
Metabolic Chaos in Kidney Disease: Unraveling Energy Dysregulation



Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Energy Metabolism in the Kidneys Under Physiological Conditions
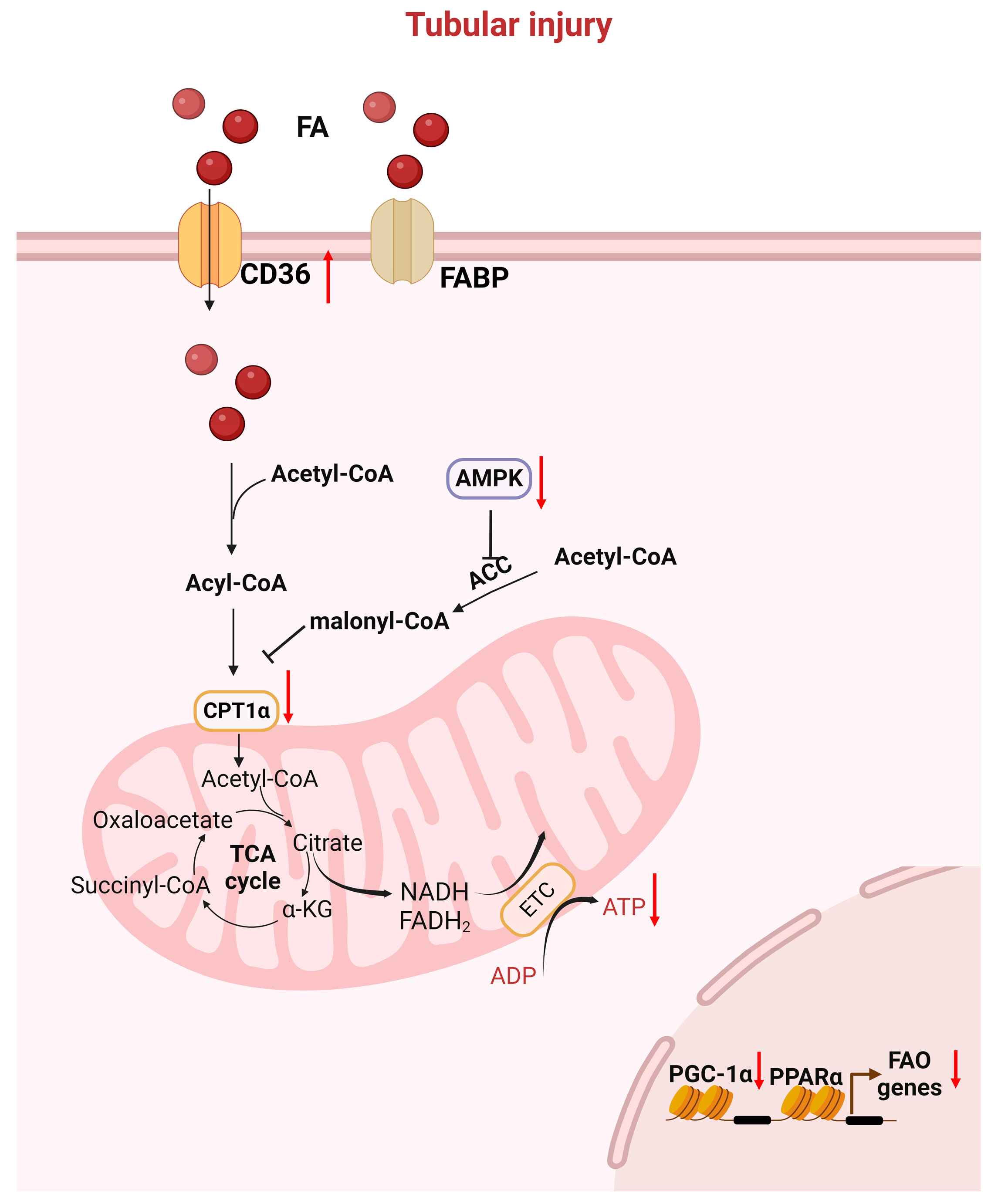
2.1. Fatty Acid Oxidation and the TCA Cycle in the Kidneys
2.2. Glucose and Amino Acid Metabolism in the Kidneys
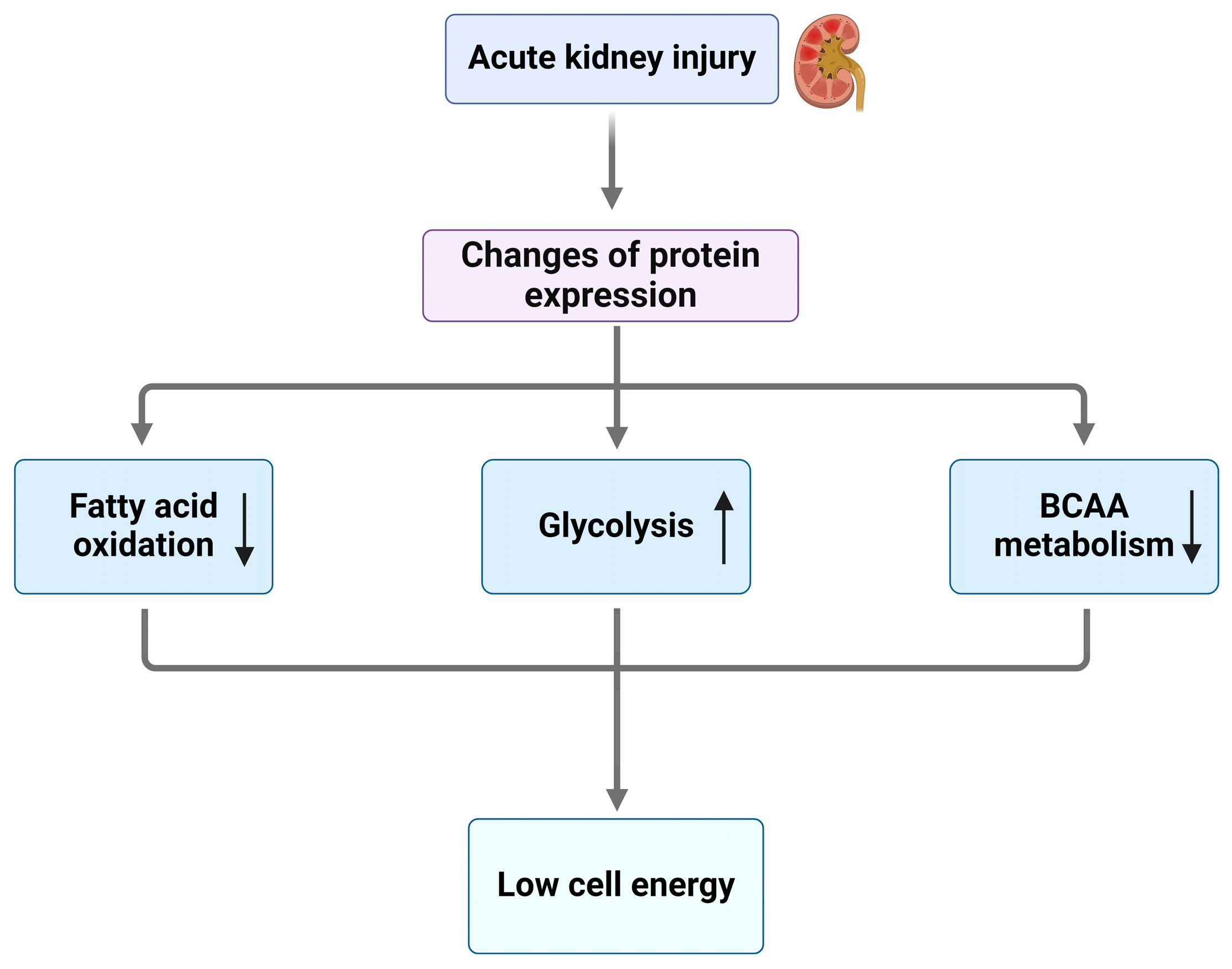
3. Energy Metabolism Dysregulation in AKI
3.1. Key Genes/Proteins Involved in FAO Defects After AKI
3.1.1. PPARs Family
3.1.2. The KLF Family
3.1.3. AMPK
3.1.4. Sirtuins Family
3.1.5. Other Genes
3.2. Key Genes/Proteins Involved in Glycolysis in AKI
4. Energy Metabolism Dysfunction in CKD
4.1. Key Genes/Proteins Involved in FAO in CKD
4.1.1. CPT1a
4.1.2. PGC1α
4.1.3. KLF and Cytokines
4.2. Key Genes/Proteins Involved in Lipid Accumulation in CKD
5. Energy Metabolism Dysfunction in the Transition from AKI to CKD
6. Diagnostic Tools and Therapies for Kidney Disease
7. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Zhu, Y.; Yang, S.; Lv, L.; Zhai, X.; Wu, G.; Qi, X.; Dong, D.; Tao, X. Research Progress on the Positive and Negative Regulatory Effects of Rhein on the Kidney: A Review of Its Molecular Targets. Molecules 2022, 27, 6572. [Google Scholar] [CrossRef] [PubMed]
- Muller, M.; Mentel, M.; van Hellemond, J.J.; Henze, K.; Woehle, C.; Gould, S.B.; Yu, R.Y.; van der Giezen, M.; Tielens, A.G.; Martin, W.F. Biochemistry and evolution of anaerobic energy metabolism in eukaryotes. Microbiol. Mol. Biol. Rev. 2012, 76, 444–495. [Google Scholar] [CrossRef] [PubMed]
- Fani, R.; Fondi, M. Origin and evolution of metabolic pathways. Phys. Life Rev. 2009, 6, 23–52. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.L.; Soeters, M.R.; Wust, R.C.I.; Houtkooper, R.H. Metabolic Flexibility as an Adaptation to Energy Resources and Requirements in Health and Disease. Endocr. Rev. 2018, 39, 489–517. [Google Scholar] [CrossRef] [PubMed]
- Miguel, V.; Shaw, I.W.; Kramann, R. Metabolism at the crossroads of inflammation and fibrosis in chronic kidney disease. Nat. Rev. Nephrol. 2024. [Google Scholar] [CrossRef]
- Gui, Y.; Palanza, Z.; Gupta, P.R.; Li, H.; Pan, Y.; Wang, Y.; Hargis, G.R.; Kreutzer, D.L.; Wang, Y.; Bastacky, S.I.; et al. Calponin 2 regulates ketogenesis to mitigate acute kidney injury. JCI Insight 2023, 8, e170521. [Google Scholar] [CrossRef]
- Gui, Y.; Wang, Y.; Palanza, Z.; Wang, J.L.; Gupta, P.; Tao, J.; Qiao, Y.; Hargis, G.; Kreutzer, D.L.; Bastacky, S.I.; et al. Calponin 2 harnesses metabolic reprogramming to determine kidney fibrosis. Mol. Metab 2023, 71, 101712. [Google Scholar] [CrossRef]
- Clark, A.J.; Saade, M.C.; Vemireddy, V.; Vu, K.Q.; Flores, B.M.; Etzrodt, V.; Ciampa, E.J.; Huang, H.; Takakura, A.; Zandi-Nejad, K.; et al. Hepatocyte nuclear factor 4alpha mediated quinolinate phosphoribosylltransferase (QPRT) expression in the kidney facilitates resilience against acute kidney injury. Kidney Int. 2023, 104, 1150–1163. [Google Scholar] [CrossRef]
- Saade, M.C.; Parikh, S.M. Energy Metabolism in CKD: Running Low on Fuel. Kidney360 2023, 4, 1014–1016. [Google Scholar] [CrossRef]
- Lee, K.; Thompson, E.A.; Gharaie, S.; Patel, C.H.; Kurzhagen, J.T.; Pierorazio, P.M.; Arend, L.J.; Thomas, A.G.; Noel, S.; Slusher, B.S.; et al. T cell metabolic reprogramming in acute kidney injury and protection by glutamine blockade. JCI Insight 2023, 8, e160345. [Google Scholar] [CrossRef]
- Fang, Y.; Chen, B.; Gong, A.Y.; Malhotra, D.K.; Gupta, R.; Dworkin, L.D.; Gong, R. The ketone body beta-hydroxybutyrate mitigates the senescence response of glomerular podocytes to diabetic insults. Kidney Int. 2021, 100, 1037–1053. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Gu, W.; Hepokoski, M.; Pham, H.; Tham, R.; Kim, Y.C.; Simonson, T.S.; Singh, P. Energy Metabolism Dysregulation in Chronic Kidney Disease. Kidney360 2023, 4, 1080–1094. [Google Scholar] [CrossRef] [PubMed]
