
SA-β-Gal in Kidney Tubules as a Predictor of Renal Outcome in Patients with Chronic Kidney Disease
 , ,
, ,
Abstract
:
1. Introduction
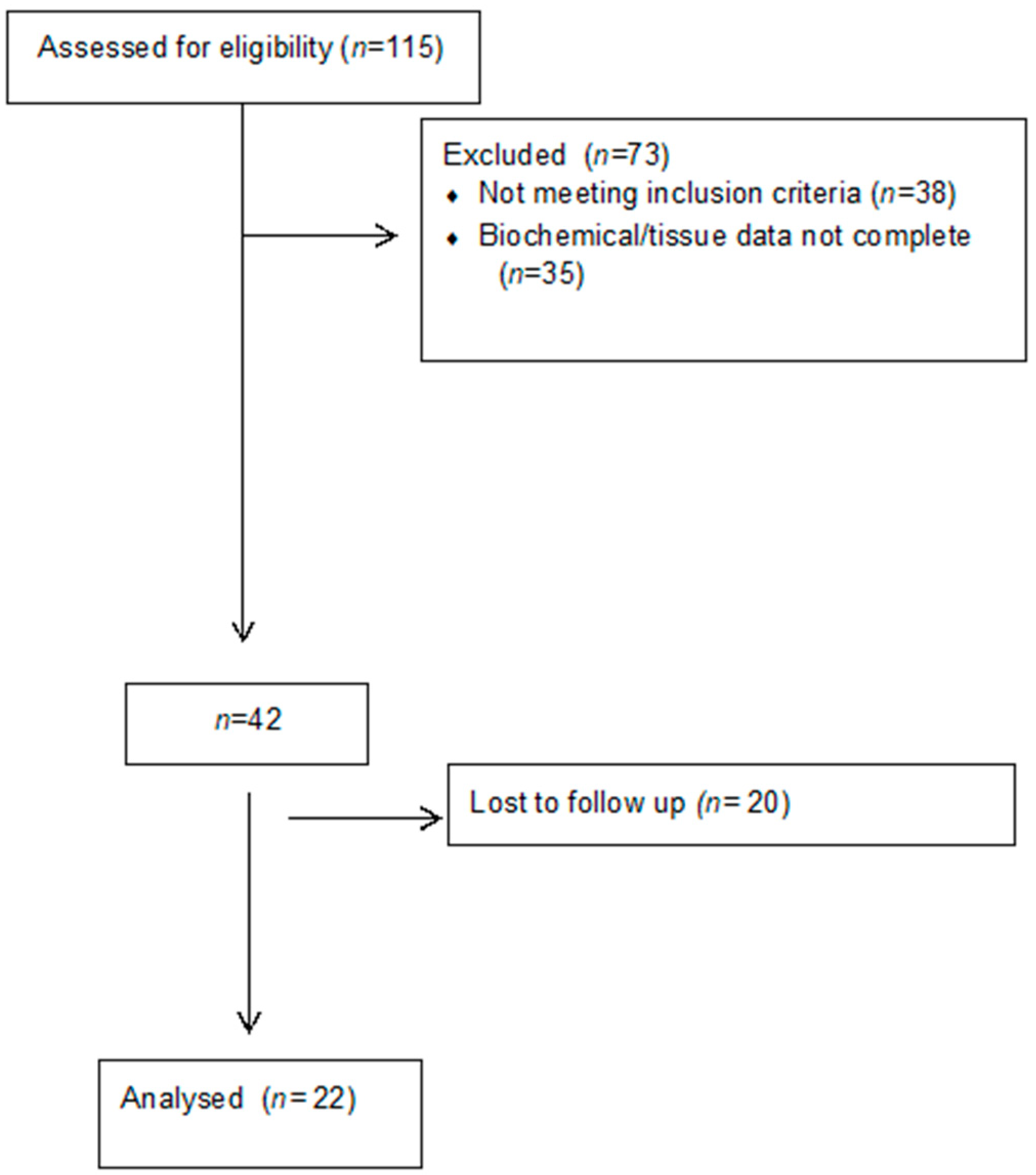
2. Materials and Methods
2.1. Tissue Samples
2.2. SA-β-Gal Activity on Cryopreserved Kidney Biopsies
2.3. Statistical Analysis
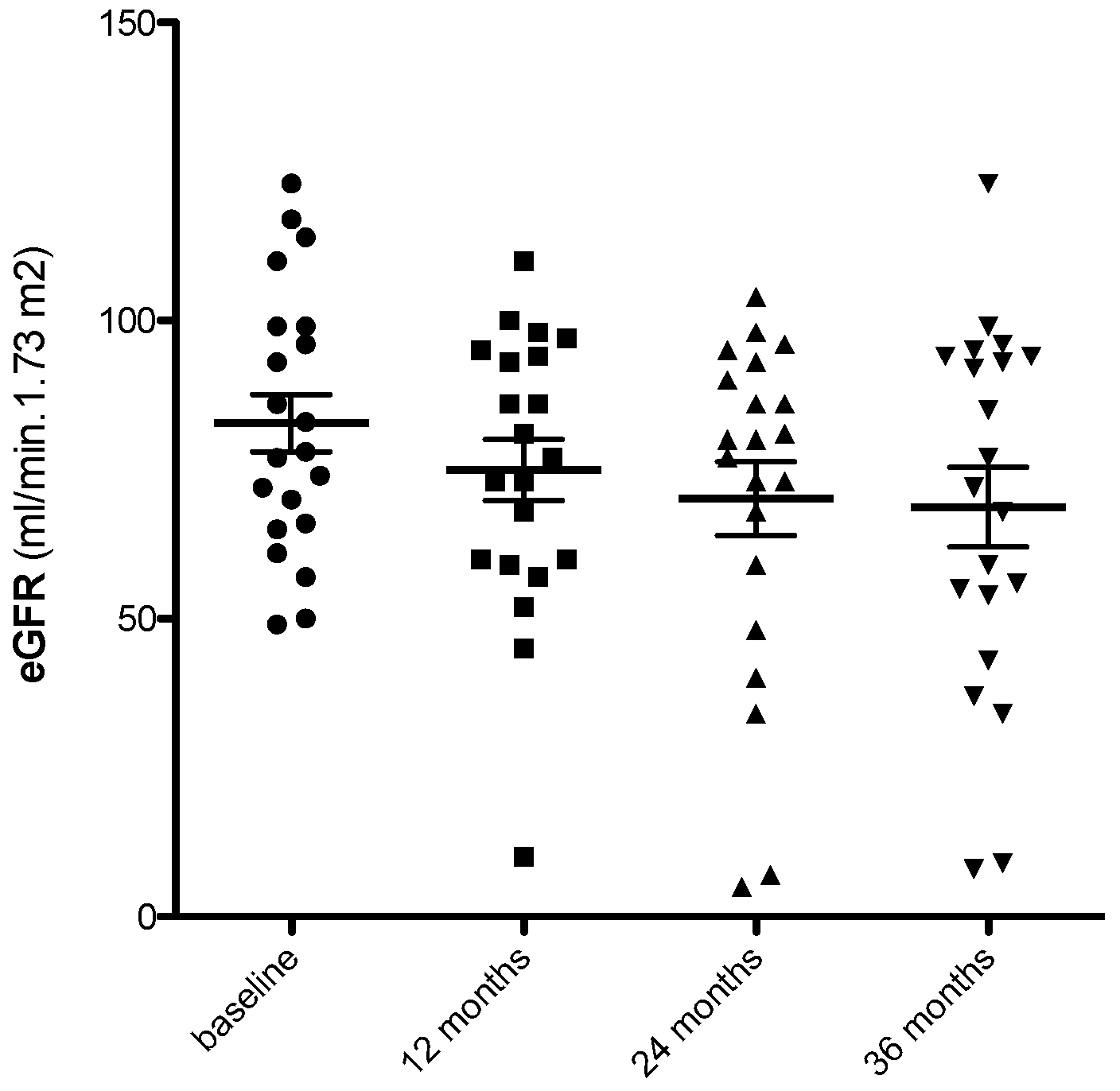
3. Results
3.1. SA-β-Gal Staining in the Tubular Compartment
3.2. Clinical Correlates of SA-β-Gal Expression in Kidney Tubules
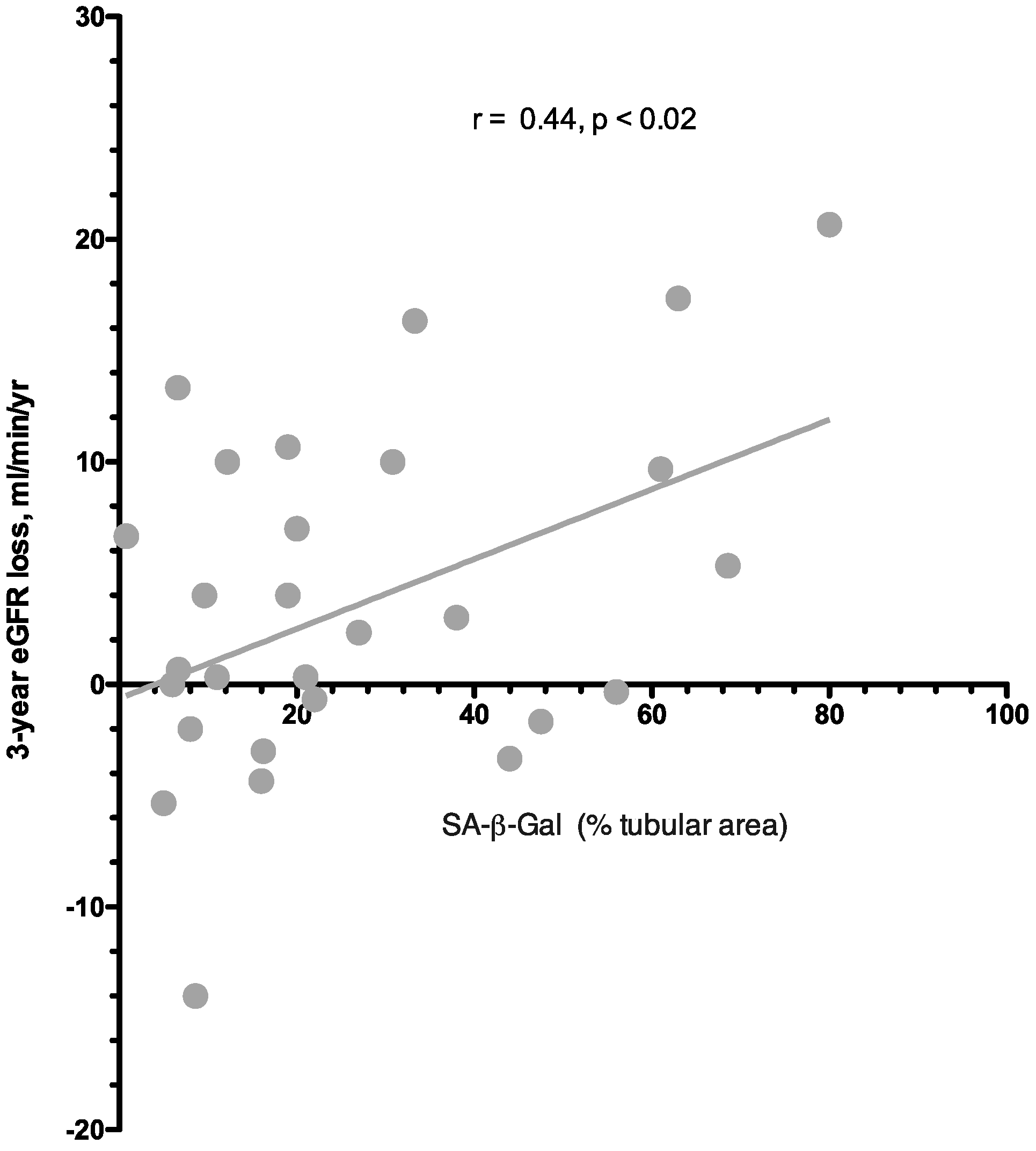
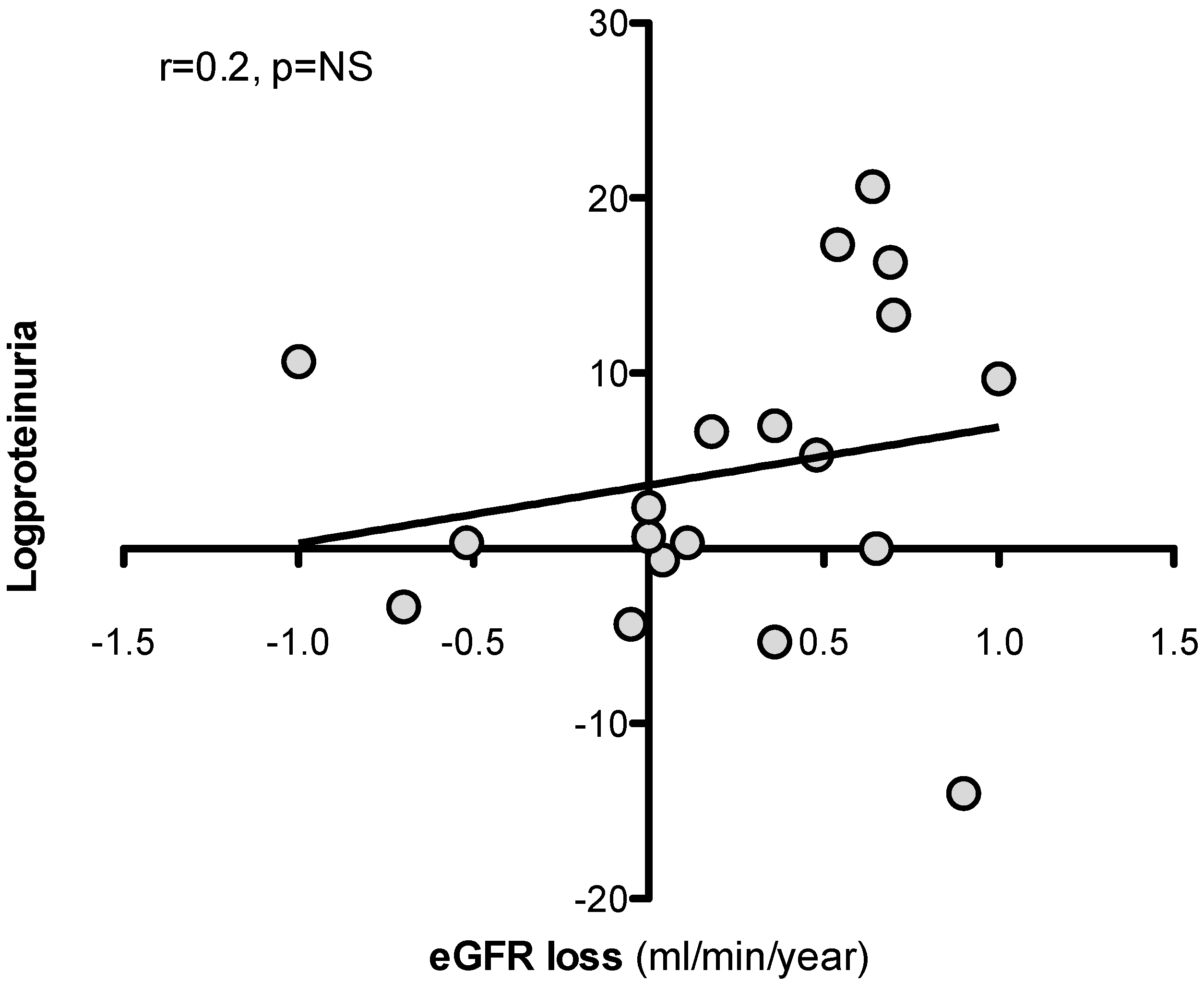
3.3. Predictors of eGFR Loss at 36 Months
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Melk, A.; Schmidt, B.M.; Vongwiwatana, A.; Rayner, D.C.; Halloran, P.F. Increased expression of senescence-associated cell cycle inhibitor p16INK4a in deteriorating renal transplants and diseased native kidney. Am. J. Transpl. 2005, 6, 1375–1382. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, R.; Melk, A. Molecular mechanisms of renal aging. Kidney Int. 2017, 92, 569–579. [Google Scholar] [CrossRef] [PubMed]
- Docherty, M.H.; Baird, D.P.; Hughes, J.; Ferenbach, D.A. Cellular Senescence and Senotherapies in the Kidney: Current Evidence and Future Directions. Front. Pharmacol. 2020, 11, 755–769. [Google Scholar] [CrossRef] [PubMed]
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O.; et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 9363–9367. [Google Scholar] [CrossRef] [PubMed]
- Coppe, J.P.; Patil, C.K.; Rodier, F.; Krtolica, A.; Beausejour, C.M.; Parrinello, S.; Campisi, J. A human-like senescence-associated secretory phenotype is conserved in mouse cells dependent on physiological oxygen. PLoS ONE 2010, 5, e9188. [Google Scholar] [CrossRef] [PubMed]
