.; Liang, J.; Lu, B.; et al. Increased Circulating Chemerin in Relation to Chronic Microvascular Complications in Patients with Type 2 Diabetes. Int. J. Endocrinol. 2019, 2019, 8693516. [Google Scholar] [CrossRef]
- Watts, S.W.; Dorrance, A.M.; Penfold, M.E.; Rourke, J.L.; Sinal, C.J.; Seitz, B.; Sullivan, T.J.; Charvat, T.T.; Thompson, J.M.; Burnett, R.; et al. Chemerin connects fat to arterial contraction. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1320–1328. [Google Scholar] [CrossRef]
- Ferland, D.J.; Seitz, B.; Darios, E.S.; Thompson, J.M.; Yeh, S.T.; Mullick, A.E.; Watts, S.W. Whole-Body but Not Hepatic Knockdown of Chemerin by Antisense Oligonucleotide Decreases Blood Pressure in Rats. J. Pharmacol. Exp. Ther. 2018, 365, 212–218. [Google Scholar] [CrossRef]
- Klionsky, D.J. Autophagy: From phenomenology to molecular understanding in less than a decade. Nat. Rev. Mol. Cell Biol. 2007, 8, 931–937. [Google Scholar] [CrossRef]
- Cecconi, F.; Levine, B. The role of autophagy in mammalian development: Cell makeover rather than cell death. Dev. Cell 2008, 15, 344–357. [Google Scholar] [CrossRef]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef] [PubMed]
- Maiuri, M.C.; Zalckvar, E.; Kimchi, A.; Kroemer, G. Self-eating and self-killing: Crosstalk between autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2007, 8, 741–752. [Google Scholar] [CrossRef]
- Kroemer, G.; Levine, B. Autophagic cell death: The story of a misnomer. Nat. Rev. Mol. Cell Biol. 2008, 9, 1004–1010. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Zhang, C.; Zhao, W. Autophagy and Hypertension. Adv. Exp. Med. Biol. 2020, 1207, 213–216. [Google Scholar] [CrossRef]
- Wang, Z.V.; Rothermel, B.A.; Hill, J.A. Autophagy in hypertensive heart disease. J. Biol. Chem. 2010, 285, 8509–8514. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Zhang, Y.; Ren, J. Acetylation in cardiovascular diseases: Molecular mechanisms and clinical implications. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165836. [Google Scholar] [CrossRef]
- Kang, S.-H.; Seok, Y.M.; Song, M.; Lee, H.-A.; Kurz, T.; Kim, I. Histone deacetylase inhibition attenuates cardiac hypertrophy and fibrosis through acetylation of mineralocorticoid receptor in spontaneously hypertensive rats. Mol. Pharmacol. 2015, 87, 782–791. [Google Scholar] [CrossRef]
- Seok, Y.M.; Lee, H.A.; Park, K.M.; Hwangbo, M.-H.; Kim, I.K. Lysine deacetylase inhibition attenuates hypertension and is accompanied by acetylation of mineralocorticoid receptor instead of histone acetylation in spontaneously hypertensive rats. Naunyn. Schmiedebergs Arch. Pharmacol. 2016, 389, 799–808. [Google Scholar] [CrossRef]
- Dikalova, A.E.; Pandey, A.; Xiao, L.; Arslanbaeva, L.; Sidorova, T.; Lopez, M.G.; Billings, F.T.; Verdin, E.; Auwerx, J.; Harrison, D.G.; et al. Mitochondrial deacetylase sirt3 reduces vascular dysfunction and hypertension while sirt3 depletion in essential hypertension is linked to vascular inflammation and oxidative stress. Circ. Res. 2020, 126, 439–452. [Google Scholar] [CrossRef]