- Goto, H.; Nakashima, H.; Mori, K.; Tanoue, K.; Ito, S.; Kearney, B.M.; Kato, A.; Nakashima, M.; Imakiire, T.; Kumagai, H.; et al. l-Carnitine pretreatment ameliorates heat stress-induced acute kidney injury by restoring mitochondrial function of tubular cells. Am. J. Physiol. Renal Physiol. 2024, 326, F338–F351. [Google Scholar] [CrossRef] [PubMed]
- Silva Barbosa, A.C.; Pfister, K.E.; Chiba, T.; Bons, J.; Rose, J.P.; Burton, J.B.; King, C.D.; O’Broin, A.; Young, V.; Zhang, B.; et al. Dicarboxylic Acid Dietary Supplementation Protects against AKI. J. Am. Soc. Nephrol. 2024, 35, 135–148. [Google Scholar] [CrossRef]
- Osborn, J.W.; Tyshynsky, R.; Vulchanova, L. Function of Renal Nerves in Kidney Physiology and Pathophysiology. Annu. Rev. Physiol. 2021, 83, 429–450. [Google Scholar] [CrossRef]
- Jacob, J.; Dannenhoffer, J.; Rutter, A. Acute Kidney Injury. Prim. Care 2020, 47, 571–584. [Google Scholar] [CrossRef]
- Zhu, Z.; Hu, J.; Chen, Z.; Feng, J.; Yang, X.; Liang, W.; Ding, G. Transition of acute kidney injury to chronic kidney disease: Role of metabolic reprogramming. Metabolism 2022, 131, 155194. [Google Scholar] [CrossRef]
- Simon, N.; Hertig, A. Alteration of Fatty Acid Oxidation in Tubular Epithelial Cells: From Acute Kidney Injury to Renal Fibrogenesis. Front. Med. (Lausanne) 2015, 2, 52. [Google Scholar] [CrossRef]
- Nakamura, M.T.; Yudell, B.E.; Loor, J.J. Regulation of energy metabolism by long-chain fatty acids. Prog. Lipid Res. 2014, 53, 124–144. [Google Scholar] [CrossRef]
- Choi, I.; Son, H.; Baek, J.H. Tricarboxylic Acid (TCA) Cycle Intermediates: Regulators of Immune Responses. Life 2021, 11, 69. [Google Scholar] [CrossRef]
- Jang, H.S.; Noh, M.R.; Kim, J.; Padanilam, B.J. Defective Mitochondrial Fatty Acid Oxidation and Lipotoxicity in Kidney Diseases. Front. Med. (Lausanne) 2020, 7, 65. [Google Scholar] [CrossRef] [PubMed]
- Schlaepfer, I.R.; Joshi, M. CPT1A-mediated Fat Oxidation, Mechanisms, and Therapeutic Potential. Endocrinology 2020, 161, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Chung, K.W.; Lee, E.K.; Lee, M.K.; Oh, G.T.; Yu, B.P.; Chung, H.Y. Impairment of PPARalpha and the Fatty Acid Oxidation Pathway Aggravates Renal Fibrosis during Aging. J. Am. Soc. Nephrol. 2018, 29, 1223–1237. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, S.; Gupta, P.; Saini, A.S.; Kaushal, C.; Sharma, S. The peroxisome proliferator-activated receptor: A family of nuclear receptors role in various diseases. J. Adv. Pharm. Technol. Res. 2011, 2, 236–240. [Google Scholar] [CrossRef]
- Broeders, N.; Abramowicz, D. Peroxisome proliferator-activated receptors (PPARs): Novel therapeutic targets in renal disease. Kidney Int. 2002, 61, 354–355. [Google Scholar] [CrossRef]
- Hong, G.; Lockhart, A.; Davis, B.; Rahmoune, H.; Baker, S.; Ye, L.; Thompson, P.; Shou, Y.; O’Shaughnessy, K.; Ronco, P.; et al. PPARgamma activation enhances cell surface ENaCalpha via up-regulation of SGK1 in human collecting duct cells. FASEB J. 2003, 17, 1966–1968. [Google Scholar] [CrossRef]
- Ke, R.; Xu, Q.; Li, C.; Luo, L.; Huang, D. Mechanisms of AMPK in the maintenance of ATP balance during energy metabolism. Cell Biol. Int. 2018, 42, 384–392. [Google Scholar] [CrossRef]
- Blenkharn, J.I.; Thompson, J.N.; Blumgart, L.H. Antibiotic prophylaxis in endoscopic retrograde cholangiopancreatography. Eur. J. Clin. Microbiol. 1986, 5, 53. [Google Scholar] [CrossRef]
- Fernie, A.R.; Carrari, F.; Sweetlove, L.J. Respiratory metabolism: Glycolysis, the TCA cycle and mitochondrial electron transport. Curr. Opin. Plant Biol. 2004, 7, 254–261. [Google Scholar] [CrossRef]
- Li, Z.; Lu, S.; Li, X. The role of metabolic reprogramming in tubular epithelial cells during the progression of acute kidney injury. Cell Mol. Life Sci. 2021, 78, 5731–5741. [Google Scholar] [CrossRef]
