
Arterial Thrombosis in Acute Respiratory Infections: An Underestimated but Clinically Relevant Problem



Abstract
:1. Introduction
2. Materials and Methods
3. Results
3.1. Clinical Aspects of the Problem
3.1.1. Myocardial Infarction and Ischemic Stroke: Potential Association with Acute Lower Respiratory Tract Infections
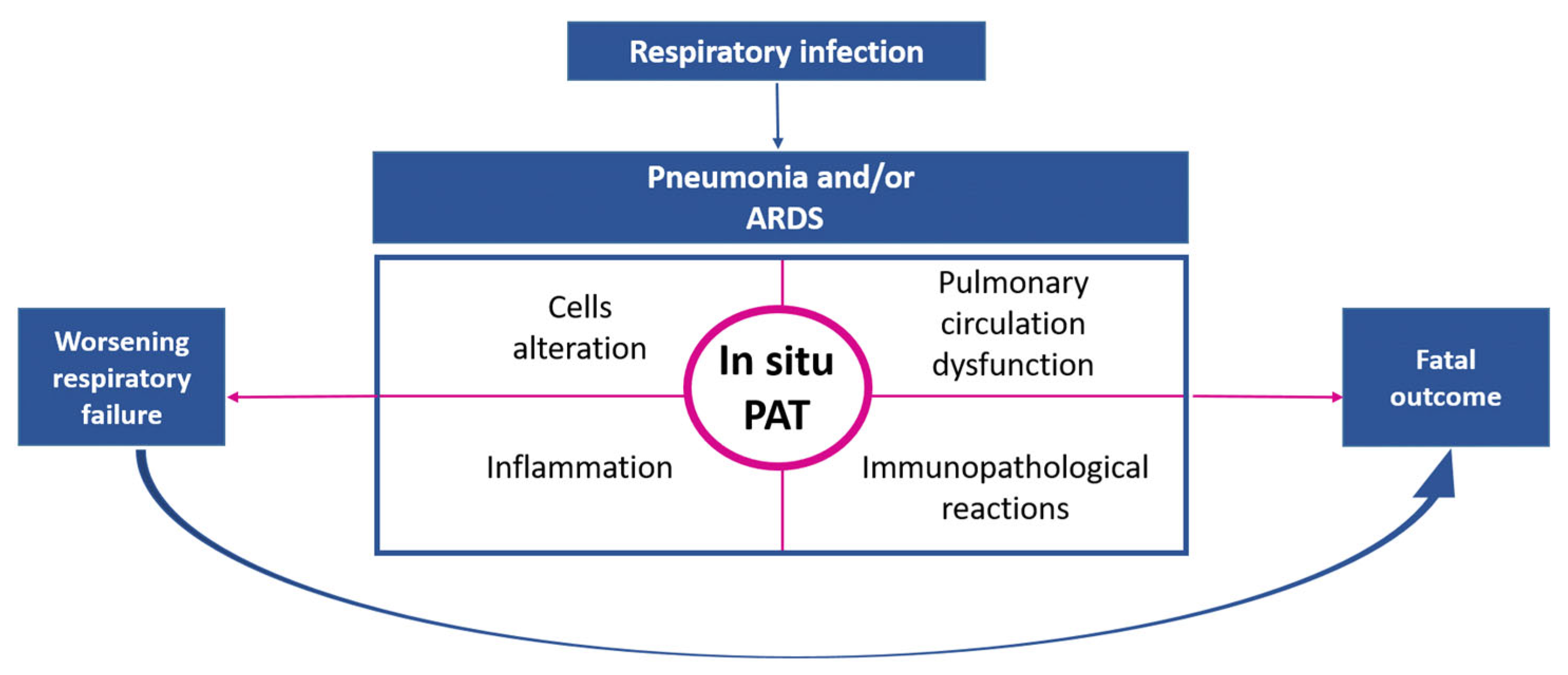
3.1.2. Pulmonary Embolism and Pulmonary Artery Thrombosis In Situ as Complications of Respiratory Infections
3.1.3. Intracardiac Thrombosis in Patients with Respiratory Infections
3.1.4. Mesenteric Thrombosis in Respiratory Infectious Diseases
3.1.5. Acute Lower Extremity Arterial Thrombosis in Severe Respiratory Infections
3.1.6. Diagnostic Aspects of Thrombotic Complications of Acute Lower Respiratory Tract Infections
3.2. Pathophysiological Aspects of the Problem
3.2.1. Role of Tissue Factor in Activation of the Coagulation System during Lung Infection
3.2.2. Thrombin and Coagulation Factor FXIIa in the Regulation of In Situ Thrombus Structure
3.2.3. Protein C Dysregulation as a Potential Mechanism of Thrombosis in Respiratory Infections
3.2.4. Platelet Activation in Severe Respiratory Infections as a Component of Thrombosis Pathophysiology
3.2.5. The Role of Neutrophils in the Pathogenesis of Thrombosis
3.2.6. Von Willebrand Factor and ADAMTS-13 in the Pathogenesis of Arterial Thrombosis In Situ
3.2.7. The Role of Co/Superinfection in Thrombotic Complications
3.2.8. Comorbidity and Thrombotic Complications in Acute Respiratory Infections
4. Future Perspectives
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Cui, S.; Chen, S.; Li, X.; Liu, S.; Wang, F. Prevalence of venous thromboembolism in patients with severe novel coronavirus pneumonia. J. Thromb. Haemost. 2020, 18, 1421–1424. [Google Scholar] [CrossRef] [PubMed]
- Guan, W.J.; Ni, Z.Y.; Hu, Y.; Liang, W.H.; Ou, C.Q.; He, J.X.; Liu, L.; Shan, H.; Lei, C.L.; Hui, D.S.C.; et al. Clinical characteristics of coronavirus disease 2019 in China. N. Engl. J. Med. 2020, 382, 1708–1720. [Google Scholar] [CrossRef] [PubMed]
- Zuckier, L.S.; Moadel, R.M.; Haramati, L.B.; Freeman, L. Diagnostic evaluation of pulmonary embolism during the COVID-19 pandemic. J. Nucl. Med. 2020, 61, 630–631. [Google Scholar] [CrossRef] [PubMed]
- Recommendations of the Russian Ministry of Health—Federal State Budgetary Institution “National Medical Research Center for Phthisiopulmonology and Infectious Diseases” of the Ministry of Health of the Russian. Available online: https://nmrc.ru/for_specialists/o-koronaviruse/rekomendatsii-minzdrava-rossii-2/ (accessed on 1 September 2024).
- Zabolotskikh, I.; Kirov, M.; Lebedinskii, K.; Protsenko, D.; Avdeev, S.; Andreenko, A.; Arsentyev, L.; Afonchikov, V.; Afukov, I.; Belkin, A. Anesthesia and intensive care for patients with COVID-19. Russian Federation of anesthesiologists and reanimatologists guidelines. Ann. Crit. Care. 2021, S1, 9–143. [Google Scholar] [CrossRef]
- COVID-19 Rapid Guideline: Managing COVID-19. Available online: https://www.nice.org.uk/guidance/ng191/resources/covid19-rapid-guideline-managing-covid19-pdf-66142077109189 (accessed on 1 September 2024).