- Childs, B.G.; Durik, M.; Baker, D.J.; Van Deursen, J.M. Cellular senescence in aging and age-related disease: From mechanisms to therapy. Nat. Med. 2015, 21, 1424–1435. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Besschetnova, T.Y.; Brooks, C.R.; Shah, J.V.; Bonventre, J.V. Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat. Med. 2010, 16, 535–543. [Google Scholar] [CrossRef]
- Melk, A.; Schmidt, B.M.; Takeuchi, O.; Sawitzki, B.; Rayner, D.C.; Halloran, P.F. Expression of p16INK4a and other cell cycle regulator and senescence associated genes in aging human kidney. Kidney Int. 2004, 65, 510–520. [Google Scholar] [CrossRef]
- Docherty, M.H.; O’Sullivan, E.D.; Bonventre, J.V.; Ferenbach, D.A. Cellular Senescence in the Kidney. J. Am. Soc. Nephrol. 2019, 30, 726–736. [Google Scholar] [CrossRef]
- Gorgoulis, V.; Adams, P.D.; Alimonti, A.; Bennett, D.C.; Bischof, O.; Bishop, C.; Campisi, J.; Collado, M.; Evangelou, K.; Ferbeyre, G.; et al. Cellular Senescence: Defining a Path Forward. Cell 2019, 179, 813–827. [Google Scholar] [CrossRef]
- Gurung, R.L.; Yiamunaa, M.; Liu, S.; Liu, J.J.; Lim, S.C. Short Leukocyte Telomere Length Predicts Albuminuria Progression in Individuals with Type 2 Diabetes. Kidney Int. Rep. 2018, 3, 592–601. [Google Scholar] [CrossRef] [PubMed]
- Krishna, D.R.; Sperker, B.; Fritz, P.; Klotz, U. Does pH 6 β-galactosidase activity indicate cell senescence? Mech. Ageing Dev. 1999, 109, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Verzola, D.; Gandolfo, M.T.; Gaetani, G.; Ferraris, A.; Mangerini, R. Accelerated senescence in the kidneys of patients with type 2 diabetic nephropathy. Am. J. Physiol. Renal. Physiol. 2008, 295, F1563–F1573. [Google Scholar] [CrossRef] [PubMed]