- Skrabic, R.; Kumric, M.; Vrdoljak, J.; Rusic, D.; Skrabic, I.; Vilovic, M.; Martinovic, D.; Duplancic, V.; Ticinovic Kurir, T.; Bozic, J. SGLT2 Inhibitors in Chronic Kidney Disease: From Mechanisms to Clinical Practice. Biomedicines 2022, 10, 2458. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Zhu, Z.; Liang, W.; Luo, Z.; Hu, J.; Feng, J.; Zhang, Z.; Luo, Q.; Yang, H.; Ding, G. Reduction of anaerobic glycolysis contributes to angiotensin II-induced podocyte injury with foot process effacement. Kidney Int. 2023, 103, 735–748. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Bai, X.; Chen, J.; Huang, M.; Hong, Q.; Ouyang, Q.; Sun, X.; Zhang, Y.; Liu, J.; Wang, X.; et al. Pyruvate kinase M2 regulates kidney fibrosis through pericyte glycolysis during the progression from acute kidney injury to chronic kidney disease. Cell Prolif. 2024, 57, e13548. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Nagothu, K.K.; Desai, V.; Lee, T.; Branham, W.; Moland, C.; Megyesi, J.K.; Crew, M.D.; Portilla, D. Transgenic expression of proximal tubule peroxisome proliferator-activated receptor-alpha in mice confers protection during acute kidney injury. Kidney Int. 2009, 76, 1049–1062. [Google Scholar] [CrossRef] [PubMed]
- Krebs, H.A.; Lund, P. Aspects of the regulation of the metabolism of branched-chain amino acids. Adv. Enzyme. Regul. 1976, 15, 375–394. [Google Scholar] [CrossRef]
- Harris, R.A.; Joshi, M.; Jeoung, N.H.; Obayashi, M. Overview of the molecular and biochemical basis of branched-chain amino acid catabolism. J. Nutr. 2005, 135, 1527S–1530S. [Google Scholar] [CrossRef]
- Chen, H.; Chen, L.; Liu, D.; Chen, D.Q.; Vaziri, N.D.; Yu, X.Y.; Zhang, L.; Su, W.; Bai, X.; Zhao, Y.Y. Combined Clinical Phenotype and Lipidomic Analysis Reveals the Impact of Chronic Kidney Disease on Lipid Metabolism. J. Proteome Res. 2017, 16, 1566–1578. [Google Scholar] [CrossRef]
- Lima, C.; Macedo, E. Urinary Biochemistry in the Diagnosis of Acute Kidney Injury. Dis. Markers. 2018, 2018, 4907024. [Google Scholar] [CrossRef]
- Bataille, A.; Galichon, P.; Chelghoum, N.; Oumoussa, B.M.; Ziliotis, M.J.; Sadia, I.; Vandermeersch, S.; Simon-Tillaux, N.; Legouis, D.; Cohen, R.; et al. Increased Fatty Acid Oxidation in Differentiated Proximal Tubular Cells Surviving a Reversible Episode of Acute Kidney Injury. Cell Physiol. Biochem. 2018, 47, 1338–1351. [Google Scholar] [CrossRef]
- Poston, J.T.; Koyner, J.L. Sepsis associated acute kidney injury. BMJ 2019, 364, k4891. [Google Scholar] [CrossRef]
- Basu, R.K. Acute Kidney Injury in Hospitalized Pediatric Patients. Pediatr. Ann. 2018, 47, e286–e291. [Google Scholar] [CrossRef] [PubMed]
- Susantitaphong, P.; Cruz, D.N.; Cerda, J.; Abulfaraj, M.; Alqahtani, F.; Koulouridis, I.; Jaber, B.L. Acute Kidney Injury Advisory Group of the American Society of Nephrology: World incidence of AKI: A meta-analysis. Clin. J. Am. Soc. Nephrol. 2013, 8, 1482–1493. [Google Scholar] [CrossRef] [PubMed]
- Arulkumaran, N.; Pollen, S.; Greco, E.; Courtneidge, H.; Hall, A.M.; Duchen, M.R.; Tam, F.W.K.; Unwin, R.J.; Singer, M. Renal Tubular Cell Mitochondrial Dysfunction Occurs Despite Preserved Renal Oxygen Delivery in Experimental Septic Acute Kidney Injury. Crit. Care Med. 2018, 46, e318–e325. [Google Scholar] [CrossRef] [PubMed]
- Masereeuw, R.; Mutsaers, H.A.; Toyohara, T.; Abe, T.; Jhawar, S.; Sweet, D.H.; Lowenstein, J. The kidney and uremic toxin removal: Glomerulus or tubule? Semin. Nephrol. 2014, 34, 191–208. [Google Scholar] [CrossRef] [PubMed]
- Lima, N.K.S.; Farias, W.R.A.; Cirilo, M.A.S.; Oliveira, A.G.; Farias, J.S.; Aires, R.S.; Muzi-Filho, H.; Paixao, A.D.O.; Vieira, L.D. Renal ischemia-reperfusion leads to hypertension and changes in proximal tubule Na(+) transport and renin-angiotensin-aldosterone system: Role of NADPH oxidase. Life Sci. 2021, 266, 118879. [Google Scholar] [CrossRef]
- Basile, D.P. The endothelial cell in ischemic acute kidney injury: Implications for acute and chronic function. Kidney Int. 2007, 72, 151–156. [Google Scholar] [CrossRef]