- Ferroni, P.; Della-Morte, D.; Palmirotta, R.; Rundek, T.; Guadagni, F.; Roselli, M. Angiogenesis and hypertension: The dual role of anti-hypertensive and anti-angiogenic therapies. Curr. Vasc. Pharmacol. 2012, 10, 479–493. [Google Scholar] [CrossRef] [PubMed]
- Piani, F.; Tossetta, G.; Cara-Fuentes, G.; Agnoletti, D.; Marzioni, D.; Borghi, C. Diagnostic and Prognostic Role of CD93 in Cardiovascular Disease: A Systematic Review. Biomolecules 2023, 13, 910. [Google Scholar] [CrossRef] [PubMed]
- Haliga, R.; Zugun-Eloae, F.; Oboroceanu, T.; Pînzariu, A.; Mocanu, V. Vitamin D and Tissular Expression of Vitamin D Receptor in Obesity. Rev. Med. Chir. Soc. Med. Nat. Iasi. 2016, 120, 404–408. [Google Scholar] [PubMed]
- McCartney, D.M.; Byrne, D.G.; Turner, M.J. Dietary contributors to hypertension in adults reviewed. Ir. J. Med. Sci. 2015, 184, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Miller, E.R., 3rd; Erlinger, T.P.; Appel, L.J. The effects of macronutrients on blood pressure and lipids: An overview of the DASH and OmniHeart trials. Curr. Atheroscler. Rep. 2006, 8, 460–465. [Google Scholar] [CrossRef] [PubMed]
- Pérez-López, F.R.; Chedraui, P.; Haya, J.; Cuadros, J.L. Effects of the Mediterranean diet on longevity and age-related morbid conditions. Maturitas 2009, 64, 67–79. [Google Scholar] [CrossRef] [PubMed]
- Houston, M. The role of nutrition and nutraceutical supplements in the treatment of hypertension. World J. Cardiol. 2014, 6, 38–66. [Google Scholar] [CrossRef] [PubMed]
- Borghi, C.; Cicero, A.F. Nutraceuticals with a clinically detectable blood pressure-lowering effect: A review of available randomized clinical trials and their meta-analyses. Br. J. Clin. Pharmacol. 2017, 83, 163–171. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Popiolek-Kalisz, J.; Fornal, E. The Effects of Quercetin Supplementation on Blood Pressure—Meta-Analysis. Curr. Probl. Cardiol. 2022, 47, 101350. [Google Scholar] [CrossRef] [PubMed]
- Ibarra, M.; Moreno, L.; Vera, R.; Cogolludo, A.; Duarte, J.; Tamargo, J.; Perez-Vizcaino, F. Effects of the flavonoid quercetin and its methylated metabolite isorhamnetin in isolated arteries from spontaneously hypertensive rats. Planta Med. 2003, 69, 995–1000. [Google Scholar] [CrossRef] [PubMed]
- Popiolek-Kalisz, J.; Blaszczak, P.; Fornal, E. Dietary Isorhamnetin Intake Is Associated with Lower Blood Pressure in Coronary Artery Disease Patients. Nutrients 2022, 14, 4586. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ellinger, S.; Reusch, A.; Stehle, P.; Helfrich, H.P. Epicatechin ingested via cocoa products reduces blood pressure in humans: A nonlinear regression model with a Bayesian approach. Am. J. Clin. Nutr. 2012, 95, 1365–1377. [Google Scholar] [CrossRef] [PubMed]
- Kluknavsky, M.; Balis, P.; Skratek, M.; Manka, J.; Bernatova, I. (-)-Epicatechin Reduces the Blood Pressure of Young Borderline Hypertensive Rats During the Post-Treatment Period. Antioxidants 2020, 9, 96. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
 : inhibition.
: inhibition.
: inhibition.