- Bikdeli, B.; Madhavan, M.V.; Jimenez, D.; Chuich, T.; Dreyfus, I.; Driggin, E.; Der Nigoghossian, C.; Ageno, W.; Madjid, M.; Guo, Y.; et al. COVID-19 and Thrombotic or Thromboembolic Disease: Implications for Prevention, Antithrombotic Therapy, and Follow-Up: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2020, 75, 2950–2973. [Google Scholar] [CrossRef]
- Nardelli, P.; Landoni, G. COVID-19-Related Thromboinflammatory Status: MicroCLOTS and Beyond (Editorial). Obs. Reanimatol. 2020, 16, 14–15. [Google Scholar] [CrossRef]
- Cheng, N.M.; Chan, Y.C.; Cheng, S.W. COVID-19 related thrombosis: A mini-review. Phlebology 2022, 37, 326–337. [Google Scholar] [CrossRef]
- Lax, S.F.; Skok, K.; Zechner, P.; Kessler, H.H.; Kaufmann, N.; Koelblinger, C.; Vander, K.; Bargfrieder, U.; Trauner, M. Pulmonary Arterial Thrombosis in COVID-19 With Fatal Outcome: Results from a Prospective, Single-Center, Clinicopathologic Case Series. Ann. Intern. Med. 2020, 173, 350–361. [Google Scholar] [CrossRef]
- Babkina, A.S.; Yadgarov, M.Y.; Volkov, A.V.; Kuzovlev, A.N.; Grechko, A.V.; Golubev, A.M. Spectrum of Thrombotic Complications in Fatal Cases of COVID-19: Focus on Pulmonary Artery Thrombosis In Situ. Viruses 2023, 15, 1681. [Google Scholar] [CrossRef]
- Violi, F.; Cangemi, R.; Calvieri, C. Pneumonia, thrombosis and vascular disease. J. Thromb. Haemost. 2014, 12, 1391–1400. [Google Scholar] [CrossRef]
- Warren-Gash, C.; Blackburn, R.; Whitaker, H.; McMenamin, J.; Hayward, A.C. Laboratory-confirmed respiratory infections as triggers for acute myocardial infarction and stroke: A self-controlled case series analysis of national linked datasets from Scotland. Eur. Respir. J. 2018, 51, 1701794. [Google Scholar] [CrossRef] [PubMed]
- Kwong, J.C.; Schwartz, K.L.; Campitelli, M.A.; Chung, H.; Crowcroft, N.S.; Karnauchow, T.; Katz, K.; Ko, D.T.; McGeer, A.J.; McNally, D.; et al. Acute myocardial infarction after laboratory-confirmed influenza infection. N. Engl. J. Med. 2018, 378, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Davidson, J.A.; Warren-Gash, C. Cardiovascular complications of acute respiratory infections: Current research and future directions. Expert Rev. Anti Infect. Ther. 2019, 17, 939–942. [Google Scholar] [CrossRef] [PubMed]
- Gropper, M.A.; Wiener-Kronish, J. The epithelium in acute lung injury/acute respiratory distress syndrome. Curr. Opin. Crit. Care. 2008, 14, 11–15. [Google Scholar] [CrossRef]
- Caldeira, D.; Nogueira-Garcia, B. Myocardial infarction and viral triggers: What do we know by now? Eur. Heart J. Suppl. 2023, 25 (Suppl. A), A12–A16. [Google Scholar] [CrossRef]
- Collins, S. Excess mortality from causes other than influenza and pneumonia during influenza epidemics. Public Health Rep. 1932, 47, 2159–2189. [Google Scholar] [CrossRef]
- Meier, C.R.; Jick, S.S.; Derby, L.E.; Vasilakis, C.; Jick, H. Acute respiratory-tract infections and risk of first-time acute myocardial infarction. Lancet 1998, 351, 1467–1471. [Google Scholar] [CrossRef]
- Clayton, T.C.; Capps, N.E.; Stephens, N.G.; Wedzicha, J.A.; Meade, T.W. Recent respiratory infection and the risk of myocardial infarction. Heart 2005, 91, 1601–1602. [Google Scholar] [CrossRef]
- Clayton, T.C.; Thompson, M.; Meade, T.W. Recent respiratory infection and risk of cardiovascular disease: Case-control study through a general practice database. Eur. Heart J. 2008, 29, 96–103. [Google Scholar] [CrossRef]
- Veizades, S.; Tso, A.; Nguyen, P.K. Infection, inflammation and thrombosis: A review of potential mechanisms mediating arterial thrombosis associated with influenza and severe acute respiratory syndrome coronavirus 2. Biol Chem. 2021, 403, 231–241. [Google Scholar] [CrossRef]
- Smeeth, L.; Thomas, S.L.; Hall, A.J.; Hubbard, R.; Farrington, P.; Vallance, P. Risk of myocardial infarction and stroke after acute infection or vaccination. N. Engl. J. Med. 2004, 351, 2611–2618. [Google Scholar] [CrossRef] [PubMed]
- Musher, D.M.; Alexandraki, I.; Graviss, E.A.; Yanbeiy, N.; Eid, A.; Inderias, L.A.; Phan, H.M.; Solomon, E. Bacteremic and nonbacteremic pneumococcal pneumonia. A prospective study. Medicine 2000, 79, 210–221. [Google Scholar] [CrossRef] [PubMed]
- Musher, D.M.; Rueda, A.M.; Kaka, A.S.; Mapara, S.M. The association between pneumococcal pneumonia and acute cardiac events. Clin. Infect. Dis. 2007, 45, 158–165. [Google Scholar] [CrossRef]
- Pieralli, F.; Vannucchi, V.; Nozzoli, C.; Augello, G.; Dentali, F.; De Marzi, G.; Uomo, G.; Risaliti, F.; Morbidoni, L.; Mazzone, A.; et al. Acute cardiovascular events in patients with community acquired pneumonia: Results from the observational prospective FADOI-ICECAP study. BMC Infect Dis. 2021, 21, 116. [Google Scholar] [CrossRef]
- Africano, H.F.; Serrano-Mayorga, C.C.; Ramirez-Valbuena, P.C.; Bustos, I.G.; Bastidas, A.; Vargas, H.A.; Gómez, S.; Rodriguez, A.; Orihuela, C.J.; Reyes, L.F. Major Adverse Cardiovascular Events During Invasive Pneumococcal Disease Are Serotype Dependent. Clin. Infect. Dis. 2021, 72, e711–e719. [Google Scholar] [CrossRef]
- Guan, X.R.; Jiang, L.X.; Ma, X.H. Relationship between Mycoplasma pneumoniae infection and acute myocardial infarction. Zhongguo Wei Zhong Bing Ji Jiu Yi Xue. 2008, 20, 236–237. (In Chinese) [Google Scholar] [PubMed]
- Momiyama, Y.; Ohmori, R.; Taniguchi, H.; Nakamura, H.; Ohsuzu, F. Association of Mycoplasma pneumoniae infection with coronary artery disease and its interaction with chlamydial infection. Atherosclerosis 2004, 176, 139–144. [Google Scholar] [CrossRef]
- Long, B.; Brady, W.J.; Koyfman, A.; Gottlieb, M. Cardiovascular complications in COVID-19. Am. J. Emerg. Med. 2020, 38, 1504–1507. [Google Scholar] [CrossRef]
- Del Prete, A.; Conway, F.; Della Rocca, D.G.; Biondi-Zoccai, G.; De Felice, F.; Musto, C.; Picichè, M.; Martuscelli, E.; Natale, A.; Versaci, F. COVID-19, Acute Myocardial Injury, and Infarction. Card. Electrophysiol. Clin. 2022, 14, 29–39. [Google Scholar] [CrossRef]
- Li, S.; Hu, M.; Zhang, R.; Xu, J.; Hou, H.; Wang, Y.; Yang, H. The association between myocardial infarction and COVID-19 related mortality: A meta-analysis based on adjusted effect estimates. Am. J. Emerg. Med. 2022, 57, 227–229. [Google Scholar] [CrossRef]
- Zuin, M.; Rigatelli, G.; Battisti, V.; Costola, G.; Roncon, L.; Bilato, C. Increased risk of acute myocardial infarction after COVID-19 recovery: A systematic review and meta-analysis. Int. J. Cardiol. 2023, 372, 138–143. [Google Scholar] [CrossRef] [PubMed]
- Piroth, L.; Cottenet, J.; Mariet, A.-S.; Bonniaud, P.; Blot, M.; Tubert-Bitter, P.; Quantin, C. Comparison of the characteristics, morbidity, and mortality of COVID-19 and seasonal influenza: A nationwide, population-based retrospective cohort study. Lancet Respir. Med. 2021, 9, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Ward, A.; Sarraju, A.; Lee, D.; Bhasin, K.; Gad, S.; Beetel, R.; Chang, S.; Bonafede, M.; Rodriguez, F.; Dash, R. COVID-19 is associated with higher risk of venous thrombosis, but not arterial thrombosis, compared with influenza: Insights from a large US cohort. PLoS ONE 2022, 17, e0261786. [Google Scholar] [CrossRef] [PubMed]
- Miller, E.C.; Elkind, M.S. Infection and Stroke: An Update on Recent Progress. Curr. Neurol. Neurosci. Rep. 2016, 16, 2. [Google Scholar] [CrossRef]
- Cornet, M.C.; Grose, C.; Vexler, Z.; Wu, Y.W.; Fullerton, H.J. The Role of Infection and Inflammation in the Pathogenesis of Pediatric Arterial Ischemic Stroke. Semin. Pediatr. Neurol. 2022, 44, 100995. [Google Scholar] [CrossRef]
- Zurrú, M.C.; Alonzo, C.; Brescacín, L.; Romano, M.; Cámera, L.A.; Waisman, G.; Cristiano, E.; Ovbiagele, B. Recent respiratory infection predicts atherothrombotic stroke: Case-control study in a Buenos Aires healthcare system. Stroke 2009, 40, 1986–1990. [Google Scholar] [CrossRef]
- Chen, L.F.; Chen, H.P.; Huang, Y.S.; Huang, K.Y.; Chou, P.; Lee, C.C. Pneumococcal pneumonia and the risk of stroke: A population-based follow-up study. PLoS ONE 2012, 7, e51452. [Google Scholar] [CrossRef]