- Barutta, F.; Bellini, S.; Gruden, G. Mechanisms of podocyte injury and implications for diabetic nephropathy. Clin. Sci. 2022, 136, 493–520. [Google Scholar] [CrossRef]
- Chen, Y.; Lin, L.; Tao, X.; Song, Y.; Cui, J.; Wan, J. The role of podocyte damage in the etiology of ischemia-reperfusion acute kidney injury and post-injury fibrosis. BMC Nephrol. 2019, 20, 106. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Thompson, C.B. Cellular metabolism and disease: What do metabolic outliers teach us? Cell 2012, 148, 1132–1144. [Google Scholar] [CrossRef]
- Afshinnia, F.; Nair, V.; Lin, J.; Rajendiran, T.M.; Soni, T.; Byun, J.; Sharma, K.; Fort, P.E.; Gardner, T.W.; Looker, H.C.; et al. Increased lipogenesis and impaired beta-oxidation predict type 2 diabetic kidney disease progression in American Indians. JCI Insight 2019, 4, e130317. [Google Scholar] [CrossRef]
- Xiong, L.; He, T.; Liu, C.; Qin, S.; Xiao, T.; Xin, W.; Wang, Y.; Ran, L.; Zhang, B.; Zhao, J. IL-37 Ameliorates Renal Fibrosis by Restoring CPT1A-Mediated Fatty Acid Oxidation in Diabetic Kidney Disease. Kidney Dis. 2023, 9, 104–117. [Google Scholar] [CrossRef] [PubMed]
- Afshinnia, F.; Rajendiran, T.M.; Soni, T.; Byun, J.; Wernisch, S.; Sas, K.M.; Hawkins, J.; Bellovich, K.; Gipson, D.; Michailidis, G.; et al. Impaired beta-Oxidation and Altered Complex Lipid Fatty Acid Partitioning with Advancing CKD. J. Am. Soc. Nephrol. 2018, 29, 295–306. [Google Scholar] [CrossRef] [PubMed]
- Baek, J.; He, C.; Afshinnia, F.; Michailidis, G.; Pennathur, S. Lipidomic approaches to dissect dysregulated lipid metabolism in kidney disease. Nat. Rev. Nephrol. 2022, 18, 38–55. [Google Scholar] [CrossRef] [PubMed]
- Piret, S.E.; Guo, Y.; Attallah, A.A.; Horne, S.J.; Zollman, A.; Owusu, D.; Henein, J.; Sidorenko, V.S.; Revelo, M.P.; Hato, T.; et al. Kruppel-like factor 6-mediated loss of BCAA catabolism contributes to kidney injury in mice and humans. Proc. Natl. Acad. Sci. USA 2021, 118, e2024414118. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Guo, X.; Cui, S.; Wu, Y.; Zhang, Y.; Shen, X.; Xie, C.; Li, J. Dephosphorylation of AMP-activated protein kinase exacerbates ischemia/reperfusion-induced acute kidney injury via mitochondrial dysfunction. Kidney Int. 2022, 101, 315–330. [Google Scholar] [CrossRef]
- Peterson, J.M.; Aja, S.; Wei, Z.; Wong, G.W. CTRP1 protein enhances fatty acid oxidation via AMP-activated protein kinase (AMPK) activation and acetyl-CoA carboxylase (ACC) inhibition. J. Biol. Chem. 2012, 287, 1576–1587. [Google Scholar] [CrossRef]
- Aranda-Rivera, A.K.; Cruz-Gregorio, A.; Aparicio-Trejo, O.E.; Pedraza-Chaverri, J. Mitochondrial Redox Signaling and Oxidative Stress in Kidney Diseases. Biomolecules 2021, 11, 1144. [Google Scholar] [CrossRef]
- Jiang, M.; Bai, M.; Lei, J.; Xie, Y.; Xu, S.; Jia, Z.; Zhang, A. Mitochondrial dysfunction and the AKI-to-CKD transition. Am. J. Physiol. Renal Physiol. 2020, 319, F1105–F1116. [Google Scholar] [CrossRef]
- Chiba, T.; Peasley, K.D.; Cargill, K.R.; Maringer, K.V.; Bharathi, S.S.; Mukherjee, E.; Zhang, Y.; Holtz, A.; Basisty, N.; Yagobian, S.D.; et al. Sirtuin 5 Regulates Proximal Tubule Fatty Acid Oxidation to Protect against AKI. J. Am. Soc. Nephrol. 2019, 30, 2384–2398. [Google Scholar] [CrossRef]
- Harley, G.; Katerelos, M.; Gleich, K.; de Souza, D.P.; Narayana, V.K.; Kemp, B.E.; Power, D.A.; Mount, P.F. Blocking AMPK signalling to acetyl-CoA carboxylase increases cisplatin-induced acute kidney injury and suppresses the benefit of metformin. Biomed. Pharmacother. 2022, 153, 113377. [Google Scholar] [CrossRef]