- Debacq-Chainiaux, F.; Erusalimsky, J.D.; Campisi, J.; Toussaint, O. Protocols to detect senescence-associated beta-galactosidase (SA-betagal) activity, a biomarker of senescent cells in culture and in vivo. Nat. Protoc. 2009, 4, 1798–1806. [Google Scholar] [CrossRef]
- Liu, J.; Yang, J.R.; He, Y.N.; Cai, G.Y.; Zhang, J.G.; Lin, L.R.; Xiao, H.S. Accelerated senescence of renal tubular epithelial cells is associated with disease progression of patients with immunoglobulin A (IgA) nephropathy. Transl. Res. 2012, 159, 454–463. [Google Scholar] [CrossRef] [PubMed]
- Verzola, D.; Saio, M.; Picciotto, D.; Viazzi, F.; Russo, E.; Cipriani, L. Cellular Senescence Is Associated with Faster Progression of Focal Segmental Glomerulosclerosis. Am. J. Nephrol. 2021, 51, 950–958. [Google Scholar] [CrossRef]
- Verzola, D.; Gandolfo, M.T.; Ferrario, F.; Rastaldi, M.P.; Villaggio, B.; Gianiorio, F. Apoptosis in the kidneys of patients with type II diabetic nephropathy. Kidney Int. 2007, 72, 1262–1272. [Google Scholar] [CrossRef]
- O’Callaghan, C.A.; Shine, B.; Lasserson, D.S. Chronic kidney disease: A large-scale population-based study of the effects of introducing the CKD-EPI formula for eGFR reporting. BMJ Open 2011, 1, e000308. [Google Scholar] [CrossRef]
- Li, M.; Yang, M.; Zhu, W.H. Advances in fluorescent sensors for β-galactosidase. Mater. Chem. Front. 2021, 5, 763–774. [Google Scholar] [CrossRef]
