
Pathogenesis and Management Strategies in Radioiodine-Refractory Differentiated Thyroid Cancer: From Molecular Mechanisms Toward Therapeutic Approaches: A Comprehensive Review

 ,
,
Abstract
:1. Introduction
Objective
2. Materials and Methods
3. Pathogenesis of RAIR-DTC
3.1. Molecular Genetic Characterisation
3.1.1. BRAF Pathogenic Variant and Rearrangement
3.1.2. NTRK Gene Fusion
3.1.3. TERT Promoter Mutation
3.1.4. RAS Mutation
3.1.5. ALK Gene Mutation and Fusion
3.1.6. RET Rearrangement
3.1.7. PAX8/PPARγ
3.1.8. SWI/SNF Complex Alteration
3.2. Regulation of Signaling Pathways
3.2.1. TSHR Pathway Activation
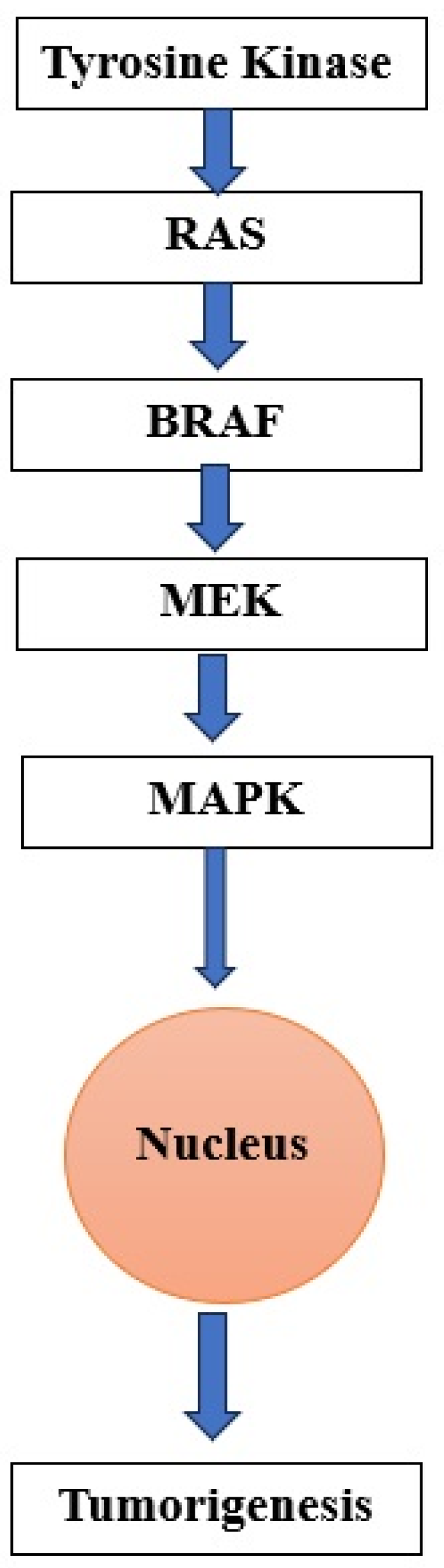
3.2.2. MAPK Pathway
3.2.3. PI3K Cascade
3.2.4. TGF-β Pathway
3.2.5. Wnt/β-Catenin Pathway
3.2.6. Notch-Related Pathway
3.3. Modulation of microRNAs
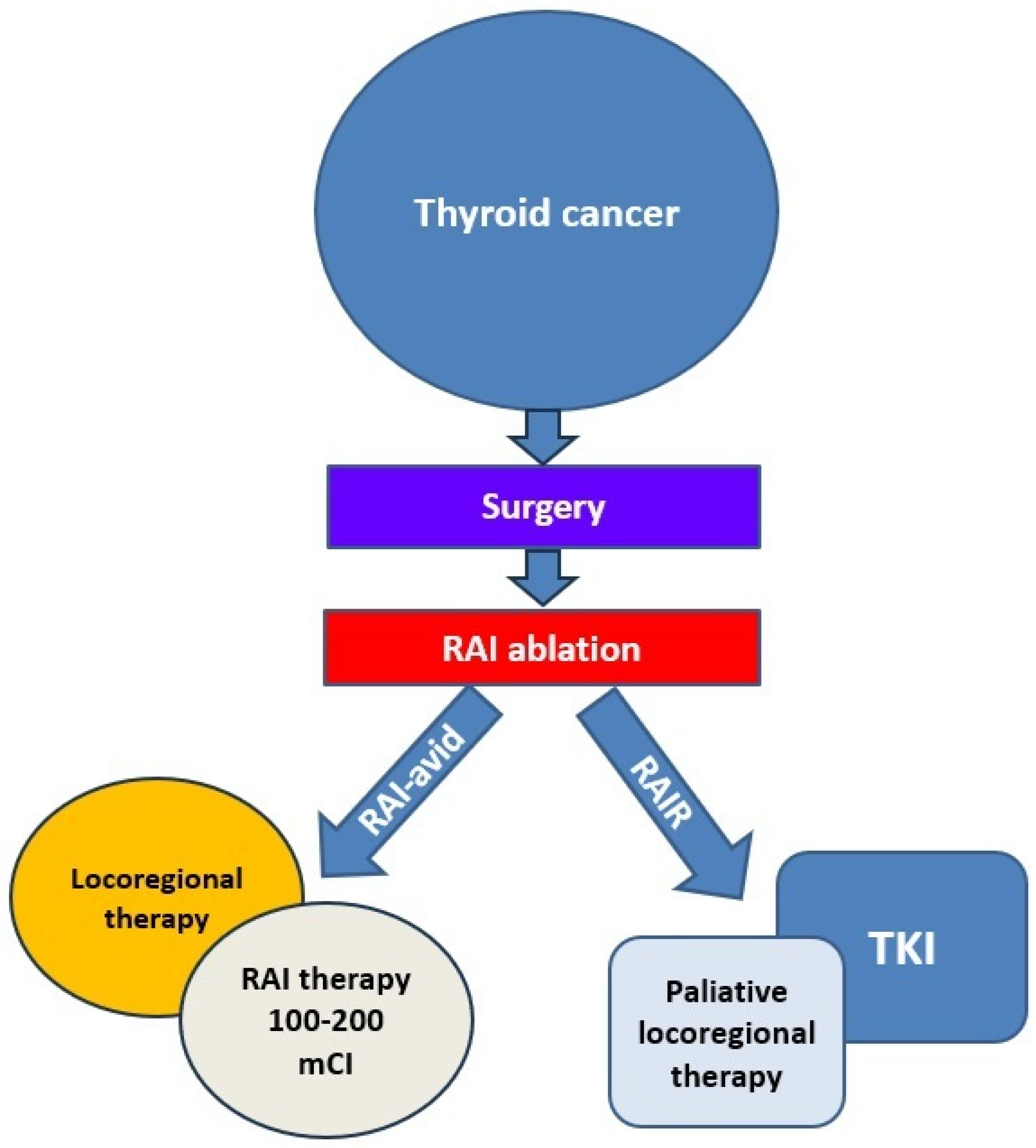
4. Management of RAIR-DTC
4.1. Monitoring
4.2. Local Treatments
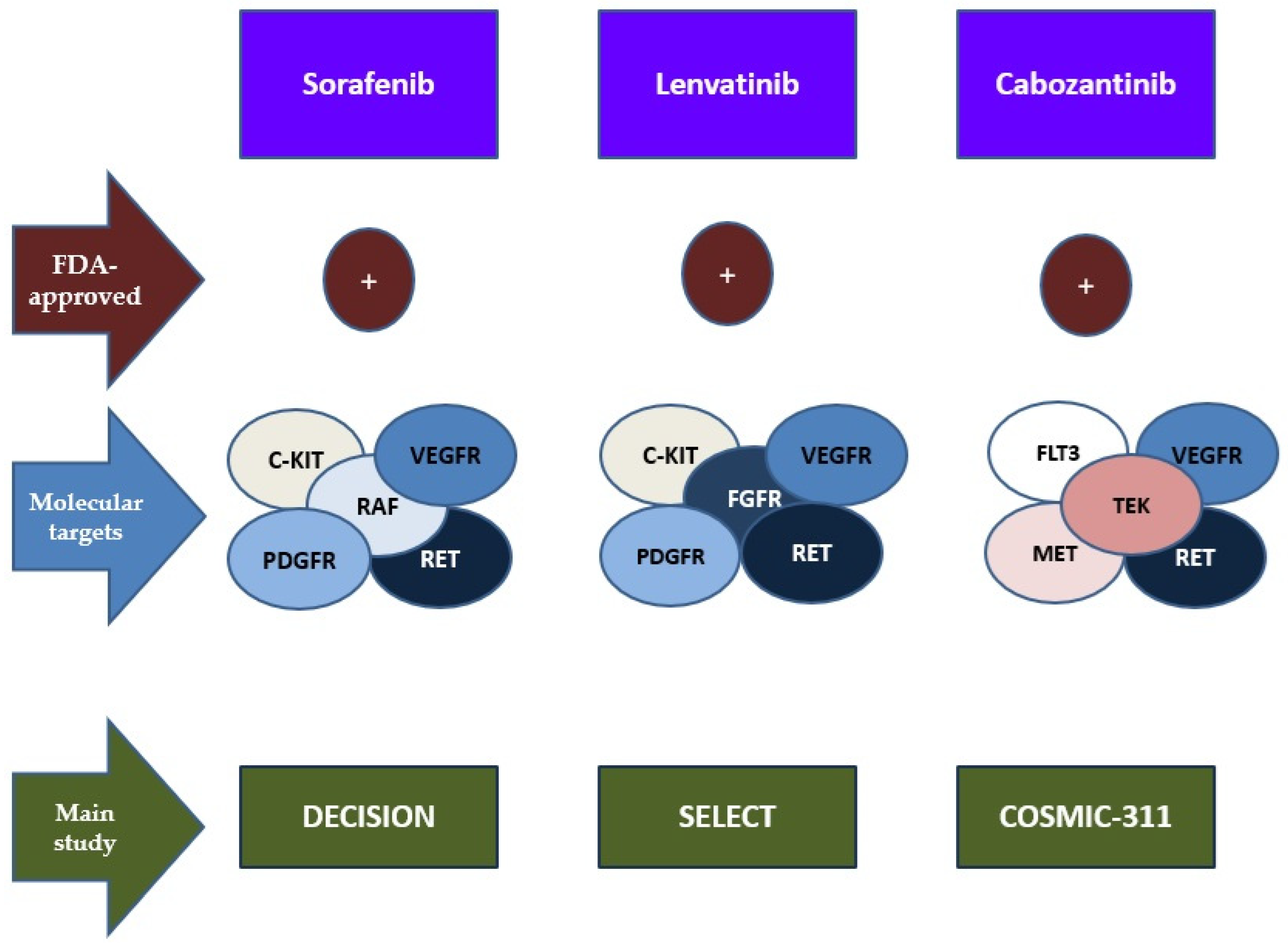
4.3. TKIs as Targeted Therapies

| Randomized Control Trial | Drug | Molecular Targets | Phase | Results: PFS | ORR |
|---|---|---|---|---|---|
| DECISION [123] | Sorafenib | VEGFR, PDGFR, c-KIT, RET, RAF | III | from 10.8 months to 5.8 months (placebo) | 12.2% (vs. 0.5%) |
| SELECT [112] | Lenvatinib | VEGFR, PDGFR, c-KIT, RET, FGFR | III | 18.3 months vs. 3.6 months (placebo) | 64.8% (vs. 1.5) |
| COSMIC-311 [121] | Cabozantinib | VEGFR, RET, c-MET, FLT3, TEK | III | 11.0 months vs. 1.9 months placebo | 15% |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
4.4. Redifferentiation Therapy
4.5. Immunotherapy
5. Conclusions and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| TC | Thyroid cancer |
| DTC | Differentiated thyroid cancer |
| RAI | Radioactive iodine |
| TSH | Thyroid-stimulating hormone |
| MAPK | Mitogen-activated protein kinase |
| PI3K/mTOR/Akt | Photoshatidyl-inositol-3-kinase/mammalian target of rapamycin/protein kinase B |
| BRAF | V-Raf mouse sarcoma virus oncogene homologous B1 |
| TGF-β | Transforming growth factor-β |
| NIS | Sodium iodine symporter |
| PTEN | Phosphatase and tensin homolog |
| ATC | Anaplastic thyroid cancer |
| TERT | Telomerase reverse transcriptase |
| RAIR-DTC | Radioiodine-refractory differentiated thyroid cancer |
| SLC5A5 | Solute carrier family 5A |
| cAMP | Cyclic adenosine monophosphate |
| NUE | NIS upstream enhances |
| PKA | Protein kinase A |
| PAX8 | Paired box gene-8 |
| Ref-1 | Redox effector factor-1 |
| CREM | aAMP-response element modulator |
| MAPKKK | Mitogen activated protein kinase kinase kinase |
| V600E | B-raf protein residue 600 from glutamic acid to valine |
| AHR | Aromatic hydrocarbon receptor |
| WT1 | Wilm tumor gene 1 |
| NTRK | Neurotrophic receptor tyrosine kinase |
| TRK | Tropomyosin receptor kinase |
| TERTp | TERT promoter |
| ALK | Anaplastic lymphoma kinase |
| CRKL-C3G | Adaptor protein-Rap guanine nucleotide exchange factor 1 |
| MEKK2/3-MEK5-ERK5 | Mitogen-activated protein kinase kinase kinase 2/3-Mitogen-activated protein kinase kinase 5-extracellular signal regulated kinase 5 |
| JAK-STAT | Janus linase-signal transducer and activator of transcription |