- Iwaki, T.; Bennion, B.G.; Stenson, E.K.; Lynn, J.C.; Otinga, C.; Djukovic, D.; Raftery, D.; Fei, L.; Wong, H.R.; Liles, W.C.; et al. PPARalpha contributes to protection against metabolic and inflammatory derangements associated with acute kidney injury in experimental sepsis. Physiol. Rep. 2019, 7, e14078. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.; Sun, M.; Wu, J.; Fang, H.; Cai, S.; An, S.; Huang, Q.; Chen, Z.; Wu, C.; Zhou, Z.; et al. SIRT1 attenuates sepsis-induced acute kidney injury via Beclin1 deacetylation-mediated autophagy activation. Cell Death Dis. 2021, 12, 217. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Zhang, L.; Chen, R.; Lu, H.; Sui, M.; Zhu, Y.; Zeng, L. SIRT3 Protects Against Acute Kidney Injury via AMPK/mTOR-Regulated Autophagy. Front. Physiol. 2018, 9, 1526. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Peng, Y.; Li, Y.; Zhou, R.; Chen, D.; Jin, W.; Xu, Q.; Xu, L.; Luo, Z.; Yang, H. Glucose metabolism pattern of peripheral blood immune cells in myasthenia gravis patients. Ann. Transl. Med. 2020, 8, 577. [Google Scholar] [CrossRef]
- Cao, F.; Li, Y.; Peng, T.; Li, Y.; Yang, L.; Hu, L.; Zhang, H.; Wang, J. PTEN in kidney diseases: A potential therapeutic target in preventing AKI-to-CKD transition. Front. Med. (Lausanne) 2024, 11, 1428995. [Google Scholar] [CrossRef]
- Kang, H.M.; Ahn, S.H.; Choi, P.; Ko, Y.A.; Han, S.H.; Chinga, F.; Park, A.S.; Tao, J.; Sharma, K.; Pullman, J.; et al. Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat. Med. 2015, 21, 37–46. [Google Scholar] [CrossRef]
- Scholz, H.; Boivin, F.J.; Schmidt-Ott, K.M.; Bachmann, S.; Eckardt, K.U.; Scholl, U.I.; Persson, P.B. Kidney physiology and susceptibility to acute kidney injury: Implications for renoprotection. Nat. Rev. Nephrol. 2021, 17, 335–349. [Google Scholar] [CrossRef]
- Lan, R.; Geng, H.; Singha, P.K.; Saikumar, P.; Bottinger, E.P.; Weinberg, J.M.; Venkatachalam, M.A. Mitochondrial Pathology and Glycolytic Shift during Proximal Tubule Atrophy after Ischemic AKI. J. Am. Soc. Nephrol. 2016, 27, 3356–3367. [Google Scholar] [CrossRef]
- Coca, S.G.; Singanamala, S.; Parikh, C.R. Chronic kidney disease after acute kidney injury: A systematic review and meta-analysis. Kidney Int. 2012, 81, 442–448. [Google Scholar] [CrossRef]
- Webster, A.C.; Nagler, E.V.; Morton, R.L.; Masson, P. Chronic Kidney Disease. Lancet 2017, 389, 1238–1252. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, C. From AKI to CKD: Maladaptive Repair and the Underlying Mechanisms. Int. J. Mol. Sci. 2022, 23, 10880. [Google Scholar] [CrossRef] [PubMed]
- Nishi, H.; Higashihara, T.; Inagi, R. Lipotoxicity in Kidney, Heart, and Skeletal Muscle Dysfunction. Nutrients 2019, 11, 1664. [Google Scholar] [CrossRef] [PubMed]
- Visconti, L.; Benvenga, S.; Lacquaniti, A.; Cernaro, V.; Bruzzese, A.; Conti, G.; Buemi, M.; Santoro, D. Lipid disorders in patients with renal failure: Role in cardiovascular events and progression of chronic kidney disease. J. Clin. Transl. Endocrinol. 2016, 6, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Gai, Z.; Wang, T.; Visentin, M.; Kullak-Ublick, G.A.; Fu, X.; Wang, Z. Lipid Accumulation and Chronic Kidney Disease. Nutrients 2019, 11, 722. [Google Scholar] [CrossRef]
- Cuevas-Delgado, P.; Miguel, V.; Ruperez, F.J.; Lamas, S.; Barbas, C. Impact of renal tubular Cpt1a overexpression on the kidney metabolome in the folic acid-induced fibrosis mouse model. Front. Mol. Biosci. 2023, 10, 1161036. [Google Scholar] [CrossRef]
- Fontecha-Barriuso, M.; Martin-Sanchez, D.; Martinez-Moreno, J.M.; Monsalve, M.; Ramos, A.M.; Sanchez-Nino, M.D.; Ruiz-Ortega, M.; Ortiz, A.; Sanz, A.B. The Role of PGC-1alpha and Mitochondrial Biogenesis in Kidney Diseases. Biomolecules 2020, 10, 347. [Google Scholar] [CrossRef]
- Cheng, C.F.; Ku, H.C.; Lin, H. PGC-1alpha as a Pivotal Factor in Lipid and Metabolic Regulation. Int. J. Mol. Sci. 2018, 19, 3447. [Google Scholar] [CrossRef]
- Li, L.; Long, J.; Mise, K.; Galvan, D.L.; Overbeek, P.A.; Tan, L.; Kumar, S.V.; Chan, W.K.; Lorenzi, P.L.; Chang, B.H.; et al. PGC1alpha is required for the renoprotective effect of lncRNA Tug1 in vivo and links Tug1 with urea cycle metabolites. Cell Rep. 2021, 36, 109510. [Google Scholar] [CrossRef]