- Chiang, C.-H.; Huang, C.-C.; Chan, W.-L.; Chen, Y.-C.; Chen, T.-J.; Lin, S.-J.; Chen, J.-W.; Leu, H.-B. Association between Mycoplasma pneumonia and increased risk of ischemic stroke: A nationwide study. Stroke 2011, 42, 2940–2943. [Google Scholar] [CrossRef]
- Boehme, A.K.; Luna, J.; Kulick, E.R.; Kamel, H.; Elkind, M.S.V. Influenza-like illness as a trigger for ischemic stroke. Ann. Clin. Transl. Neurol. 2018, 5, 456–463. [Google Scholar] [CrossRef]
- Luo, W.; Liu, X.; Bao, K.; Huang, C. Ischemic stroke associated with COVID-19: A systematic review and meta-analysis. J. Neurol. 2022, 269, 1731–1740. [Google Scholar] [CrossRef]
- Babkina, A.S.; Yadgarov, M.Y.; Lyubomudrov, M.A.; Ostrova, I.V.; Volkov, A.V.; Kuzovlev, A.N.; Grechko, A.V.; Golubev, A.M. Morphologic Findings in the Cerebral Cortex in COVID-19: Association of Microglial Changes with Clinical and Demographic Variables. Biomedicines 2023, 11, 1407. [Google Scholar] [CrossRef] [PubMed]
- Beach, T.G.; Sue, L.I.; Intorcia, A.J.; Glass, M.J.; Walker, J.E.; Arce, R.; Nelson, C.M.; Serrano, G.E. Acute Brain Ischemia, Infarction and Hemorrhage in Subjects Dying with or without Autopsy-Proven Acute Pneumonia. medRxiv 2021. medRxiv:2021.03.22.21254139. [Google Scholar] [CrossRef]
- Merkler, A.E.; Parikh, N.S.; Mir, S.; Gupta, A.; Kamel, H.; Lin, E.; Lantos, J.; Schenck, E.J.; Goyal, P.; Bruce, S.S.; et al. Risk of Ischemic Stroke in Patients with Coronavirus Disease 2019 (COVID-19) vs Patients with Influenza. JAMA Neurol. 2020, 77, 1366–1372. [Google Scholar] [CrossRef] [PubMed]
- Smeeth, L.; Cook, C.; Thomas, S.; Hall, A.J.; Hubbard, R.; Vallance, P. Risk of deep vein thrombosis and pulmonary embolism after acute infection in a community setting. Lancet 2006, 367, 1075–1079. [Google Scholar] [CrossRef] [PubMed]
- Clayton, T.C.; Gaskin, M.; Meade, T.W. Recent respiratory infection and risk of venous thromboembolism: Case-control study through a general practice database. Int. J. Epidemiol. 2011, 40, 819–827. [Google Scholar] [CrossRef]
- Ribeiro, D.D.; Lijfering, W.M.; Van Hylckama Vlieg, A.; Rosendaal, F.R.; Cannegieter, S.C. Pneumonia and risk of venous thrombosis: Results from the MEGA study. J. Thromb. Haemost. 2012, 10, 1179–1182. [Google Scholar] [CrossRef]
- Cattaneo, M.; Bertinato, E.M.; Birocchi, S.; Brizio, C.; Malavolta, D.; Manzoni, M.; Muscarella, G.; Orlandi, M. Pulmonary embolism or pulmonary thrombosis in COVID-19? Is the recommendation to use high-dose heparin for thromboprophylaxis justified? Thromb. Haemost. 2020, 120, 1230–1232. [Google Scholar] [CrossRef]
- Birocchi, S.; Manzoni, M.; Podda, G.M.; Casazza, G.; Cattaneo, M. High rates of pulmonary artery occlusions in COVID-19. A meta-analysis. Eur. J. Clin. Investig. 2021, 51, e13433. [Google Scholar] [CrossRef]
- Poissy, J.; Goutay, J.; Caplan, M.; Parmentier, E.; Duburcq, T.; Lassalle, F.; Jeanpierre, E.; Rauch, A.; Labreuche, J.; Susen, S. Pulmonary Embolism in Patients With COVID-19: Awareness of an Increased Prevalence. Circulation 2020, 142, 184–186. [Google Scholar] [CrossRef]
- Cao, Y.; Geng, C.; Li, Y.; Zhang, Y. In situ Pulmonary Artery Thrombosis: A Previously Overlooked Disease. Front. Pharmacol. 2021, 8, 671589. [Google Scholar] [CrossRef]
- Porembskaya, O.; Toropova, Y.; Tomson, V.; Lobastov, K.; Laberko, L.; Kravchuk, V.; Saiganov, S.; Brill, A. Pulmonary Artery Thrombosis: A Diagnosis That Strives for Its Independence. Int. J. Mol. Sci. 2020, 21, 5086. [Google Scholar] [CrossRef] [PubMed]
- Benns, M.; Reilly, P.; Kim, P. Early Pulmonary Embolism after Injury: A Different Clinical Entity? Injury 2014, 45, 241–244. [Google Scholar] [CrossRef] [PubMed]
- Paffrath, T.; Wafaisade, A.; Lefering, R.; Simanski, C.; Bouillon, B.; Spanholtz, T.; Wutzler, S.; Maegele, M. Trauma Registry of DGU Venous Thromboembolism after Severe Trauma: Incidence, Risk Factors and Outcome. Injury 2010, 41, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Van Lange-Velde, K.; Srámek, A.; Vincken, P.W.; van Rooden, J.K.; Rosendaal, F.R.; Cannegieter, S.C. Finding the origin of pulmonary emboli with a total-body magnetic resonance direct thrombus imaging technique. Haematologica 2013, 98, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Desborough, M.J.R.; Doyle, A.J.; Griffiths, A.; Retter, A.; Breen, K.A.; Hunt, B.J. Image-proven thromboembolism in patients with severe COVID-19 in a tertiary critical care unit in the United Kingdom. Thromb. Res. 2020, 193, 1–4. [Google Scholar] [CrossRef]
- Frantzeskaki, F.; Armaganidis, A.; Orfanos, S.E. Immunothrombosis in Acute Respiratory Distress Syndrome: Cross Talks between Inflammation and Coagulation. Respiration 2017, 93, 212–225. [Google Scholar] [CrossRef]
- Ng, K.H.L.; Wu, A.K.L.; Cheng, V.C.C.; Tang, B.S.F.; Chan, C.Y.; Yung, C.Y.; Luk, S.H.; Lee, T.W.; Chow, L.; Yuen, K.Y. Pulmonary artery thrombosis in a patient with severe acute respiratory syndrome. Postgrad. Med. J. 2005, 81, e3. [Google Scholar] [CrossRef]
- Harms, P.W.; Schmidt, L.A.; Smith, L.B.; Newton, D.W.; Pletneva, M.A.; Walters, L.L.; Tomlins, S.A.; Fisher-Hubbard, A.; Napolitano, L.M.; Park, P.K.; et al. Autopsy findings in eight patients with fatal H1N1 influenza. Am. J. Clin. Pathol. 2010, 1, 27–35. [Google Scholar] [CrossRef]
- Masana, M.; Martinez, L.I.; Gil, M.; Bueno, G.; Llagostera, S. Thoracic Aortic Mural Thrombus, Right Ventricular Clot and Pulmonary Embolism in a Patient With COVID-19 Pneumonia. Vasc. Endovasc. Surg. 2021, 55, 273–276. [Google Scholar] [CrossRef]
- Sethi, S.S.; Zilinyi, R.; Green, P.; Eisenberger, A.; Brodie, D.; Agerstrand, C.; Takeda, K.; Kirtane, A.J.; Parikh, S.A.; Rosenzweig, E.B. Right Ventricular Clot in Transit in COVID-19: Implications for the Pulmonary Embolism Response Team. JACC Case Rep. 2020, 2, 1391–1396. [Google Scholar] [CrossRef]
- Kashi, M.; Jacquin, A.; Dakhil, B.; Zaimi, R.; Mahé, E.; Tella, E.; Bagan, P. Severe arterial thrombosis associated with Covid-19 infection. Thromb. Res. 2020, 192, 75–77. [Google Scholar] [CrossRef] [PubMed]
- Afari, H.; Tefera, L.; Rosovsky, R.P. Case of right ventricular and aortic thrombi in a patient with severe COVID-19. BMJ Case Rep. 2021, 14, e240745. [Google Scholar] [CrossRef] [PubMed]
- Petrov, A.; Romera, J.P.D.G.; Wilbring, M.; Alexiou, K.; Kappert, U.; Matschke, K.E.; Tugtekin, S.-M. Aortic Thrombosis following COVID-19: A Systematic Review. Thorac. Cardiovasc. Surg. 2022, 70, 323–332. [Google Scholar] [CrossRef] [PubMed]
- Spreadbury, M.; Gubberud, E.; Halvorsen, H.; Bjørkum, J.; Butter, A. Multiple arterial thromboemboli after COVID-19. Multiple arterielle tromboembolier etter covid-19. Tidsskr. Nor. Laegeforen. 2021, 141, 0289. [Google Scholar] [CrossRef]
- Tinelli, G.; Minelli, F.; Sica, S.; Tshomba, Y. Complete aortic thrombosis in SARS-CoV-2 infection. Eur. Heart J. 2021, 42, 2314. [Google Scholar] [CrossRef]
- Nagashima, M.; Higaki, T.; Satoh, H.; Nakano, T. Cardiac thrombus associated with Mycoplasma pneumoniae infection. Interact. Cardiovasc. Thorac. Surg. 2010, 11, 849–851. [Google Scholar] [CrossRef]
- Kobayashi, K.; Yano, S.; Shishido, S.; Tokushima, T. Invasive pulmonary aspergillosis with thrombosis in the left atrium. Intern. Med. 2001, 40, 250–254. [Google Scholar] [CrossRef]
- Ishiguro, T.; Matsuo, K.; Fujii, S.; Takayanagi, N. Acute thrombotic vascular events complicating influenza-associated pneumonia. Respir. Med. Case Rep. 2019, 28, 100884. [Google Scholar] [CrossRef]
- Sousa, C.; Almeida, P.; Gonçalves, A.; Rodrigues, J.; Rangel, I.; Macedo, F.; Maciel, M.J. Large right ventricular thrombus. Acta Med. Port. 2014, 27, 390–393. [Google Scholar] [CrossRef]
- Segovia, F.D.; Ream, S.; Dang, T.; Chaganti, B.T.; Ortega, A.J.; Rhee, S.; Borges, J.C. COVID-19-Associated Superior Mesenteric Artery Thrombosis and Acute Intestinal Ischemia. Cureus 2022, 14, e27722. [Google Scholar] [CrossRef]
- Carmo Filho, A.; Cunha, B.D.S. Inferior mesenteric vein thrombosis and COVID-19. Rev. Soc. Bras. Med. Trop. 2020, 53, e20200412. [Google Scholar] [CrossRef] [PubMed]