| STRN | Recurrent striatal protein |
| NGS | Next-generation sequencing |
| TKR | Tyrosine kinase membrane receptor |
| GDNK | Glial cell line-derived neurotrophic factor |
| GFL | GDNF family ligand |
| DSBs | DNA double-strand breaks |
| PPAR-γ | Peroxisome proliferator-activated receptor gamma |
| PPFP | PAX8-PPAR-γ fusion protein |
| TPO | Thyroid peroxidase |
| TG | Thyroglobulin |
| TSHR | Thyroid-stimulating hormone receptor |
| SWI/SNF | SWItch/sucrose nonfermentable |
| BAF | BRG1/BRM related factor |
| PBAF | Polybromine-related factor |
| ncBAf | Atypical BAF |
| TF | Transcription factors |
| GPCR | G protein-coupled receptor |
| PD-L1 | Tumor programmed death-ligand 1 |
| ERK | Extracellular-signal-regulated kinase |
| JNK/SAPK | Jun kinase |
| MEK | Mitogen-activated protein kinase/extracellular signal-regulated kinase kinase |
| IGF-2 | Insulin-like growth factor 2 |
| IR-A | Insulin receptor subtype A |
| IIGFs | Insulin/insulin-like growth factor systems |
| DDRs | Discoid domain receptors |
| RUNX2 | Runt-related transcription factor 2 |
| mTORC2 | mTOR complex 2 |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
| NOX4 | NADPH oxidase 4 |
| ROS | Reactive oxygen species |
| TCF | T cell factor |
| CSCs | Cancer stem cells |
| LSD1 | Lysine-specific histone demethylase 1 A |
| APC2 | Adenomatous polyposis coli 2 |
| DKK1 | Dickkopf-related protein 1 |
| Notch1-4 | Notch receptors |
| miRNAs | MicroRNAs |
| 3′-UTR | 3′-untranslated region |
| fT4 | Free thyroxine |
| TKIs | Tyrosine kinase inhibitors |
| EBRT | External-beam radiation therapy |
| TACE | Trans-arterial chemoembolization |
| VEGFR1/2/3 | Vascular endothelial growth factor receptor 1, 2, 3 |
| c-KIT | Cellular kit |
| PDGFR | Platelet-derived growth factor receptor |
| FLT | Fms-like tyrosine kinase |
| FDA | Food and Drug Administration |
| PR | Partial response |
| PFS | Progression-free survival |
| OS | Overall survival |
| FGFR | Fibroblast growth factor receptor |
| c-MET | Mesenchymal–epithelial transition factor |
| MTC | Medullary thyroid carcinoma |
| EGFR | Epidermal growth factor receptor |
| ORR | Overall response rate |
| RAF | Rapidly accelerated fibrosarcoma |
| MKIs | Multi-kinase inhibitors |
| CTLA-4 | Cytotoxic T-lymphocyte antigen 4 |
References
- Fugazzola, L.; Elisei, R.; Fuhrer, D.; Jarzab, B.; Leboulleux, S.; Newbold, K.; Smit, J. 2019 European Thyroid Association Guidelines for the Treatment and Follow-Up of Advanced Radioiodine-Refractory Thyroid Cancer. Eur. Thyroid J. 2019, 8, 227–245. [Google Scholar] [CrossRef] [PubMed]
- Shobab, L.; Gomes-Lima, C.; Zeymo, A.; Feldman, R.; Jonklaas, J.; Wartofsky, L.; Burman, K.D. Clinical, Pathological, and Molecular Profiling of Radioactive Iodine Refractory Differentiated Thyroid Cancer. Thyroid 2019, 29, 1262–1268. [Google Scholar] [CrossRef] [PubMed]
- Durante, C.; Haddy, N.; Baudin, E.; Leboulleux, S.; Hartl, D.; Travagli, J.P.; Caillou, B.; Ricard, M.; Lumbroso, J.D.; De Vathaire, F.; et al. Long-Term Outcome of 444 Patients with Distant Metastases from Papillary and Follicular Thyroid Carcinoma: Benefits and Limits of Radioiodine Therapy. J. Clin. Endocrinol. Metab. 2006, 91, 2892–2899. [Google Scholar] [CrossRef] [PubMed]