- Han, S.S.; Yu, M.Y.; Yoo, K.D.; Lee, J.P.; Kim, D.K.; Kim, Y.S.; Yang, S.H. Loss of KLF15 accelerates chronic podocyte injury. Int. J. Mol. Med. 2018, 42, 1593–1602. [Google Scholar] [CrossRef]
- Chen, L.; Sha, M.L.; Chen, F.T.; Jiang, C.Y.; Li, D.; Xu, C.L.; Pan, D.S.; Xu, Z.J.; Tang, Q.L.; Xia, S.J.; et al. Upregulation of KLF14 expression attenuates kidney fibrosis by inducing PPARalpha-mediated fatty acid oxidation. Free Radic. Biol. Med. 2023, 195, 132–144. [Google Scholar] [CrossRef]
- Yang, X.; Okamura, D.M.; Lu, X.; Chen, Y.; Moorhead, J.; Varghese, Z.; Ruan, X.Z. CD36 in chronic kidney disease: Novel insights and therapeutic opportunities. Nat. Rev. Nephrol. 2017, 13, 769–781. [Google Scholar] [CrossRef] [PubMed]
- Coburn, C.T.; Hajri, T.; Ibrahimi, A.; Abumrad, N.A. Role of CD36 in membrane transport and utilization of long-chain fatty acids by different tissues. J. Mol. Neurosci. 2001, 16, 117–121; discussion 151–117. [Google Scholar] [CrossRef] [PubMed]
- Luangrath, V.; Brodeur, M.R.; Rhainds, D.; Brissette, L. Mouse CD36 has opposite effects on LDL and oxidized LDL metabolism in vivo. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1290–1295. [Google Scholar] [CrossRef] [PubMed]
- van Herpen, N.A.; Schrauwen-Hinderling, V.B. Lipid accumulation in non-adipose tissue and lipotoxicity. Physiol. Behav. 2008, 94, 231–241. [Google Scholar] [CrossRef]
- Li, L.O.; Klett, E.L.; Coleman, R.A. Acyl-CoA synthesis, lipid metabolism and lipotoxicity. Biochim. Biophys. Acta 2010, 1801, 246–251. [Google Scholar] [CrossRef]
- Devarajan, P. Update on mechanisms of ischemic acute kidney injury. J. Am. Soc. Nephrol. 2006, 17, 1503–1520. [Google Scholar] [CrossRef]
- Chen, D.Q.; Chen, H.; Chen, L.; Vaziri, N.D.; Wang, M.; Li, X.R.; Zhao, Y.Y. The link between phenotype and fatty acid metabolism in advanced chronic kidney disease. Nephrol. Dial. Transpl. 2017, 32, 1154–1166. [Google Scholar] [CrossRef]
- Wei, X.; Hou, Y.; Long, M.; Jiang, L.; Du, Y. Advances in energy metabolism in renal fibrosis. Life Sci. 2023, 312, 121033. [Google Scholar] [CrossRef]
- Liu, S.; Gui, Y.; Wang, M.S.; Zhang, L.; Xu, T.; Pan, Y.; Zhang, K.; Yu, Y.; Xiao, L.; Qiao, Y.; et al. Serum integrative omics reveals the landscape of human diabetic kidney disease. Mol. Metab. 2021, 54, 101367. [Google Scholar] [CrossRef]
- Yang, J.; Zhang, M.; Ran, F.; Gou, X.; Ma, Y.; Wu, X. Screening for Early Biomarkers of Cisplatin-Induced Acute Kidney Injury in Rats Through Serum Metabolomics Technology. J. Coll. Physicians Surg. Pak. 2024, 34, 936–941. [Google Scholar]
- Singh, A.; Siddiqui, M.A.; Pandey, S.; Azim, A.; Sinha, N. Unveiling Pathophysiological Insights: Serum Metabolic Dysregulation in Acute Respiratory Distress Syndrome Patients with Acute Kidney Injury. J. Proteome Res. 2024, 23, 4216–4228. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Gordaliza, E.; Gonzalez-Nicolas, M.A.; Lazaro, A.; Barbas, C.; Gomez-Gomez, M.M.; Lopez-Gonzalvez, A. Untargeted metabolomics analysis of serum and urine unveils the protective effect of cilastatin on altered metabolic pathways during cisplatin-induced acute kidney injury. Biochem. Pharmacol. 2024, 227, 116435. [Google Scholar] [CrossRef] [PubMed]
- Wee, H.N.; Liu, J.J.; Ching, J.; Kovalik, J.P.; Lim, S.C. The Kynurenine Pathway in Acute Kidney Injury and Chronic Kidney Disease. Am. J. Nephrol. 2021, 52, 771–787. [Google Scholar] [CrossRef] [PubMed]
- Brinkley, G.; Nam, H.; Shim, E.; Kirkman, R.; Kundu, A.; Karki, S.; Heidarian, Y.; Tennessen, J.M.; Liu, J.; Locasale, J.W.; et al. Teleological role of L-2-hydroxyglutarate dehydrogenase in the kidney. Dis. Model Mech. 2020, 13, dmm045898. [Google Scholar] [CrossRef] [PubMed]
- Dalamaga, M. Clinical metabolomics: Useful insights, perspectives and challenges. Metabol. Open 2024, 22, 100290. [Google Scholar] [CrossRef]
- Gao, P.; Huang, X.; Fang, X.Y.; Zheng, H.; Cai, S.L.; Sun, A.J.; Zhao, L.; Zhang, Y. Application of metabolomics in clinical and laboratory gastrointestinal oncology. World J. Gastrointest. Oncol. 2021, 13, 536–549. [Google Scholar] [CrossRef]