- Gebran, A.; El Moheb, M.; Argandykov, D.; Mashbari, H.; Gartland, R.M.; Hwabejire, J.O.; Velmahos, G.C.; Kaafarani, H.M. Mesenteric Ischemia in Patients with Coronavirus 2019: A Scoping Review. Surg. Infect. 2022, 23, 781–786. [Google Scholar] [CrossRef] [PubMed]
- Navarro-Martínez, S.; Diez Ares, J.Á.; Peris Tomás, N.; Gonzálvez Guardiola, P.; Pérez-Rubio, Á. Superior mesenteric vein thrombosis as the only manifestation of SARS-CoV-2 infection. Cir. Esp. 2022, 100, 244–246. [Google Scholar] [CrossRef] [PubMed]
- Sureshkumar, N.B.; Surendran, S.A. Acute Mesenteric Ischemia in COVID-19 While Receiving Prophylactic Enoxaparin. Clin. Med. Res. 2022, 20, 236–240. [Google Scholar] [CrossRef]
- Collins, M.H.; McGinn, M.K.; Weber, D.J. Mesenteric Thrombosis Complicating Influenza B Infection. Am. J. Med. 2016, 129, e17–e18. [Google Scholar] [CrossRef]
- Wilson, M.L.; Menjivar, E.; Kalapatapu, V.; Hand, A.P.; Garber, J.; Ruiz, M.A. Mycoplasma pneumoniae associated with hemolytic anemia, cold agglutinins, and recurrent arterial thrombosis. South Med. J. 2007, 100, 215–217. [Google Scholar] [CrossRef]
- Bao, X.L.; Tang, N.; Wang, Y.Z. Severe Klebsiella pneumoniae pneumonia complicated by acute intra-abdominal multiple arterial thrombosis and bacterial embolism: A case report. World J. Clin. Cases. 2022, 10, 11101–11110. [Google Scholar] [CrossRef]
- Fournier, M.; Faille, D.; Dossier, A.; Mageau, A.; Roland, P.N.; Ajzenberg, N.; Borie, R.; Bouadma, L.; Bunel, V.; Castier, Y.; et al. Arterial thrombotic events in adult inpatients with COVID-19. Mayo Clin. Proc. 2021, 96, 295–303. [Google Scholar] [CrossRef]
- Bilaloglu, S.; Aphinyanaphongs, Y.; Jones, S.; Iturrate, E.; Hochman, J.; Berger, J.S. Thrombosis in hospitalized patients with COVID-19 in a New York City health system. JAMA 2020, 324, 799–801. [Google Scholar] [CrossRef]
- Attisani, L.; Pucci, A.; Luoni, G.; Luzzani, L.; Pegorer, M.A.; Settembrini, A.M.; Bissacco, D.; Wohlauer, M.V.; Piffaretti, G.; Bellosta, R. COVID-19 and acute limb ischemia: A systematic review. J. Cardiovasc. Surg. 2021, 62, 542–547. [Google Scholar] [CrossRef]
- Sánchez, J.B.; Alcalde, J.D.C.; Isidro, R.R.; Luna, C.Z.; Cubas, W.S.; Charres, A.C.; Gutiérrez, J.E.; Ochoa, J.D.; Arias, P.F. Acute limb ischemia in a Peruvian cohort infected by COVID-19. Ann. Vasc. Surg. 2021, 72, 196–204. [Google Scholar] [CrossRef] [PubMed]
- Thompson, O.; Pierce, D.; Whang, D.; O’Malley, M.; Geise, B.; Malhotra, U. Acute limb ischemia as sole initial manifestation of SARS-CoV-2 infection. J. Vasc. Surg. Cases Innov. Tech. 2020, 6, 511–513. [Google Scholar] [CrossRef] [PubMed]
- Rinaldo, J.E.; Perez, H. Ischemic necrosis of both lower extremities as a result of the microembolism syndrome complicating the adult respiratory distress syndrome caused by Escherichia coli pneumonia and septicemia. Am. Rev. Respir. Dis. 1982, 126, 932–935. [Google Scholar] [CrossRef] [PubMed]
- Joo, C.U.; Kim, J.S.; Han, Y.M. Mycoplasma pneumoniae induced popliteal artery thrombosis treated with urokinase. Postgrad. Med. J. 2001, 77, 723–724. [Google Scholar] [CrossRef]
- Ducloux, G.; Defaux, D.; Folliot, J.P.; Laurent, J.M. Thrombose artérielle fémorale en rapport avec une infection à Mycoplasma pneumoniae [Femoral artery thrombosis associated with Mycoplasma pneumoniae infection]. LARC Med. 1981, 1, 42–44. [Google Scholar]
- Peyton, B.D.; Cutler, B.S.; Stewart, F.M. Spontaneous tibial artery thrombosis associated with varicella pneumonia and free protein S deficiency. J. Vasc. Surg. 1998, 27, 563–567. [Google Scholar] [CrossRef]
- Paparoupa, M.; Spineli, L.; Framke, T.; Ho, H.; Schuppert, F.; Gillissen, A. Pulmonary Embolism in Pneumonia: Still a Diagnostic Challenge? Results of a Case-Control Study in 100 Patients. Dis. Markers 2016, 2016, 8682506. [Google Scholar] [CrossRef]
- Hanelin, J.; Eyler, W.R. Pulmonary artery thrombosis; roentgen manifestations. Radiology 1951, 5, 689–703. [Google Scholar] [CrossRef] [PubMed]
- Baranga, L.; Khanuja, S.; Scott, J.A.; Provancha, I.; Gosselin, M.; Walsh, J.; Arancibia, R.; Bruno, M.A.; Waite, S. In Situ Pulmonary Arterial Thrombosis: Literature Review and Clinical Significance of a Distinct Entity. AJR Am. J. Roentgenol. 2023, 1, 57–68. [Google Scholar] [CrossRef]
- Le Gal, G.; Righini, M.; Parent, F.; van Strijen, M.; Couturaud, F. Diagnosis and management of subsegmental pulmonary embolism. J. Thromb. Haemost. 2006, 4, 724–731. [Google Scholar] [CrossRef]
- Trunz, L.M.; Lee, P.; Lange, S.M.; Pomeranz, C.L.; Needleman, L.; Ford, R.W.; Karambelkar, A.; Sundaram, B. Imaging approach to COVID-19 associated pulmonary embolism. Int. J. Clin. Pract. 2021, 75, e14340. [Google Scholar] [CrossRef] [PubMed]
- Fang, C.; Xu, L. Chlamydia psittaci Pneumonia-Induced Pulmonary Thrombosis: A Case Report. Infect. Drug Resist. 2023, 16, 7063–7069. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, R.; Kreutz, R.P.; Bliden, K.P.; Tantry, U.S.; Gurbel, P.A. Personalizing Antithrombotic Therapy in COVID-19: Role of Thromboelastography and Thromboelastometry. Thromb Haemost. 2020, 120, 1594–1596. [Google Scholar] [CrossRef] [PubMed]
- El-Qutob, D.; Alvarez-Arroyo, L.; Barreda, I.; Nieto, M.; Pin, M.; Poveda-Andrés, J.L.; Carrera-Hueso, F.J. High incidence of pulmonary thromboembolism in hospitalized SARS-CoV-2 infected patients despite thrombo-prophylaxis. Heart Lung 2022, 53, 77–82. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ge, Y.; Liu, C.; Wang, N.; Xu, J.; Zhu, X.; Su, C.; Li, H.; Zhang, H.; Li, Z.; Li, H.; et al. Elevated Plasma D-Dimer in Adult Community-Acquired Pneumonia Patients is Associated with an Increased Inflammatory Reaction and Lower Survival. Clin. Lab. 2019, 65, 180720. [Google Scholar] [CrossRef] [PubMed]
- Crowther, M.A.; Cook, D.J.; Griffith, L.E.; Devereaux, P.J.; Rabbat, C.C.; Clarke, F.J.; Hoad, N.; McDonald, E.; Meade, M.O.; Guyatt, G.H.; et al. Deep venous thrombosis: Clinically silent in the intensive care unit. J. Crit. Care 2005, 20, 334–340. [Google Scholar] [CrossRef]
- Umairi, R.A.; Adawi, K.A.; Khoori, M.A.; Lawati, A.A.; Jose, S. COVID-19-Associated Thrombotic Complication: Is It Pulmonary Embolism or In Situ Thrombosis? Radiol. Res. Pract. 2023, 2023, 3844069. [Google Scholar] [CrossRef]
- Van Dam, L.; Kroft, L.; van der Wal, L.; Cannegieter, S.; Eikenboom, J.; de Jonge, E.; Huisman, M.; Klok, F. Clinical and computed tomography characteristics of COVID-19 associated acute pulmonary embolism: A different phenotype of thrombotic disease? Thromb. Res. 2020, 193, 86–89. [Google Scholar] [CrossRef]
- Liu, Z.B.; Xie, W.M.; Zhai, Z.G.; Cao, B.; Wan, J. Pulmonary aspergillosis with in-situ pulmonary artery thrombosis: To anti-coagulate or not? Chin. Med. J. 2019, 14, 1742–1744. [Google Scholar] [CrossRef]
- Emad, Y.; Ragab, Y.; Farber, H.W.; Erkan, D.; Ibrahim, O.; Kindermann, M.; Tekavec-Trkanjec, J.; Jayakrishnan, B.; El-Shaarawy, N.; Rasker, J.J.; et al. Pulmonary embolism versus pulmonary vasculitis in Hughes-Stovin syndrome: Characteristic computed tomography pulmonary angiographic findings and diagnostic and therapeutic implications. HSS International Study Group. Thromb. Res. 2024, 239, 109040. [Google Scholar] [CrossRef]
- Katz, D.S.; Loud, P.A.; Bruce, D.; Gittleman, A.M.; Mueller, R.; Klippenstein, D.L.; Grossman, Z.D. Combined CT venography and pulmonary angiography: A comprehensive review. Radiographics 2002, 22, S3–S24. [Google Scholar] [CrossRef] [PubMed]
- Parker, J.A.; Coleman, R.E.; Grady, E.; Royal, H.D.; Siegel, B.A.; Stabin, M.G.; Sostman, H.D.; Hilson, A.J.; Society of Nuclear Medicine. SNM practice guideline for lung scintigraphy 4.0. J. Nucl. Med. Technol. 2012, 1, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Moore, A.J.E.; Wachsmann, J.; Chamarthy, M.R.; Panjikaran, L.; Tanabe, Y.; Rajiah, P. Imaging of acute pulmonary embolism: An update. Cardiovasc. Diagn. Ther. 2018, 3, 225–243. [Google Scholar] [CrossRef] [PubMed]