- Muñoz-Espín, D.; Serrano, M. Cellular senescence: From physiology to pathology. Nat. Rev. Mol. Cell Biol. 2014, 15, 482–496. [Google Scholar] [CrossRef]
- Fagagna, F.D.A.D.; Reaper, P.M.; Clay-Farrace, L.; Fiegler, H.; Carr, P.; Von Zglinicki, T. A DNA damage checkpoint response in telomere-initiated senescence. Nature 2003, 426, 194–198. [Google Scholar] [CrossRef] [PubMed]
- Miwa, S.; Kashyap, S.; Chini, E.; von Zglinicki, T. Mitochondrial dysfunction in cell senescence and aging. J. Clin. Investig. 2022, 132, e158447. [Google Scholar] [CrossRef]
- Chiu, C.L. Does telomere shortening precede the onset of hypertension in spontaneously hypertensive mice? Hypertension 2008, 52, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Cicalese, S.M.; da Silva, J.F.; Priviero, F.; Webb, R.C.; Eguchi, S.; Tostes, R.C. Vascular Stress Signaling in Hypertension. Circ. Res. 2021, 128, 969–992. [Google Scholar] [CrossRef] [PubMed]
- Westhoff, J.H.; Hilgers, K.F.; Steinbach, M.P.; Hartner, A.; Klanke, B.; Amann, K.; Melk, A. Hypertension induces somatic cellular senescence in rats and humans by induction of cell cycle inhibitor p16INK4a. Hypertension 2008, 52, 123–129. [Google Scholar] [CrossRef]
- Garibotto, G.; Carta, A.; Picciotto, D.; Viazzi, F.; Verzola, D. Toll-like receptor-4 signaling mediates inflammation and tissue injury in diabetic nephropathy. J. Nephrol. 2017, 30, 719–727. [Google Scholar] [CrossRef]
- White, W.E.; Yaqoob, M.M.; Harwood, S.M. Aging and uremia: Is there cellular and molecular crossover? World J. Nephrol. 2015, 4, 19–30. [Google Scholar] [CrossRef]