- Nervo, A.; Retta, F.; Ragni, A.; Piovesan, A.; Gallo, M.; Arvat, E. Management of Progressive Radioiodine-Refractory Thyroid Carcinoma: Current Perspective. Cancer Manag. Res. 2022, 14, 3047–3062. [Google Scholar] [CrossRef] [PubMed]
- Saïe, C.; Wassermann, J.; Mathy, E.; Chereau, N.; Leenhardt, L.; Tezenas du Montcel, S.; Buffet, C. Impact of age on survival in radioiodine refractory differentiated thyroid cancer patients. Eur. J. Endocrinol. 2021, 184, 667–676. [Google Scholar] [CrossRef]
- Brenner, H. Long-term survival rates of cancer patients achieved by the end of the 20th century: A period analysis. Lancet 2002, 360, 1131–1135. [Google Scholar] [CrossRef]
- Cooper, D.S.; Doherty, G.M.; Haugen, B.R.; Kloos, R.T.; Lee, S.L.; Mandel, S.J.; Mazzaferri, E.L.; McIver, B.; Pacini, F.; Schlumberger, M.; et al. Revised American Thyroid Association Management Guidelines for Patients with Thyroid Nodules and Differentiated Thyroid Cancer. Thyroid 2009, 19, 1167–1214. [Google Scholar] [CrossRef]
- Saftencu, M.; Braicu, C.; Cojocneanu, R.; Buse, M.; Irimie, A.; Piciu, D.; Berindan-Neagoe, I. Gene Expression Patterns Unveil New Insights in Papillary Thyroid Cancer. Medicina 2019, 55, 500. [Google Scholar] [CrossRef]
- Filetti, S.; Durante, C.; Hartl, D.; Leboulleux, S.; Locati, L.D.; Newbold, K.; Papotti, M.G.; Berruti, A. Thyroid cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2019, 30, 1856–1883. [Google Scholar] [CrossRef]
- Mazzaferri, E.L. An Overview of the Management of Papillary and Follicular Thyroid Carcinoma. Thyroid 1999, 9, 421–427. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, N.; Akbani, R.; Aksoy, B.A.; Ally, A.; Arachchi, H.; Asa, S.L.; Auman, J.T.; Balasundaram, M.; Balu, S.; Baylin, S.B.; et al. Integrated Genomic Characterization of Papillary Thyroid Carcinoma. Cell 2014, 159, 676–690. [Google Scholar] [CrossRef]
- Kim, K.H.; Kang, D.W.; Kim, S.H.; Seong, I.O.; Kang, D.Y. Mutations of the BRAF Gene in Papillary Thyroid Carcinoma in a Korean Population. Yonsei Med. J. 2004, 45, 818–821. [Google Scholar] [CrossRef]
- Soares, P.; Trovisco, V.; Rocha, A.S.; Lima, J.; Castro, P.; Preto, A.; Máximo, V.; Botelho, T.; Seruca, R.; Sobrinho-Simões, M. BRAF mutations and RET/PTC rearrangements are alternative events in the etiopathogenesis of PTC. Oncogene 2003, 22, 4578–4580. [Google Scholar] [CrossRef] [PubMed]
- Nikiforova, M.N.; Kimura, E.T.; Gandhi, M.; Biddinger, P.W.; Knauf, J.A.; Basolo, F.; Zhu, Z.; Giannini, R.; Salvatore, G.; Fusco, A.; et al. BRAF Mutations in Thyroid Tumors Are Restricted to Papillary Carcinomas and Anaplastic or Poorly Differentiated Carcinomas Arising from Papillary Carcinomas. J. Clin. Endocrinol. Metab. 2003, 88, 5399–5404. [Google Scholar] [CrossRef] [PubMed]
- Fugazzola, L.; Mannavola, D.; Cirello, V.; Vannucchi, G.; Muzza, M.; Vicentini, L.; Beck-Peccoz, P. BRAF mutations in an Italian cohort of thyroid cancers. Clin. Endocrinol. 2004, 61, 239–243. [Google Scholar] [CrossRef] [PubMed]