- Anderson, D.R.; Barnes, D.C. Computerized tomographic pulmonary angiography versus ventilation perfusion lung scanning for the diagnosis of pulmonary embolism. Curr. Opin. Pulm. Med. 2009, 5, 425–429. [Google Scholar] [CrossRef] [PubMed]
- Bang, J.I.; Lee, W.W.; Cho, S.G.; Choi, M.; Song, Y.S. The Diagnostic Accuracy of SPECT Imaging in Patients with Suspected Pulmonary Embolism: Systematic Review and Network Meta-analysis. Clin. Nucl. Med. 2024, 7, 637–643. [Google Scholar] [CrossRef]
- Yilmaz, F.; Tunçez, A.; Önner, H.; Gedik, G.K.; Uysal, E.; Körez, M.K. Diagnostic compatibility of V/Q SPECT and CTPA, which are non-invasive diagnostic methods, for the detection of CTEPH, which is a treatable cause of pulmonary hypertension. Hell J. Nucl. Med. 2022, 2, 168–176. [Google Scholar] [CrossRef]
- Anderson, D.R.; Kahn, S.R.; Rodger, M.A.; Kovacs, M.J.; Morris, T.; Hirsch, A.; Lang, E.; Stiell, I.; Kovacs, G.; Dreyer, J.; et al. Computed tomographic pulmonary angiography vs ventilation-perfusion lung scanning in patients with suspected pulmonary embolism: A randomized controlled trial. JAMA 2007, 23, 2743–2753. [Google Scholar] [CrossRef]
- Tessonnier, L.; Guedj, E.; Cano, A.; Cammilleri, S.; Dragulescu, A.; Chabrol, B.; Mundler, O. Multiple distal pulmonary arterial thromboses revealed by lung scintigraphy in a patient with homocystinuria and normal multidetector CT pulmonary angiography. Clin. Nucl. Med. 2009, 34, 42–43. [Google Scholar] [CrossRef]
- De Bruyn, V.H.; Bergmann, R.; Keyt, B.A.; Bennett, W.F.; Sobel, B.E. Scintigraphic visualization of pulmonary thrombi with 123I-YPACK-TNK-tPA. Coron. Artery Dis. 1995, 9, 715–721. [Google Scholar]
- Lim, B.; Yao, Y.; Huang, A.L.; Yap, M.L.; Flierl, U.; Palasubramaniam, J.; Zaldivia, M.T.K.; Wang, X.; Peter, K. A Unique Recombinant Fluoroprobe Targeting Activated Platelets Allows In Vivo Detection of Arterial Thrombosis and Pulmonary Embolism Using a Novel Three-Dimensional Fluorescence Emission Computed Tomography (FLECT) Technology. Theranostics 2017, 5, 1047–1061. [Google Scholar] [CrossRef]
- Yende, S.; D’Angelo, G.; Mayr, F.; Kellum, J.A.; Weissfeld, L.; Kaynar, A.M.; Young, T.; Irani, K.; Angus, D.C. Elevated hemostasis markers after pneumonia increases one-year risk of all-cause and cardiovascular deaths. PLoS ONE 2011, 6, e22847. [Google Scholar] [CrossRef] [PubMed]
- Bone, R.C.; Francis, P.B.; Pierce, A.K. Intravascular coagulation associated with the adult respiratory distress syndrome. Am. J. Med. 1976, 61, 585–589. [Google Scholar] [CrossRef] [PubMed]
- Saldeen, T. Intravascular coagulation in the lungs in experimental fat embolism. Acta Chir. Scand. 1969, 135, 653–662. [Google Scholar] [PubMed]
- Tang, N.; Li, D.; Wang, X.; Sun, Z. Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J. Thromb. Haemost. 2020, 4, 844–847. [Google Scholar] [CrossRef]
- Spiezia, L.; Boscolo, A.; Poletto, F. COVID-19-related severe hypercoagulability in patients admitted to intensive care unit for acute respiratory failure. Thromb. Haemost. 2020, 6, 998–1000. [Google Scholar] [CrossRef]
- Ranucci, M.; Ballotta, A.; Di Dedda, U. The procoagulant pattern of patients with COVID-19 acute respiratory distress syndrome. J. Thromb. Haemost. 2020, 7, 1747–1751. [Google Scholar] [CrossRef]
- Hoechter, D.J.; Becker-Pennrich, A.; Langrehr, J.; Bruegel, M.; Zwissler, B.; Schaefer, S.; Spannagl, M.; Hinske, L.C.; Zoller, M. Higher procoagulatory potential but lower DIC score in COVID-19 ARDS patients compared to non-COVID-19 ARDS patients. Thromb. Res. 2020, 196, 186–192. [Google Scholar] [CrossRef]
- Spiezia, L.; Campello, E.; Cola, M.; Poletto, F.; Cerruti, L.; Poretto, A.; Simion, C.; Cattelan, A.; Vettor, R.; Simioni, P. More Severe Hypercoagulable State in Acute COVID-19 Pneumonia as Compared with Other Pneumonia. Mayo Clin. Proc. Innov. Qual. Outcomes 2020, 6, 696–702. [Google Scholar] [CrossRef]
- Mitchell, W.B. Thromboinflammation in COVID-19 acute lung injury. Paediatr. Respir. Rev. 2020, 35, 20–24. [Google Scholar] [CrossRef]
- Ochoa, C.D.; Wu, S.; Stevens, T. New developments in lung endothelial heterogeneity: Von Willebrand factor, P-selectin, and the Weibel-Palade body. Semin Thromb. Hemost. 2010, 36, 301–308. [Google Scholar] [CrossRef]
- Libby, P.; Lüscher, T. COVID-19 is, in the end, an endothelial disease. Eur. Heart J. 2020, 41, 3038–3044. [Google Scholar] [CrossRef] [PubMed]
- Rubins, J.B.; Duane, P.G.; Charboneau, D.; Janoff, E.N. Toxicity of pneumolysin to pulmonary endothelial cells in vitro. Infect Immun. 1992, 60, 1740–1746. [Google Scholar] [CrossRef] [PubMed]
- Gibson, R.L.; Soderland, C.; Henderson, W.R., Jr.; Chi, E.Y.; Rubens, C.E. Group B streptococci (GBS) injure lung endothelium in vitro: GBS invasion and GBS-induced eicosanoid production is greater with microvascular than with pulmonary artery cells. Infect. Immun. 1995, 63, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Birkun, A.A.; Zagoruĺko, A.K. Ultrastructural changes in the endothelium of lung capillaries during experimental pneumonia. Cor. Vasa. 1991, 33, 75–81. [Google Scholar] [PubMed]
- Spadaro, S.; Fogagnolo, A.; Campo, G.; Zucchetti, O.; Verri, M.; Ottaviani, I.; Tunstall, T.; Grasso, S.; Scaramuzzo, V.; Murgolo, F.; et al. Markers of endothelial and epithelial pulmonary injury in mechanically ventilated COVID-19 ICU patients. Crit. Care 2021, 25, 74. [Google Scholar] [CrossRef] [PubMed]
- Glas, G.J.; Van Der Sluijs, K.F.; Schultz, M.J.; Hofstra, J.J.; Van Der Poll, T.; Levi, M. Bronchoalveolar hemostasis in lung injury and acute respiratory distress syndrome. J. Thromb. Haemost. 2013, 11, 17–25. [Google Scholar] [CrossRef]
- Ryu, K.H.; Hindman, B.J.; Reasoner, D.K.; Dexter, F. Heparin reduces neurological impairment after cerebral arterial air embolism in the rabbit. Stroke 1996, 27, 303–310. [Google Scholar] [CrossRef]
- Sims, P.J.; Wiedmer, T. Induction of cellular procoagulant activity by the membrane attack complex of complement. Semin. Cell Biol. 1995, 6, 275–282. [Google Scholar] [CrossRef]
- Keragala, C.B.; Draxler, D.F.; McQuilten, Z.K.; Medcalf, R.L. Haemostasis and innate immunity—A complementary relationship: A review of the intricate relationship between coagulation and complement pathways. Br. J. Haematol. 2018, 180, 782–798. [Google Scholar] [CrossRef]
- Lüscher, E.F. Induction of platelet aggregation by immune complexes. Ser. Haematol. 1970, 3, 121–129. [Google Scholar]
- Penny, R.; Castaldi, P.A.; Whitsed, H.M. Inflammation and haemostasis in paraproteinaemias. Br. J. Haematol. 1971, 20, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Lisiewicz, J. Rola leukocytów w biomorfozie zakrzepów i krzepnieciu krwi [The role of leukocytes in the biomorphosis of thrombosis and blood coagulation]. Acta Physiol. Pol. 1971, 22, 785–789. [Google Scholar] [PubMed]
- Levine, P.H.; Weinger, R.S.; Simon, J.; Scoon, K.L.; Krinsky, N.I. Leukocyte-platelet interaction. Release of hydrogen peroxide by granulocytes as a modulator of platelet reactions. J. Clin. Investig. 1976, 57, 955–963. [Google Scholar] [CrossRef] [PubMed]
- Eilertsen, K.E.; Østerud, B. Tissue factor: (patho)physiology and cellular biology. Blood Coagul. Fibrinolysis 2004, 15, 521–538. [Google Scholar] [CrossRef]
- Bastarache, J.A.; Wang, L.; Geiser, T.; Wang, Z.; Albertine, K.H.; Matthay, M.A.; Ware, L.B. The alveolar epithelium can initiate the extrinsic coagulation cascade through expression of tissue factor. Thorax 2007, 62, 608–616. [Google Scholar] [CrossRef]
- Levi, M.; van der Poll, T.; Schultz, M. New insights into pathways that determine the link between infection and thrombosis. Neth. J. Med. 2012, 70, 114–120. [Google Scholar]
- Bastarache, J.A.; Fremont, R.D.; Kropski, J.A.; Bossert, F.R.; Ware, L.B. Procoagulant alveolar microparticles in the lungs of patients with acute respiratory distress syndrome. Am. J. Physiol. Lung Cell Mol Physiol. 2009, 297, L1035–L1041. [Google Scholar] [CrossRef]
- Maris, N.; de Vos, A.; Bresser, P.; van der Zee, J.; Meijers, J.; Lijnen, R.; Levi, M.; Jansen, H.; van der Poll, T. Activation of coagulation and inhibition of fibrinolysis in the lung after inhalation of lipopolysaccharide by healthy volunteers. Thromb. Haemost. 2005, 93, 1036–1040. [Google Scholar] [CrossRef]