: inhibition.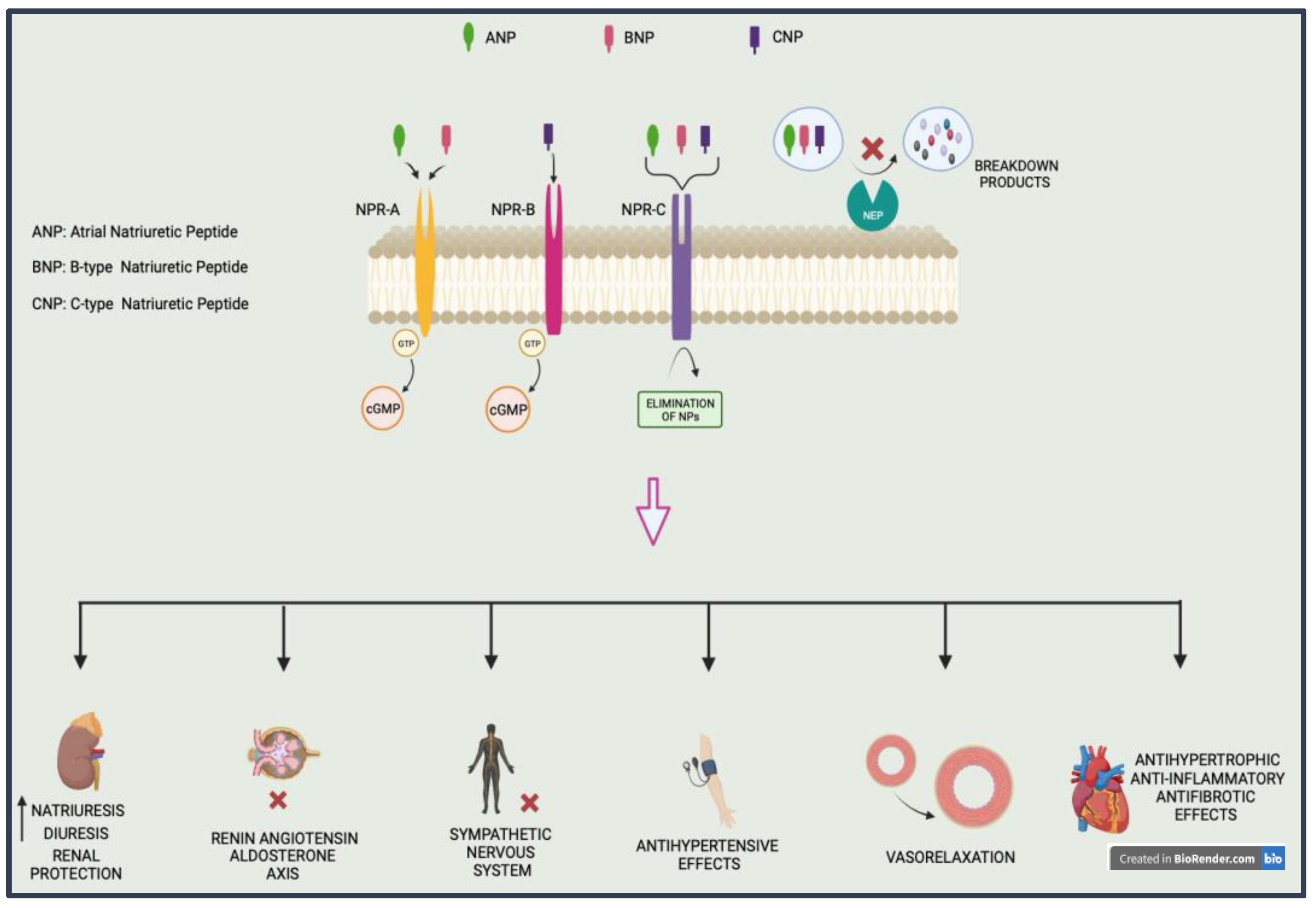
 —inhibition of.
—inhibition of.
—inhibition of.
—inhibition of.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Popa, I.P.; Clim, A.; Pînzariu, A.C.; Lazăr, C.I.; Popa, Ș.; Tudorancea, I.M.; Moscalu, M.; Șerban, D.N.; Șerban, I.L.; Costache-Enache, I.-I.; et al. Arterial Hypertension: Novel Pharmacological Targets and Future Perspectives. J. Clin. Med. 2024, 13, 5927. https://doi.org/10.3390/jcm13195927
Popa IP, Clim A, Pînzariu AC, Lazăr CI, Popa Ș, Tudorancea IM, Moscalu M, Șerban DN, Șerban IL, Costache-Enache I-I, et al. Arterial Hypertension: Novel Pharmacological Targets and Future Perspectives. Journal of Clinical Medicine. 2024; 13(19):5927. https://doi.org/10.3390/jcm13195927
Chicago/Turabian StylePopa, Irene Paula, Andreea Clim, Alin Constantin Pînzariu, Cristina Iuliana Lazăr, Ștefan Popa, Ivona Maria Tudorancea, Mihaela Moscalu, Dragomir N. Șerban, Ionela Lăcrămioara Șerban, Irina-Iuliana Costache-Enache, and et al. 2024. "Arterial Hypertension: Novel Pharmacological Targets and Future Perspectives" Journal of Clinical Medicine 13, no. 19: 5927. https://doi.org/10.3390/jcm13195927