- Trovisco, V.; Vieira de Castro, I.; Soares, P.; Máximo, V.; Silva, P.; Magalhães, J.; Abrosimov, A.; Guiu, X.M.; Sobrinho-Simões, M. BRAF mutations are associated with some histological types of papillary thyroid carcinoma. J. Pathol. 2004, 202, 247–251. [Google Scholar] [CrossRef]
- Cappola, A.R.; Mandel, S.J. Molecular Testing in Thyroid Cancer. JAMA 2013, 309, 1529–1530. [Google Scholar] [CrossRef]
- Biondi, B.; Filetti, S.; Schlumberger, M. Thyroid-hormone therapy and thyroid cancer: A reassessment. Nat. Clin. Pract. Endocrinol. Metab. 2005, 1, 32–40. [Google Scholar] [CrossRef]
- Kimura, E.T.; Nikiforova, M.N.; Zhu, Z.; Knauf, J.A.; Nikiforov, Y.E.; Fagin, J.A. High prevalence of BRAF mutations in thyroid cancer: Genetic evidence for constitutive activation of the RET/PTC-RAS-BRAF signaling pathway in papillary thyroid carcinoma. Cancer Res. 2003, 63, 1454–1457. [Google Scholar]
- Kim, M.; Jeon, M.J.; Oh, H.-S.; Park, S.; Kim, T.Y.; Shong, Y.K.; Kim, W.B.; Kim, K.; Kim, W.G.; Song, D.E. BRAF and RAS Mutational Status in Noninvasive Follicular Thyroid Neoplasm with Papillary-Like Nuclear Features and Invasive Subtype of Encapsulated Follicular Variant of Papillary Thyroid Carcinoma in Korea. Thyroid 2018, 28, 504–510. [Google Scholar] [CrossRef] [PubMed]
- Nikiforov, Y.E.; Seethala, R.R.; Tallini, G.; Baloch, Z.W.; Basolo, F.; Thompson, L.D.R.; Barletta, J.A.; Wenig, B.M.; Al Ghuzlan, A.; Kakudo, K.; et al. Nomenclature Revision for Encapsulated Follicular Variant of Papillary Thyroid Carcinoma. JAMA Oncol. 2016, 2, 1023–1029. [Google Scholar] [CrossRef] [PubMed]
- Prete, A.; Borges de Souza, P.; Censi, S.; Muzza, M.; Nucci, N.; Sponziello, M. Update on Fundamental Mechanisms of Thyroid Cancer. Front. Endocrinol. 2020, 11, 102. [Google Scholar] [CrossRef] [PubMed]
- Al Rasheed, M.R.H.; Xu, B. Molecular Alterations in Thyroid Carcinoma. Surg. Pathol. Clin. 2019, 12, 921–930. [Google Scholar] [CrossRef] [PubMed]
- Oikonomou, E.; Koustas, E.; Goulielmaki, M.; Pintzas, A. BRAF vs. RAS oncogenes: Are mutations of the same pathway equal? Differential signalling and therapeutic implications. Oncotarget 2014, 5, 11752–11777. [Google Scholar] [CrossRef]
- Ratajczak, M.; Gaweł, D.; Godlewska, M. Novel Inhibitor-Based Therapies for Thyroid Cancer—An Update. Int. J. Mol. Sci. 2021, 22, 11829. [Google Scholar] [CrossRef]
- Aashiq, M.; Silverman, D.A.; Na’ara, S.; Takahashi, H.; Amit, M. Radioiodine-Refractory Thyroid Cancer: Molecular Basis of Redifferentiation Therapies, Management, and Novel Therapies. Cancers 2019, 11, 1382. [Google Scholar] [CrossRef]
- Yu, Q.; Zhang, X.; Li, L.; Zhang, C.; Huang, J.; Huang, W. Molecular basis and targeted therapies for radioiodine refractory thyroid cancer. Asia Pac. J. Clin. Oncol. 2023, 19, 279–289. [Google Scholar] [CrossRef]
- Liu, J.; Liu, Y.; Lin, Y.; Liang, J. Radioactive Iodine-Refractory Differentiated Thyroid Cancer and Redifferentiation Therapy. Endocrinol. Metab. 2019, 34, 215–225. [Google Scholar] [CrossRef]