- Rhee, J.J.; Jardine, M.J.; Chertow, G.M.; Mahaffey, K.W. Dedicated kidney disease-focused outcome trials with sodium-glucose cotransporter-2 inhibitors: Lessons from CREDENCE and expectations from DAPA-HF, DAPA-CKD, and EMPA-KIDNEY. Diabetes Obes. Metab. 2020, 22 (Suppl. 1), 46–54. [Google Scholar] [CrossRef]
- Rossing, P.; Caramori, M.L.; Chan, J.C.; Heerspink, H.J.; Hurst, C.; Khunti, K.; Liew, A.; Michos, E.D.; Navaneethan, S.D.; Olowu, W.A.; et al. Executive summary of the KDIGO 2022 Clinical Practice Guideline for Diabetes Management in Chronic Kidney Disease: An update based on rapidly emerging new evidence. Kidney Int. 2022, 102, 990–999. [Google Scholar] [CrossRef]
- Sis, B.; Tasanarong, A.; Dadras, F.; Solez, K.; Halloran, P.F. Accelerated expression of senescence associated cell cycle inhibitor p16INK4A in kidneys with glomerular disease. Kidney Int. 2007, 71, 218–226. [Google Scholar] [CrossRef]
- Xu, M.; Pirtskhalava, T.; Farr, J.N.; Weigand, B.M.; Palmer, A.K.; Weivoda, M.M.; Kirkland, J.L. Senolytics improve physical function and increase lifespan in old age. Nat. Med. 2018, 24, 1246–1256. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.; Eide, I.A.; Tannæs, T.M.; Waldum-Grevbo, B.; Jenssen, T.; Svensson, M. Marine n-3 Polyunsaturated Fatty Acids and Cellular Senescence Markers in Incident Kidney Transplant Recipients: The Omega-3 Fatty Acids in Renal Transplantation (ORENTRA) Randomized Clinical Trial. Kidney Med. 2021, 3, 1041–1049. [Google Scholar] [CrossRef] [PubMed]
- Koppelstaetter, C.; Leierer, J.; Rudnicki, M.; Kerschbaum, J.; Kronbichler, A.; Melk, A.; Mayer, G.; Perco, P. Computational Drug Screening Identifies Compounds Targeting Renal Age-associated Molecular Profiles. Comput. Struct. Biotechnol. J. 2019, 17, 843–853. [Google Scholar] [CrossRef] [PubMed]