- Hoogerwerf, J.J.; de Vos, A.F.; Bresser, P.; van der Zee, J.S.; Pater, J.M.; de Boer, A.; Tanck, M.; Lundell, D.L.; Her-Jenh, C.; Draing, C.; et al. Lung inflammation induced by lipoteichoic acid or lipopolysaccharide in humans. Am. J. Respir. Crit. Care Med. 2008, 178, 34–41. [Google Scholar] [CrossRef]
- Aras, O.; Shet, A.; Bach, R.R.; Hysjulien, J.L.; Slungaard, A.; Hebbel, R.P.; Escolar, G.; Jilma, B.; Key, N.S. Induction of microparticle- and cell-associated intravascular tissue factor in human endotoxemia. Blood 2004, 103, 4545–4553. [Google Scholar] [CrossRef]
- Subramaniam, S.; Kothari, H.; Bosmann, M. Tissue factor in COVID-19-associated coagulopathy. Thromb. Res. 2022, 220, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Mackman, N.; Grover, S.P.; Antoniak, S. Tissue factor expression, extracellular vesicles, and thrombosis after infection with the respiratory viruses influenza A virus and coronavirus. J. Thromb. Haemost. 2021, 19, 2652–2658. [Google Scholar] [CrossRef] [PubMed]
- Rijneveld, A.W.; Weijer, S.; Bresser, P.; Florquin, S.; Vlasuk, G.P.; Rote, W.E.; Spek, C.A.; Reitsma, P.H.; van der Zee, J.S.; Levi, M.; et al. Local activation of the tissue factor-factor VIIa pathway in patients with pneumonia and the effect of inhibition of this pathway in murine pneumococcal pneumonia. Crit. Care Med. 2006, 34, 1725–1730. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.L.; Welty-Wolf, K.; Carraway, M.S.; Ezban, M.; Ghio, A.; Suliman, H.; Piantadosi, C.A. Extrinsic coagulation blockade attenuates lung injury and proinflammatory cytokine release after intratracheal lipopolysaccharide. Am. J. Respir. Cell Mol. Biol. 2002, 26, 650–658. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Wang, Y.; Liu, B.; Erkan, D.; Ibrahim, O.; Kindermann, M.; Tekavec-Trkanjec, J.; Jayakrishnan, B.; El-Shaarawy, N.; Rasker, J.J. SN50 attenuates alveolar hypercoagulation and fibrinolysis inhibition in acute respiratory distress syndrome mice through inhibiting NF-κB p65 translocation. Respir. Res. 2020, 21, 130. [Google Scholar] [CrossRef]
- Yang, H.; Qian, H.; Liu, B.; Wu, Y.; Cheng, Y.; Zheng, X.; Li, X.; Yang, G.; He, T.; Li, S.; et al. Triptolide dose-dependently improves LPS-induced alveolar hypercoagulation and fibrinolysis inhibition through NF-κB inactivation in ARDS mice. Biomed. Pharmacother. 2021, 139, 111569. [Google Scholar] [CrossRef]
- Antoniak, S.; Tatsumi, K.; Hisada, Y.; Milner, J.J.; Neidich, S.D.; Shaver, C.M.; Pawlinski, R.; Beck, M.A.; Bastarache, J.A.; Mackman, N. Tissue factor deficiency increases alveolar hemorrhage and death in influenza A virus-infected mice. J. Thromb. Haemost. 2016, 14, 1238–1248. [Google Scholar] [CrossRef]
- Jin, X.; Duan, Y.; Bao, T.; Gu, J.; Chen, Y.; Li, Y.; Mao, S.; Chen, Y.; Xie, W. The values of coagulation function in COVID-19 patients. PLoS ONE 2020, 15, e0241329. [Google Scholar] [CrossRef]
- Okamoto, H.; Muraki, I.; Okada, H.; Tomita, H.; Suzuki, K.; Takada, C.; Wakayama, Y.; Kuroda, A.; Fukuda, H.; Kawasaki, Y.; et al. Recombinant Antithrombin Attenuates Acute Respiratory Distress Syndrome in Experimental Endotoxemia. Am. J. Pathol. 2021, 191, 1526–1536. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kipnis, E.; Guery, B.P.; Tournoys, A.; Leroy, X.; Robriquet, L.; Fialdes, P.; Neviere, R.; Fourrier, F. Massive alveolar thrombin activation in Pseudomonas aeruginosa-induced acute lung injury. Shock 2004, 21, 444–451. [Google Scholar] [CrossRef] [PubMed]
- Hofstra, J.J.; Cornet, A.D.; de Rooy, B.F.; Vlaar, A.P.; van der Poll, T.; Levi, M.; Zaat, S.A.; Schultz, M.J. Nebulized antithrombin limits bacterial outgrowth and lung injury in Streptococcus pneumoniae pneumonia in rats. Crit. Care 2009, 13, R145. [Google Scholar] [CrossRef] [PubMed]
- Camprubí-Rimblas, M.; Tantinyà, N.; Guillamat-Prats, R.; Bringué, J.; Puig, F.; Gómez, M.N.; Blanch, L.; Artigas, A. Effects of nebulized antithrombin and heparin on inflammatory and coagulation alterations in an acute lung injury model in rats. J. Thromb. Haemost. 2020, 18, 571–583. [Google Scholar] [CrossRef] [PubMed]
- Wygrecka, M.; Birnhuber, A.; Seeliger, B.; Michalick, L.; Pak, O.; Schultz, A.-S.; Schramm, F.; Zacharias, M.; Gorkiewicz, G.; David, S.; et al. Altered fibrin clot structure and dysregulated fibrinolysis contribute to thrombosis risk in severe COVID-19. Blood Adv. 2022, 6, 1074–1087. [Google Scholar] [CrossRef] [PubMed]
- Kurachi, K.; Davie, E.W. Activation of human factor XI (plasma thromboplastin antecedent) by factor XIIa (activated Hageman factor). Biochemistry 1977, 16, 5831–5839. [Google Scholar] [CrossRef]
- Konings, J.; Govers-Riemslag, J.W.P.; Philippou, H.; Mutch, N.J.; Borissoff, J.I.; Allan, P.; Mohan, S.; Tans, G.; Cate, H.T.; Ariëns, R.A.S. Factor XIIa regulates the structure of the fibrin clot independently of thrombin generation through direct interaction with fibrin. Blood 2011, 118, 3942–3951. [Google Scholar] [CrossRef]
- Hess, R.; Wujak, L.; Hesse, C.; Sewald, K.; Jonigk, D.; Warnecke, G.; Fieguth, H.G.; de Maat, S.; Maas, C.; Bonella, F.; et al. Coagulation factor XII regulates inflammatory responses in human lungs. Thromb. Haemost. 2017, 117, 1896–1907. [Google Scholar] [CrossRef]
- Kenne, E.; Renné, T. Factor XII: A drug target for safe interference with thrombosis and inflammation. Drug Discov. Today 2014, 19, 1459–1464. [Google Scholar] [CrossRef] [PubMed]
- Worm, M.; Köhler, E.C.; Panda, R.; Long, A.; Butler, L.M.; Stavrou, E.X.; Nickel, K.F.; Fuchs, T.A.; Renné, T. The factor XIIa blocking antibody 3F7: A safe anticoagulant with anti-inflammatory activities. Ann. Transl. Med. 2015, 3, 247. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Griffin, J.H.; Fernandez, J.A.; Gale, A.J.; Mosnier, L.O. Activated protein C. J. Thromb. Haemost. 2007, 5 (Suppl. 1), 73–80. [Google Scholar] [CrossRef]
- Isshiki, T.; Sakamoto, S.; Kinoshita, A.; Sugino, K.; Kurosaki, A.; Homma, S. Recombinant human soluble thrombomodulin treatment for acute exacerbation of idiopathic pulmonary fibrosis: A retrospective study. Respiration 2015, 89, 201–207. [Google Scholar] [CrossRef]
- Ware, L.B.; Fang, X.; Matthay, M.A. Protein C and thrombomodulin in human acute lung injury. Am. J. Physiol. Lung Cell Mol. Physiol. 2003, 285, L514–L521. [Google Scholar] [CrossRef] [PubMed]
- Esmon, C.T. Inflammation and the activated protein C anticoagulant pathway. Semin. Thromb. Hemost. 2006, 32 (Suppl. 1), 49–60. [Google Scholar] [CrossRef] [PubMed]
- Eckle, I.; Seitz, R.; Egbring, R.; Kolb, G.; Havemann, K. Protein C degradation in vitro by neutrophil elastase. Biol. Chem. Hoppe Seyler. 1991, 372, 1007–1013. [Google Scholar] [CrossRef] [PubMed]
- Choi, G.; Schultz, M.J.; Levi, M.; van der Poll, T.; Millo, J.L.; Garrard, C.S. Protein C in pneumonia. Thorax 2005, 60, 705–706. [Google Scholar] [CrossRef] [PubMed]
- Choi, G.; Wolthuis, E.K.; Bresser, P.; Levi, M.; van der Poll, T.; Dzoljic, M.; Vroom, M.B.; Schultz, M.J. Mechanical ventilation with lower tidal volumes and positive end-expiratory pressure prevents alveolar coagulation in patients without lung injury. Anesthesiology 2006, 105, 689–695. [Google Scholar] [CrossRef]
- Yamamoto, K.; Loskutoff, D.J. Extrahepatic expression and regulation of protein C in the mouse. Am. J. Pathol. 1998, 15, 547–555. [Google Scholar] [CrossRef]
- Warkentin, T.E.; Pai, M. Shock, acute disseminated intravascular coagulation, and microvascular thrombosis: Is ‘shock liver’ the unrecognized provocateur of ischemic limb necrosis? J. Thromb. Haemost. 2016, 14, 231–235. [Google Scholar] [CrossRef]
- Levi, M.; Dörffler-Melly, J.; Reitsma, P.; Büller, H.; Florquin, S.; van der Poll, T.; Carmeliet, P. Aggravation of endotoxin-induced disseminated intravascular coagulation and cytokine activation in heterozygous protein-C-deficient mice. Blood 2003, 101, 4823–4827. [Google Scholar] [CrossRef]