- Shen, H.; Zhu, R.; Liu, Y.; Hong, Y.; Ge, J.; Xuan, J.; Niu, W.; Yu, X.; Qin, J.J.; Li, Q. Radioiodine-refractory differentiated thyroid cancer: Molecular mechanisms and therapeutic strategies for radioiodine resistance. Drug Resist. Updates 2024, 72, 101013. [Google Scholar] [CrossRef]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Valvo, V.; Nucera, C. Coding Molecular Determinants of Thyroid Cancer Development and Progression. Endocrinol. Metab. Clin. N. Am. 2019, 48, 37–59. [Google Scholar] [CrossRef] [PubMed]
- Spitzweg, C.; Bible, K.C.; Hofbauer, L.C.; Morris, J.C. Advanced radioiodine-refractory differentiated thyroid cancer: The sodium iodide symporter and other emerging therapeutic targets. Lancet Diabetes Endocrinol. 2014, 2, 830–842. [Google Scholar] [CrossRef]
- Song, E.; Jin, M.; Jang, A.; Jeon, M.J.; Song, D.E.; Yoo, H.J.; Kim, W.B.; Shong, Y.K.; Kim, W.G. Mutation in Genes Encoding Key Functional Groups Additively Increase Mortality in Patients with BRAFV600E-Mutant Advanced Papillary Thyroid Carcinoma. Cancers 2021, 13, 5846. [Google Scholar] [CrossRef]
- Efanov, A.A.; Brenner, A.V.; Bogdanova, T.I.; Kelly, L.M.; Liu, P.; Little, M.P.; Wald, A.I.; Hatch, M.; Zurnadzy, L.Y.; Nikiforova, M.N.; et al. Investigation of the Relationship between Radiation Dose and Gene Mutations and Fusions in Post-Chernobyl Thyroid Cancer. J. Natl. Cancer Inst. 2018, 110, 371–378, Erratum in J. Natl. Cancer Inst. 2018, 110, 685. https://doi.org/10.1093/jnci/djy066. [Google Scholar] [CrossRef]
- Cocco, E.; Scaltriti, M.; Drilon, A. NTRK fusion-positive cancers and TRK inhibitor therapy. Nat. Rev. Clin. Oncol. 2018, 15, 731–747. [Google Scholar] [CrossRef]
- Prasad, M.L.; Vyas, M.; Horne, M.J.; Virk, R.K.; Morotti, R.; Liu, Z.; Tallini, G.; Nikiforova, M.N.; Christison-Lagay, E.R.; Udelsman, R.; et al. NTRK fusion oncogenes in pediatric papillary thyroid carcinoma in northeast United States. Cancer 2016, 122, 1097–1107. [Google Scholar] [CrossRef]
- Bodnar, A.G.; Ouellette, M.; Frolkis, M.; Holt, S.E.; Chiu, C.P.; Morin, G.B.; Harley, C.B.; Shay, J.W.; Lichtsteiner, S.; Wright, W.E. Extension of life-span by introduction of telomerase into normal human cells. Science 1998, 279, 349–352. [Google Scholar] [CrossRef] [PubMed]
- Hafezi, F.; Perez Bercoff, D. The Solo Play of TERT Promoter Mutations. Cells 2020, 9, 749. [Google Scholar] [CrossRef]
- Liu, Y.; Li, Z.; Tang, X.; Li, M.; Shi, F. Association between hTERT Polymorphisms and Female Papillary Thyroid Carcinoma. Recent Pat. Anticancer Drug Discov. 2019, 14, 268–279. [Google Scholar] [CrossRef]
- Yang, X.; Li, J.; Li, X.; Liang, Z.; Gao, W.; Liang, J.; Cheng, S.; Lin, Y. TERT Promoter Mutation Predicts Radioiodine-Refractory Character in Distant Metastatic Differentiated Thyroid Cancer. J. Nucl. Med. 2017, 58, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Jang, E.K.; Song, D.E.; Sim, S.Y.; Kwon, H.; Choi, Y.M.; Jeon, M.J.; Han, J.M.; Kim, W.G.; Kim, T.Y.; Shong, Y.K.; et al. NRAS codon 61 mutation is associated with distant metastasis in patients with follicular thyroid carcinoma. Thyroid 2014, 24, 1275–1281. [Google Scholar] [CrossRef]
- Zou, M.; Baitei, E.Y.; Alzahrani, A.S.; BinHumaid, F.S.; Alkhafaji, D.; Al-Rijjal, R.A.; Meyer, B.F.; Shi, Y. Concomitant RAS, RET/PTC, or BRAF mutations in advanced stage of papillary thyroid carcinoma. Thyroid 2014, 24, 1256–1266. [Google Scholar] [CrossRef] [PubMed]