- Yoo, K.H.; Yim, H.E.; Bae, E.S. Angiotensin inhibition and cellular senescence in the developing rat kidney. Exp. Mol. Pathol. 2020, 117, 104551. [Google Scholar] [CrossRef]
- Kim, S.R.; Zou, H.X.; Tang, H.; Puranik, A.S.; Abumoawad, A.M.; Zhu, X.Y. Increased cellular senescence in the murine and human stenotic kidney: Effect of mesenchymal stem cells. J. Cell Physiol. 2021, 236, 1332–1344. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Number of Subjects | 22 |
| Age (years) | 50 ± 3 |
| Gender (M/F) | 9/13 |
| BMI (Kg/m2) | 28 ± 1.7 |
| Proteinuria (g/day) | 1.9 (0.75, 4.42) |
| Estimated GFR (mL/min 1.73 m2) | 80 ± 5.7 |
| eGFR loss (mL/min/year) | 4.0 ± 1.93 |
| Follow up (years) | 3 |
| SBP (mHg) | 138 ± 7 |
| DBP (mHg) | 84 ± 4 |
| Total Cholesterol (mg/dL) | 222 (200, 259) |
| Triglycerides (mg/dL) | 200 (100, 254) |
| HDL-C (mg/dL) | 61 ± 5 |
| LDL (mg/dL) | 139 (84, 345) |
| IgA nephropathy (n) | 5 |
| Focal segmental glomerulosclerosis | 5 |
| membranous glomerulonephritis | 5 |
| diabetic kidney disease | 2 |
| hypertensive nephroangiosclerosis | 2 |
| minimal change disease | 1 |
| stage II lupus nephritis | 1 |
| chronic glomerulonephritis | 1 |
| RAAS blockade (n) | 22 |
| Statins | 16 |
| Steroids/immunosuppressive treatment | 13 |
| Clinical Characteristics | SA-β-Gal (% Tubular Area) | |
|---|---|---|
| R | P | |
| Age (years) | 0.08 | NS |
| Serum creatinine (mg/dL) | 0.05 | NS |
| eGFR (mL/min.1.73 m2) | −0.28 | NS |
| BMI (kg/m2) | 0.10 | NS |
| logProteinuria (g/day) | 0.27 | NS |