- Kager, L.M.; Wiersinga, W.J.; Roelofs, J.J.; Meijers, J.C.M.; Zeerleder, S.S.; Esmon, C.T.; Veer, C.V.; van der Poll, T. Endogenous protein C has a protective role during Gram-negative pneumosepsis (melioidosis). J. Thromb. Haemost. 2013, 11, 282–292. [Google Scholar] [CrossRef]
- Wójcik, K.; Bazan-Socha, S.; Celejewska-Wójcik, N.; Górka, K.; Lichołai, S.; Polok, K.; Stachura, T.; Zaręba, L.; Dziedzic, R.; Gradzikiewicz, A.; et al. Decreased protein C activity, lower ADAMTS13 antigen and free protein S levels accompanied by unchanged thrombin generation potential in hospitalized COVID-19 patients. Thromb. Res. 2023, 223, 80–86. [Google Scholar] [CrossRef]
- Neyrinck, A.P.; Liu, K.D.; Howard, J.P.; Matthay, M.A. Protective mechanisms of activated protein C in severe inflammatory disorders. Br. J. Pharmacol. 2009, 4, 1034–1047. [Google Scholar] [CrossRef] [PubMed]
- Robriquet, L.; Collet, F.; Tournoys, A.; Prangère, T.; Nevière, R.; Fourrier, F.; Guery, B.P. Intravenous administration of activated protein C in Pseudomonas-induced lung injury: Impact on lung fluid balance and the inflammatory response. Respir. Res. 2006, 7, 41. [Google Scholar] [CrossRef] [PubMed]
- Cerletti, C.; Tamburrelli, C.; Izzi, B.; Gianfagna, F.; de Gaetano, G. Platelet-leukocyte interactions in thrombosis. Thromb. Res. 2012, 129, 263–266. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, J.R.; Foster, T.J.; Cox, D. The interaction of bacterial pathogens with platelets. Nat. Rev. Microbiol. 2006, 4, 445–457. [Google Scholar] [CrossRef] [PubMed]
- Claushuis, T.A.M.; Van Der Veen, A.I.; Horn, J.; Schultz, M.J.; Houtkooper, R.H.; Veer, C.V.; Van Der Poll, T. Platelet toll-like receptor expression and activation induced by lipopolysaccharide and sepsis. Platelets 2019, 30, 296–304. [Google Scholar] [CrossRef]
- Palankar, R.; Kohler, T.P.; Krauel, K.; Wesche, J.; Hammerschmidt, S.; Greinacher, A. Platelets kill bacteria by bridging innate and adaptive immunity via platelet factor 4 and FcγRIIA. J. Thromb. Haemost. 2018, 16, 1187–1197. [Google Scholar] [CrossRef]
- Tunjungputri, R.N.; de Jonge, M.I.; de Greeff, A.; van Selm, S.; Buys, H.; Harders-Westerveen, J.F.; Stockhofe-Zurwieden, N.; Urbanus, R.T.; de Groot, P.G.; Smith, H.E.; et al. Invasive pneumococcal disease leads to activation and hyperreactivity of platelets. Thromb. Res. 2016, 144, 123–126. [Google Scholar] [CrossRef]
- Cangemi, R.; Carnevale, R.; Nocella, C.; Calvieri, C.; Cammisotto, V.; Novo, M.; Castellani, V.; D’Amico, A.; Zerbinati, C.; Stefanini, L.; et al. Glucocorticoids impair platelet thromboxane biosynthesis in community-acquired pneumonia. Pharmacol. Res. 2018, 131, 66–74. [Google Scholar] [CrossRef]
- Feldman, C. Cardiac complications in community-acquired pneumonia and COVID-19. Afr. J. Thorac. Crit. Care Med. 2020, 26, 1–5. [Google Scholar] [CrossRef]
- Sugiyama, M.G.; Gamage, A.; Zyla, R.; Armstrong, S.M.; Advani, S.; Advani, A.; Wang, C.; Lee, W.L. Influenza Virus Infection Induces Platelet-Endothelial Adhesion Which Contributes to Lung Injury. J. Virol. 2015, 90, 1812–1823. [Google Scholar] [CrossRef]
- McMullen, P.D.; Cho, J.H.; Miller, J.L.; Husain, A.N.; Pytel, P.; Krausz, T. A Descriptive and Quantitative Immunohistochemical Study Demonstrating a Spectrum of Platelet Recruitment Patterns Across Pulmonary Infections Including COVID-19. Am. J. Clin. Pathol. 2021, 155, 354–363. [Google Scholar] [CrossRef] [PubMed]
- Heikkinen, T.; Järvinen, A. The common cold. Lancet 2003, 361, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Kreutz, R.P.; Bliden, K.P.; Tantry, U.S.; Gurbel, P.A. Viral respiratory tract infections increase platelet reactivity and activation: An explanation for the higher rates of myocardial infarction and stroke during viral illness. J. Thromb. Haemost. 2005, 3, 2108–2109. [Google Scholar] [CrossRef] [PubMed]
- Consolo, F.; DELLA Valle, P.; Saracino, M.; Bonora, M.; Donadoni, G.; Ciceri, F.; Tresoldi, M.; D’angelo, A.; Landoni, G.; Zangrillo, A. Platelet activation state in early stages of COVID-19. Minerva Anestesiol. 2022, 6, 472–478. [Google Scholar] [CrossRef] [PubMed]
- Campo, G.; Contoli, M.; Fogagnolo, A.; Sega, F.V.D.; Zucchetti, O.; Ronzoni, L.; Verri, M.; Fortini, F.; Pavasini, R.; Morandi, L.; et al. Over time relationship between platelet reactivity, myocardial injury and mortality in patients with SARS-CoV-2-associated respiratory failure. Platelets 2021, 4, 560–567. [Google Scholar] [CrossRef]
- Iba, T.; Levy, J.H.; Connors, J.M.; Warkentin, T.E.; Thachil, J.; Levi, M. The unique characteristics of COVID-19 coagulopathy. Crit. Care 2020, 24, 360. [Google Scholar] [CrossRef]
- Turpie, A.G.; Esmon, C. Venous and arterial thrombosis--pathogenesis and the rationale for anticoagulation. Thromb. Haemost. 2011, 105, 586–596. [Google Scholar] [CrossRef]
- Jamasbi, J.; Ayabe, K.; Goto, S.; Nieswandt, B.; Peter, K.; Siess, W. Platelet receptors as therapeutic targets: Past, present and future. Thromb. Haemost. 2017, 7, 1249–1257. [Google Scholar] [CrossRef]
- Porembskaya, O.; Zinserling, V.; Tomson, V.; Toropova, Y.; Starikova, E.A.; Maslei, V.V.; Bulavinova, N.I.; Kirik, O.V.; Syrtsova, M.A.; Laberko, L.; et al. Neutrophils Mediate Pulmonary Artery Thrombosis In Situ. Int. J. Mol. Sci. 2022, 23, 5829. [Google Scholar] [CrossRef]
- Khismatullin, R.R.; Ponomareva, A.A.; Nagaswami, C.; Ivaeva, R.A.; Montone, K.T.; Weisel, J.W.; Litvinov, R.I. Pathology of lung-specific thrombosis and inflammation in COVID-19. J. Thromb. Haemost. 2021, 19, 3062–3072. [Google Scholar] [CrossRef]
- Quartuccio, L.; Sonaglia, A.; Casarotto, L.; McGonagle, D.; Di Loreto, C.; Pegolo, E. Clinical, laboratory and immunohistochemical characterization of in situ pulmonary arterial thrombosis in fatal COVID-19. Thromb. Res. 2022, 219, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, T.A.; Brill, A.; Wagner, D.D. Neutrophil extracellular trap (NET) impact on deep vein thrombosis. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1777–1783. [Google Scholar] [CrossRef] [PubMed]
- Radermecker, C.; Detrembleur, N.; Guiot, J.; Cavalier, E.; Henket, M.; d’Emal, C.; Vanwinge, C.; Cataldo, D.; Oury, C.; Delvenne, P.; et al. Neutrophil extracellular traps infiltrate the lung airway, interstitial, and vascular compartments in severe COVID-19. J. Exp. Med. 2020, 217, e20201012. [Google Scholar] [CrossRef] [PubMed]
- Al-Kuraishy, H.M.; Al-Gareeb, A.I.; Al-Hussaniy, H.A.; Al-Harcan, N.A.H.; Alexiou, A.; Batiha, G.E. Neutrophil Extracellular Traps (NETs) and Covid-19: A new frontiers for therapeutic modality. Int. Immunopharmacol. 2022, 104, 108516. [Google Scholar] [CrossRef]
- Liu, X.; Li, T.; Chen, H.; Yuan, L.; Ao, H. Role and intervention of PAD4 in NETs in acute respiratory distress syndrome. Respir. Res. 2024, 25, 63. [Google Scholar] [CrossRef]
- Franchini, M.; Lippi, G. Von Willebrand factor and thrombosis. Ann. Hematol. 2006, 85, 415–423. [Google Scholar] [CrossRef]
- Manz, X.D.; Bogaard, H.J.; Aman, J. Regulation of VWF (Von Willebrand Factor) in Inflammatory Thrombosis. Arterioscler. Thromb. Vasc. Biol. 2022, 42, 1307–1320. [Google Scholar] [CrossRef]
- Tripodi, A.; Rossi, S.C.; Clerici, M.; Merati, G.; Scalambrino, E.; Mancini, I.; Baronciani, L.; Boscarino, M.; Monzani, V.; Peyvandi, F. Pro-coagulant imbalance in patients with community acquired pneumonia assessed on admission and one month after hospital discharge. Clin. Chem. Lab. Med. 2021, 59, 1699–1708. [Google Scholar] [CrossRef]
- Doevelaar, A.A.; Bachmann, M.; Hölzer, B.; Seibert, F.S.; Rohn, B.J.; Bauer, F.; Witzke, O.; Dittmer, U.; Bachmann, M.; Yilmaz, S.; et al. von Willebrand Factor Multimer Formation Contributes to Immunothrombosis in Coronavirus Disease 2019. Crit. Care Med. 2021, 49, e512–e520. [Google Scholar] [CrossRef]
- Pham, T.T.; Punsawad, C.; Glaharn, S.; De Meyer, S.F.; Viriyavejakul, P.; Van den Steen, P.E. Release of endothelial activation markers in lungs of patients with malaria-associated acute respiratory distress syndrome. Malar. J. 2019, 18, 395. [Google Scholar] [CrossRef]