- Schmidt, D.R.; Patel, R.; Kirsch, D.G.; Lewis, C.A.; Vander Heiden, M.G.; Locasale, J.W. Metabolomics in cancer research and emerging applications in clinical oncology. CA Cancer J. Clin. 2021, 71, 333–358. [Google Scholar] [CrossRef]
- Di Minno, A.; Gelzo, M.; Caterino, M.; Costanzo, M.; Ruoppolo, M.; Castaldo, G. Challenges in Metabolomics-Based Tests, Biomarkers Revealed by Metabolomic Analysis, and the Promise of the Application of Metabolomics in Precision Medicine. Int. J. Mol. Sci. 2022, 23, 5213. [Google Scholar] [CrossRef]
- Samanta, D.; Semenza, G.L. Metabolic adaptation of cancer and immune cells mediated by hypoxia-inducible factors. Biochim. Biophys. Acta Rev. Cancer 2018, 1870, 15–22. [Google Scholar] [CrossRef]
- Gomez, H.; Kellum, J.A.; Ronco, C. Metabolic reprogramming and tolerance during sepsis-induced AKI. Nat. Rev. Nephrol. 2017, 13, 143–151. [Google Scholar] [CrossRef]
- Ghezzi, C.; Loo, D.D.F.; Wright, E.M. Physiology of renal glucose handling via SGLT1, SGLT2 and GLUT2. Diabetologia 2018, 61, 2087–2097. [Google Scholar] [CrossRef] [PubMed]
- Palmer, B.F.; Clegg, D.J. Kidney-Protective Effects of SGLT2 Inhibitors. Clin. J. Am. Soc. Nephrol. 2023, 18, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Cai, T.; Ke, Q.; Fang, Y.; Wen, P.; Chen, H.; Yuan, Q.; Luo, J.; Zhang, Y.; Sun, Q.; Lv, Y.; et al. Sodium-glucose cotransporter 2 inhibition suppresses HIF-1alpha-mediated metabolic switch from lipid oxidation to glycolysis in kidney tubule cells of diabetic mice. Cell Death Dis. 2020, 11, 390. [Google Scholar] [CrossRef] [PubMed]
- Juszczak, F.; Vlassembrouck, M.; Botton, O.; Zwakhals, T.; Decarnoncle, M.; Tassin, A.; Caron, N.; Decleves, A.E. Delayed Exercise Training Improves Obesity-Induced Chronic Kidney Disease by Activating AMPK Pathway in High-Fat Diet-Fed Mice. Int. J. Mol. Sci. 2020, 22, 350. [Google Scholar] [CrossRef] [PubMed]
- Li, X.J.; Suo, P.; Wang, Y.N.; Zou, L.; Nie, X.L.; Zhao, Y.Y.; Miao, H. Arachidonic acid metabolism as a therapeutic target in AKI-to-CKD transition. Front. Pharmacol. 2024, 15, 1365802. [Google Scholar] [CrossRef]
- Yang, Z.; Wang, X.; He, Y.; Qi, L.; Yu, L.; Xue, B.; Shi, H. The full capacity of AICAR to reduce obesity-induced inflammation and insulin resistance requires myeloid SIRT1. PLoS ONE 2012, 7, e49935. [Google Scholar] [CrossRef]
- Jeon, S.M. Regulation and function of AMPK in physiology and diseases. Exp. Mol. Med. 2016, 48, e245. [Google Scholar] [CrossRef]
- Szeto, H.H.; Schiller, P.W. Novel therapies targeting inner mitochondrial membrane--from discovery to clinical development. Pharm. Res. 2011, 28, 2669–2679. [Google Scholar] [CrossRef]
- Szeto, H.H.; Liu, S.; Soong, Y.; Wu, D.; Darrah, S.F.; Cheng, F.Y.; Zhao, Z.; Ganger, M.; Tow, C.Y.; Seshan, S.V. Mitochondria-targeted peptide accelerates ATP recovery and reduces ischemic kidney injury. J. Am. Soc. Nephrol. 2011, 22, 1041–1052. [Google Scholar] [CrossRef]
- Xu, S.; Gao, Y.; Zhang, Q.; Wei, S.; Chen, Z.; Dai, X.; Zeng, Z.; Zhao, K.S. SIRT1/3 Activation by Resveratrol Attenuates Acute Kidney Injury in a Septic Rat Model. Oxid Med. Cell Longev. 2016, 2016, 7296092. [Google Scholar] [CrossRef]
- Virmani, M.A.; Cirulli, M. The Role of l-Carnitine in Mitochondria, Prevention of Metabolic Inflexibility and Disease Initiation. Int. J. Mol. Sci. 2022, 23, 2717. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Fu, Z.; Wang, C.; Xu, J.; Ma, H.; Jiang, M.; Xu, T.; Feng, X.; Zhang, W. ZLN005 protects against ischemia-reperfusion-induced kidney injury by mitigating oxidative stress through the restoration of mitochondrial fatty acid oxidation. Am. J. Transl. Res. 2021, 13, 10014–10037. [Google Scholar] [PubMed]
- Ventura-Clapier, R.; Garnier, A.; Veksler, V. Transcriptional control of mitochondrial biogenesis: The central role of PGC-1alpha. Cardiovasc. Res. 2008, 79, 208–217. [Google Scholar] [CrossRef] [PubMed]