| logLDL-Cholesterol (mg/dL) | 0.06 | NS |
| logTriglycerides (mg/dL) | 0.11 | NS |
| logTotal Cholesterol | 0.19 | NS |
| SBP (mmHg) | 0.15 | NS |
| DBP (mmHg) | −0.09 | NS |
| Parameter | eGFR Loss at 36 Months (mL/min.1.73 m2) | |
|---|---|---|
| r | p | |
| Age (years) | 0.19 | NS |
| logproteinuria (g/day) | 0.20 | NS |
| BMI (kg/m2) | 0.09 | NS |
| logLDL-Cholesterol (mg/dL) | 0.01 | NS |
| logTriglycerides (mg/dL) | 0.003 | NS |
| logTotal Cholesterol | 0.06 | NS |
| Immunosuppressive treatment | 0.10 | NS |
| SBP/DBP (mmHg) | 0.1 | NS |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Esposito, P.; Picciotto, D.; Verzola, D.; Garibotto, G.; Parodi, E.L.; Sofia, A.; Costigliolo, F.; Gaggero, G.; Zanetti, V.; Saio, M.; et al. SA-β-Gal in Kidney Tubules as a Predictor of Renal Outcome in Patients with Chronic Kidney Disease. J. Clin. Med. 2024, 13, 322. https://doi.org/10.3390/jcm13020322
Esposito P, Picciotto D, Verzola D, Garibotto G, Parodi EL, Sofia A, Costigliolo F, Gaggero G, Zanetti V, Saio M, et al. SA-β-Gal in Kidney Tubules as a Predictor of Renal Outcome in Patients with Chronic Kidney Disease. Journal of Clinical Medicine. 2024; 13(2):322. https://doi.org/10.3390/jcm13020322
Chicago/Turabian StyleEsposito, Pasquale, Daniela Picciotto, Daniela Verzola, Giacomo Garibotto, Emanuele Luigi Parodi, Antonella Sofia, Francesca Costigliolo, Gabriele Gaggero, Valentina Zanetti, Michela Saio, and et al. 2024. "SA-β-Gal in Kidney Tubules as a Predictor of Renal Outcome in Patients with Chronic Kidney Disease" Journal of Clinical Medicine 13, no. 2: 322. https://doi.org/10.3390/jcm13020322