- Kelly, L.M.; Barila, G.; Liu, P.; Evdokimova, V.N.; Trivedi, S.; Panebianco, F.; Gandhi, M.; Carty, S.E.; Hodak, S.P.; Luo, J.; et al. Identification of the transforming STRN-ALK fusion as a potential therapeutic target in the aggressive forms of thyroid cancer. Proc. Natl. Acad. Sci. USA 2014, 111, 4233–4238. [Google Scholar] [CrossRef]
- Lee, H.; Krishnan, V.; Wirth, L.J.; Nucera, C.; Venturina, M.; Sadow, P.M.; Mita, A.; Sacks, W. Case Report of CCDC149-ALK Fusion: A Novel Genetic Alteration and a Clinically Relevant Target in Metastatic Papillary Thyroid Carcinoma. Thyroid 2022, 32, 1580–1585. [Google Scholar] [CrossRef]
- Subbiah, V.; Yang, D.; Velcheti, V.; Drilon, A.; Meric-Bernstam, F. State-of-the-Art Strategies for Targeting RET-Dependent Cancers. J. Clin. Oncol. 2020, 38, 1209–1221. [Google Scholar] [CrossRef]
- Ishizaka, Y.; Itoh, F.; Tahira, T.; Ikeda, I.; Sugimura, T.; Tucker, J.; Fertitta, A.; Carrano, A.V.; Nagao, M. Human ret proto-oncogene mapped to chromosome 10q11.2. Oncogene 1989, 4, 1519–1521. [Google Scholar] [PubMed]
- Takahashi, M.; Buma, Y.; Iwamoto, T.; Inaguma, Y.; Ikeda, H.; Hiai, H. Cloning and expression of the ret proto-oncogene encoding a tyrosine kinase with two potential transmembrane domains. Oncogene 1988, 3, 571–578. [Google Scholar]
- Chi, X.; Michos, O.; Shakya, R.; Riccio, P.; Enomoto, H.; Licht, J.D.; Asai, N.; Takahashi, M.; Ohgami, N.; Kato, M.; et al. Ret-dependent cell rearrangements in the Wolffian duct epithelium initiate ureteric bud morphogenesis. Dev. Cell 2009, 17, 199–209. [Google Scholar] [CrossRef]
- Dacic, S.; Luvison, A.; Evdokimova, V.; Kelly, L.; Siegfried, J.M.; Villaruz, L.C.; Socinski, M.A.; Nikiforov, Y.E. RET rearrangements in lung adenocarcinoma and radiation. J. Thorac. Oncol. 2014, 9, 118–120. [Google Scholar] [CrossRef]
- Romei, C.; Ciampi, R.; Elisei, R. A comprehensive overview of the role of the RET proto-oncogene in thyroid carcinoma. Nat. Rev. Endocrinol. 2016, 12, 192–202. [Google Scholar] [CrossRef] [PubMed]
- Raman, P.; Koenig, R.J. Pax-8-PPAR-γ fusion protein in thyroid carcinoma. Nat. Rev. Endocrinol. 2014, 10, 616–623. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Hoang, L.; Ji, J.X.; Huntsman, D.G. SWI/SNF Complex Mutations in Gynecologic Cancers: Molecular Mechanisms and Models. Annu. Rev. Pathol. 2020, 15, 467–492. [Google Scholar] [CrossRef] [PubMed]
- Saqcena, M.; Leandro-Garcia, L.J.; Maag, J.L.; Tchekmedyian, V.; Krishnamoorthy, G.P.; Tamarapu, P.P.; Tiedje, V.; Reuter, V.; Knauf, J.A.; de Stanchina, E.; et al. SWI/SNF Complex Mutations Promote Thyroid Tumor Progression and Insensitivity to Redifferentiation Therapies. Cancer Discov. 2021, 11, 1158–1175. [Google Scholar] [CrossRef]
- Szkudlinski, M.W.; Fremont, V.; Ronin, C.; Weintraub, B.D. Thyroid-stimulating hormone and thyroid-stimulating hormone receptor structure-function relationships. Physiol. Rev. 2002, 82, 473–502. [Google Scholar] [CrossRef]
- Morgan, S.J.; Neumann, S.; Marcus-Samuels, B.; Gershengorn, M.C. Thyrotropin and Insulin-Like Growth Factor 1 Receptor Crosstalk Upregulates Sodium-Iodide Symporter Expression in Primary Cultures of Human Thyrocytes. Thyroid 2016, 26, 1794–1803. [Google Scholar] [CrossRef]