- Clark, A.J.; Parikh, S.M. Targeting energy pathways in kidney disease: The roles of sirtuins, AMPK, and PGC1alpha. Kidney Int. 2021, 99, 828–840. [Google Scholar] [CrossRef] [PubMed]
- Mitrofanova, A.; Merscher, S.; Fornoni, A. Kidney lipid dysmetabolism and lipid droplet accumulation in chronic kidney disease. Nat. Rev. Nephrol. 2023, 19, 629–645. [Google Scholar] [CrossRef]
- Xiao, Q.; Yu, X.; Yu, X.; Liu, S.; Jiang, J.; Cheng, Y.; Lin, H.; Wang, Y.; Zhang, X.; Ye, X.; et al. An integrated network pharmacology and cell metabolomics approach to reveal the role of rhein, a novel PPARalpha agonist, against renal fibrosis by activating the PPARalpha-CPT1A axis. Phytomedicine 2022, 102, 154147. [Google Scholar] [CrossRef]
- Li, S.; Basnakian, A.; Bhatt, R.; Megyesi, J.; Gokden, N.; Shah, S.V.; Portilla, D. PPAR-alpha ligand ameliorates acute renal failure by reducing cisplatin-induced increased expression of renal endonuclease G. Am. J. Physiol. Renal. Physiol. 2004, 287, F990–F998. [Google Scholar] [CrossRef]
- Hou, X.; Shen, Y.H.; Li, C.; Wang, F.; Zhang, C.; Bu, P.; Zhang, Y. PPARalpha agonist fenofibrate protects the kidney from hypertensive injury in spontaneously hypertensive rats via inhibition of oxidative stress and MAPK activity. Biochem. Biophys. Res. Commun. 2010, 394, 653–659. [Google Scholar] [CrossRef]
- Heerspink, H.J.L.; Stefansson, B.V.; Correa-Rotter, R.; Chertow, G.M.; Greene, T.; Hou, F.F.; Mann, J.F.E.; McMurray, J.J.V.; Lindberg, M.; Rossing, P.; et al. Dapagliflozin in Patients with Chronic Kidney Disease. N. Engl. J. Med. 2020, 383, 1436–1446. [Google Scholar] [CrossRef]
- Packer, M.; Anker, S.D.; Butler, J.; Filippatos, G.; Pocock, S.J.; Carson, P.; Januzzi, J.; Verma, S.; Tsutsui, H.; Brueckmann, M.; et al. Cardiovascular and Renal Outcomes with Empagliflozin in Heart Failure. N. Engl. J. Med. 2020, 383, 1413–1424. [Google Scholar] [CrossRef]
- El-Damanawi, R.; Stanley, I.K.; Staatz, C.; Pascoe, E.M.; Craig, J.C.; Johnson, D.W.; Mallett, A.J.; Hawley, C.M.; Milanzi, E.; Hiemstra, T.F.; et al. Metformin for preventing the progression of chronic kidney disease. Cochrane Database Syst. Rev. 2024, 6, CD013414. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Chen, H.; Zhou, D.; Zhang, J.; Chen, Y.; Liu, Q.; Ai, D.; Zhu, H.; Chu, L.; Ren, W.; et al. Comparative genomic analysis of esophageal squamous cell carcinoma between Asian and Caucasian patient populations. Nat. Commun. 2017, 8, 1533. [Google Scholar] [CrossRef] [PubMed]
- Lalau, J.D.; Kajbaf, F.; Bennis, Y.; Hurtel-Lemaire, A.S.; Belpaire, F.; De Broe, M.E. Metformin Treatment in Patients With Type 2 Diabetes and Chronic Kidney Disease Stages 3A, 3B, or 4. Diabetes Care 2018, 41, 547–553. [Google Scholar] [CrossRef] [PubMed]
- Corchia, A.; Wynckel, A.; Journet, J.; Moussi Frances, J.; Skandrani, N.; Lautrette, A.; Zafrani, L.; Lewandowski, E.; Reboul, P.; Vrigneaud, L.; et al. Metformin-related lactic acidosis with acute kidney injury: Results of a French observational multicenter study. Clin. Toxicol. 2020, 58, 375–382. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gupta, P.; Zhu, S.; Gui, Y.; Zhou, D. Metabolic Chaos in Kidney Disease: Unraveling Energy Dysregulation. J. Clin. Med. 2024, 13, 6772. https://doi.org/10.3390/jcm13226772
Gupta P, Zhu S, Gui Y, Zhou D. Metabolic Chaos in Kidney Disease: Unraveling Energy Dysregulation. Journal of Clinical Medicine. 2024; 13(22):6772. https://doi.org/10.3390/jcm13226772
Chicago/Turabian StyleGupta, Priya, Saiya Zhu, Yuan Gui, and Dong Zhou. 2024. "Metabolic Chaos in Kidney Disease: Unraveling Energy Dysregulation" Journal of Clinical Medicine 13, no. 22: 6772. https://doi.org/10.3390/jcm13226772
APA StyleGupta, P., Zhu, S., Gui, Y., & Zhou, D. (2024). Metabolic Chaos in Kidney Disease: Unraveling Energy Dysregulation. Journal of Clinical Medicine, 13(22), 6772. https://doi.org/10.3390/jcm13226772