- Favaloro, E.J.; Henry, B.M.; Lippi, G. Increased VWF and Decreased ADAMTS-13 in COVID-19: Creating a Milieu for (Micro)Thrombosis. Semin. Thromb. Hemost. 2021, 47, 400–418. [Google Scholar] [CrossRef] [PubMed]
- Babkina, A.S.; Ostrova, I.V.; Yadgarov, M.Y.; Kuzovlev, A.N.; Grechko, A.V.; Volkov, A.V.; Golubev, A.M. The Role of Von Willebrand Factor in the Pathogenesis of Pulmonary Vascular Thrombosis in COVID-19. Viruses 2022, 14, 211. [Google Scholar] [CrossRef] [PubMed]
- Lüttge, M.; Fulde, M.; Talay, S.R.; Nerlich, A.; Rohde, M.; Preissner, K.T.; Hammerschmidt, S.; Steinert, M.; Mitchell, T.J.; Chhatwal, G.S.; et al. Streptococcus pneumoniae induces exocytosis of Weibel-Palade bodies in pulmonary endothelial cells. Cell Microbiol. 2012, 14, 210–225. [Google Scholar] [CrossRef] [PubMed]
- Martens, C.P.; Peetermans, M.; Vanassche, T.; Verhamme, P.; Jacquemin, M.; Martinod, K. Peptidylarginine deiminase 4 and ADAMTS13 activity in Staphylococcusaureus bacteraemia. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 2023, 378, 20230042. [Google Scholar] [CrossRef] [PubMed]
- Golubev, A.M.; Moroz, V.V.; Lysenko, D.V.; Kuzovlev, A.N.; Ostapchenko, D.A. Artificial Ventilation-Induced Acute Lung Lesion: Experimental, Morphological Study. Gen. Reanimatol. 2006, 2, 8–12. [Google Scholar] [CrossRef]
- Cepkova, M.; Brady, S.; Sapru, A.; Matthay, M.A.; Church, G. Biological markers of lung injury before and after the institution of positive pressure ventilation in patients with acute lung injury. Crit. Care 2006, 10, R126. [Google Scholar] [CrossRef]
- Ware, L.B.; Conner, E.R.; Matthay, M.A. von Willebrand factor antigen is an independent marker of poor outcome in patients with early acute lung injury. Crit. Care Med. 2001, 29, 2325–2331. [Google Scholar] [CrossRef]
- Yiming, M.T.; Lederer, D.J.; Sun, L.; Huertas, A.; Issekutz, A.C.; Bhattacharya, S. Platelets enhance endothelial adhesiveness in high tidal volume ventilation. Am. J. Respir. Cell Mol. Biol. 2008, 39, 569–575. [Google Scholar] [CrossRef]
- Onodera, Y.; Mitani, S.; Hosoda, C.; Takabayashi, Y.; Sakata, A.; Kawasaki, R.; Mori, R.; Ohshima, C.; Nishio, K.; Sugimoto, M.; et al. Regulation of von Willebrand factor by ADAMTS13 ameliorates lipopolysaccharide-induced lung injury in mice. Int. J. Hematol. 2023, 118, 699–710. [Google Scholar] [CrossRef]
- Zhao, Y.M.; Jiang, M.; Ji, S.D.; He, Y.; Shen, F.; Li, X.M.; Ruan, C.G. Anti-human VWF monoclonal antibody SZ-123 prevents arterial thrombus formation by inhibiting VWF-collagen and VWF-platelet interactions in Rhesus monkeys. Biochem. Pharmacol. 2013, 7, 945–953. [Google Scholar] [CrossRef]
- Chen, J.; Reheman, A.; Gushiken, F.C.; Nolasco, L.; Fu, X.; Moake, J.L.; Ni, H.; López, J.A. N-acetylcysteine reduces the size and activity of von Willebrand factor in human plasma and mice. J. Clin. Investig. 2011, 2, 593–603. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Shea, S.M.; Ku, D.N. Lysis of arterial thrombi by perfusion of N,N’-Diacetyl-L-cystine (DiNAC). PLoS ONE 2021, 16, e0247496. [Google Scholar] [CrossRef] [PubMed]
- Elabbadi, A.; Turpin, M.; Gerotziafas, G.T.; Teulier, M.; Voiriot, G.; Fartoukh, M. Bacterial coinfection in critically ill COVID-19 patients with severe pneumonia. Infection 2021, 49, 559–562. [Google Scholar] [CrossRef] [PubMed]
- Qiu, X.; Liu, M.; Wang, Q.; Zhang, Y.; Kong, L.; Zhou, L. Thrombosis in Critically Ill Influenza Patients: Incidence and Risk Factors. Clin. Appl. Thromb. Hemost. 2024, 30, 10760296241278615. [Google Scholar] [CrossRef]
- Walters, K.A.; D’Agnillo, F.; Sheng, Z.M.; Kindrachuk, J.; Schwartzman, L.M.; Kuestner, R.E.; Chertow, D.S.; Golding, B.T.; Taubenberger, J.K.; Kash, J.C. 1918 pandemic influenza virus and Streptococcus pneumoniae co-infection results in activation of coagulation and widespread pulmonary thrombosis in mice and humans. J. Pathol. 2016, 238, 85–97. [Google Scholar] [CrossRef]
- Narayana Moorthy, A.N.; Narasaraju, T.; Rai, P.; Perumalsamy, R.; Tan, K.B.; Wang, S.; Engelward, B.; Chow, V.T.K. In vivo and in vitro studies on the roles of neutrophil extracellular traps during secondary pneumococcal pneumonia after primary pulmonary influenza infection. Front. Immunol. 2013, 4, 56. [Google Scholar] [CrossRef]
- Babkina, A.S.; Golubev, A.M.; Ostrova, I.V.; Volkov, A.V.; Kuzovlev, A.N. Brain Morphological Changes in COVID-19. Gen. Reanimatol. 2021, 17, 4–15. [Google Scholar] [CrossRef]
- Mattila, K.J.; Valtonen, V.V.; Nieminen, M.S.; Asikainen, S. Role of infection as a risk factor for atherosclerosis, myocardial infarction, and stroke. Clin. Infect. Dis. 1998, 26, 719–734. [Google Scholar] [CrossRef]
- De Lourdes Higuchi, M.; Ramires, J.A. Infectious agents in coronary atheromas: A possible role in the pathogenesis of plaque rupture and acute myocardial infarction. Rev. Inst. Med. Trop. Sao Paulo 2002, 44, 217–224. [Google Scholar] [CrossRef]
- Gattinoni, L.; Chiumello, D.; Caironi, P.; Busana, M.; Romitti, F.; Brazzi, L.; Camporota, L. COVID-19 pneumonia: Different respiratory treatments for different phenotypes? Intensive Care Med. 2020, 46, 1099–1102. [Google Scholar] [CrossRef]
- Hariri, L.; Hardin, C.C. Covid-19, Angiogenesis, and ARDS Endotypes. N. Engl. J. Med. 2020, 383, 182–183. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Search Strategy |
|---|
| (respiratory infection) AND (thrombosis) |
| ((respiratory infection) AND (thrombosis)) NOT (COVID-19) |
| (pneumonia) AND (thrombosis) |
| ((pneumonia) OR (respiratory infection)) AND (stroke) |
| ((pneumonia) OR (respiratory infection)) AND (myocardial infarction) |
| ((pneumonia) OR (respiratory infection)) AND (pulmonary embolism) |
| ((pneumonia) OR (respiratory infection)) AND (arterial thrombosis in situ) |
| ((pneumonia) OR (respiratory infection)) AND (cardiac thrombosis) |
| ((pneumonia) OR (respiratory infection)) AND (mesenteric thrombosis) |
| ((pneumonia) OR (respiratory infection)) AND (lower extremity thrombosis) |
| (pulmonary artery thrombosis) AND (diagnostic) |
| ((thrombosis pulmonary) OR (pulmonary embolism)) AND (imaging) |
| ((pneumonia) OR (respiratory infection)) AND (pathogenesis) AND (thrombosis) |
| (therapeutic target) AND (immunothrombosis) |
| (thrombosis) OR (immunothrombosis) OR (immunothrombosis) OR ((thromboinflammation) AND (ARDS)) |
| Thrombotic Complication | Etiology of Infectious Diseases |
|---|---|
| Myocardial infarction | Influenza viruses [14,15,17,18,22,34,35], Streptococcus pneumoniae [24,25,27], Mycoplasma pneumoniae [28,29], Chlamydia pneumoniae [29], SARS-CoV-2 [17,22,30,31,32,33,34,35] |
| Ischemic stroke | Streptococcus pneumoniae [39], Mycoplasma pneumoniae [40], Influenza viruses [35,45], SARS-CoV-2 [11,35,42,43,45] |
| Pulmonary artery thrombosis | SARS-CoV-2 [11,50,51,57], SARS [59], Influenza viruses [60] |
| Intracardiac thrombosis | SARS-CoV-2 [11,61,62,64], Influenza viruses [70], Mycoplasma pneumoniae [68], Aspergillus [69] |
| Mesenteric thrombosis | SARS-CoV-2 [72,73,74,75,76], Influenza B [77], Mycoplasma pneumoniae [78], Klebsiella pneumoniae [79] |
| Acute lower extremity arterial thrombosis | SARS-CoV-2 [80,81,82,83,84], varicella pneumonia [88], pneumonia caused by Escherichia coli [85], Mycoplasma pneumoniae [86,87] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Babkina, A.S.; Pisarev, M.V.; Grechko, A.V.; Golubev, A.M. Arterial Thrombosis in Acute Respiratory Infections: An Underestimated but Clinically Relevant Problem. J. Clin. Med. 2024, 13, 6007. https://doi.org/10.3390/jcm13196007
Babkina AS, Pisarev MV, Grechko AV, Golubev AM. Arterial Thrombosis in Acute Respiratory Infections: An Underestimated but Clinically Relevant Problem. Journal of Clinical Medicine. 2024; 13(19):6007. https://doi.org/10.3390/jcm13196007
Chicago/Turabian StyleBabkina, Anastasiya S., Mikhail V. Pisarev, Andrey V. Grechko, and Arkady M. Golubev. 2024. "Arterial Thrombosis in Acute Respiratory Infections: An Underestimated but Clinically Relevant Problem" Journal of Clinical Medicine 13, no. 19: 6007. https://doi.org/10.3390/jcm13196007