- Chu, Y.D.; Yeh, C.T. The Molecular Function and Clinical Role of Thyroid Stimulating Hormone Receptor in Cancer Cells. Cells 2020, 9, 1730. [Google Scholar] [CrossRef]
- Wu, Z.; Xi, Z.; Xiao, Y.; Zhao, X.; Li, J.; Feng, N.; Hu, L.; Zheng, R.; Zhang, N.; Wang, S.; et al. TSH-TSHR axis promotes tumor immune evasion. J. Immunother. Cancer 2022, 10, e004049. [Google Scholar] [CrossRef]
- Hou, P.; Bojdani, E.; Xing, M. Induction of thyroid gene expression and radioiodine uptake in thyroid cancer cells by targeting major signaling pathways. J. Clin. Endocrinol. Metab. 2010, 95, 820–828. [Google Scholar] [CrossRef]
- Zhang, W.; Liu, H.T. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002, 12, 9–18. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, D.; Murugan, A.K.; Liu, Z.; Xing, M. Histone deacetylation of NIS promoter underlies BRAF V600E-promoted NIS silencing in thyroid cancer. Endocr. Relat. Cancer 2014, 21, 161–173. [Google Scholar] [CrossRef] [PubMed]
- Nagarajah, J.; Le, M.; Knauf, J.A.; Ferrandino, G.; Montero-Conde, C.; Pillarsetty, N.; Bolaender, A.; Irwin, C.; Krishnamoorthy, G.P.; Saqcena, M.; et al. Sustained ERK inhibition maximizes responses of BrafV600E thyroid cancers to radioiodine. J. Clin. Investig. 2016, 126, 4119–4124. [Google Scholar] [CrossRef]
- Borrelli, N.; Panebianco, F.; Condello, V.; Barletta, J.A.; Kaya, C.; Yip, L.; Nikiforova, M.N.; Nikiforov, Y.E. Characterization of Activating Mutations of the MEK1 Gene in Papillary Thyroid Carcinomas. Thyroid 2019, 29, 1279–1285. [Google Scholar] [CrossRef]
- Petrulea, M.S.; Plantinga, T.S.; Smit, J.W.; Georgescu, C.E.; Netea-Maier, R.T. PI3K/Akt/mTOR: A promising therapeutic target for non-medullary thyroid carcinoma. Cancer Treat. Rev. 2015, 41, 707–713. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Dong, H.; Yang, Y.; Qian, Y.; Liu, J.; Li, Z.; Guan, H.; Chen, Z.; Li, C.; Zhang, K.; et al. Upregulation of long noncoding RNA MALAT1 in papillary thyroid cancer and its diagnostic value. Future Oncol. 2018, 14, 3015–3022. [Google Scholar] [CrossRef] [PubMed]
- Vella, V.; Nicolosi, M.L.; Cantafio, P.; Massimino, M.; Lappano, R.; Vigneri, P.; Ciuni, R.; Gangemi, P.; Morrione, A.; Malaguarnera, R.; et al. DDR1 regulates thyroid cancer cell differentiation via IGF-2/IR-A autocrine signaling loop. Endocr. Relat. Cancer 2019, 26, 197–214. [Google Scholar] [CrossRef]
- Chen, H.; Ghori-Javed, F.Y.; Rashid, H.; Adhami, M.D.; Serra, R.; Gutierrez, S.E.; Javed, A. Runx2 regulates endochondral ossification through control of chondrocyte proliferation and differentiation. J. Bone Miner. Res. 2014, 29, 2653–2665. [Google Scholar] [CrossRef]
- Sancisi, V.; Borettini, G.; Maramotti, S.; Ragazzi, M.; Tamagnini, I.; Nicoli, D.; Piana, S.; Ciarrocchi, A. Runx2 isoform I controls a panel of proinvasive genes driving aggressiveness of papillary thyroid carcinomas. J. Clin. Endocrinol. Metab. 2012, 97, E2006–E2015. [Google Scholar] [CrossRef]
- Cohen-Solal, K.A.; Boregowda, R.K.; Lasfar, A. RUNX2 and the PI3K/AKT axis reciprocal activation as a driving force for tumor progression. Mol. Cancer 2015, 14, 137. [Google Scholar] [CrossRef]
- Wang, W.; Shen, T.; Dong, B.; Creighton, C.J.; Meng, Y.; Zhou, W.; Shi, Q.; Zhou, H.; Zhang, Y.; Moore, D.D.; et al. MAPK4 overexpression promotes tumor progression via noncanonical activation of AKT/mTOR signaling. J. Clin. Investig. 2019, 129, 1015–1029. [Google Scholar] [CrossRef]
- de Souza, E.C.; Padron, A.S.; Braga, W.M.; de Andrade, B.M.; Vaisman, M.; Nasciutti, L.E.; Ferreira, A.C.; de Carvalho, D.P. MTOR downregulates iodide uptake in thyrocytes. J. Endocrinol. 2010, 206, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Azouzi, N.; Cailloux, J.; Cazarin, J.M.; Knauf, J.A.; Cracchiolo, J.; Al Ghuzlan, A.; Hartl, D.; Polak, M.; Carré, A.; El Mzibri, M.; et al. NADPH Oxidase NOX4 Is a Critical Mediator of BRAFV600E-Induced Downregulation of the Sodium/Iodide Symporter in Papillary Thyroid Carcinomas. Antioxid. Redox Signal. 2017, 26, 864–877. [Google Scholar] [CrossRef] [PubMed]
- Weyemi, U.; Caillou, B.; Talbot, M.; Ameziane-El-Hassani, R.; Lacroix, L.; Lagent-Chevallier, O.; Al Ghuzlan, A.; Roos, D.; Bidart, J.M.; Virion, A.; et al. Intracellular expression of reactive oxygen species-generating NADPH oxidase NOX4 in normal and cancer thyroid tissues. Endocr. Relat. Cancer 2010, 17, 27–37. [Google Scholar] [CrossRef]
- Liu, J.; Xiao, Q.; Xiao, J.; Niu, C.; Li, Y.; Zhang, X.; Zhou, Z.; Shu, G.; Yin, G. Wnt/β-catenin signalling: Function, biological mechanisms, and therapeutic opportunities. Signal Transduct. Target. Ther. 2022, 7, 3. [Google Scholar] [CrossRef]
- Sastre-Perona, A.; Santisteban, P. Role of the wnt pathway in thyroid cancer. Front. Endocrinol. 2012, 3, 31. [Google Scholar] [CrossRef]
- Zhang, W.; Ruan, X.; Li, Y.; Zhi, J.; Hu, L.; Hou, X.; Shi, X.; Wang, X.; Wang, J.; Ma, W.; et al. KDM1A promotes thyroid cancer progression and maintains stemness through the Wnt/β-catenin signaling pathway. Theranostics 2022, 12, 1500–1517. [Google Scholar] [CrossRef] [PubMed]
- Antonelli, A.; La Motta, C. Novel therapeutic clues in thyroid carcinomas: The role of targeting cancer stem cells. Med. Res. Rev. 2017, 37, 1299–1317. [Google Scholar] [CrossRef]
- Zou, M.; BinEssa, H.A.; Al-Malki, Y.H.; Al-Yahya, S.; Al-Alwan, M.; Al-Jammaz, I.; Khabar, K.S.; Almohanna, F.; Assiri, A.M.; Meyer, B.F.; et al. β-Catenin Attenuation Inhibits Tumor Growth and Promotes Differentiation in a BRAFV600E-Driven Thyroid Cancer Animal Model. Mol. Cancer Ther. 2021, 20, 1603–1613. [Google Scholar] [CrossRef]
- Leow, P.C.; Tian, Q.; Ong, Z.Y.; Yang, Z.; Ee, P.L. Antitumor activity of natural compounds, curcumin and PKF118-310, as Wnt/β-catenin antagonists against human osteosarcoma cells. Investig. New Drugs 2010, 28, 766–782. [Google Scholar] [CrossRef]
- Lan, L.; Basourakos, S.; Cui, D.; Zuo, X.; Deng, W.; Huo, L.; Chen, L.; Zhang, G.; Deng, L.; Shi, B.; et al. Inhibiting β-catenin expression promotes efficiency of radioiodine treatment in aggressive follicular thyroid cancer cells probably through mediating NIS localization. Oncol. Rep. 2017, 37, 426–434. [Google Scholar] [CrossRef]
- Ferretti, E.; Tosi, E.; Po, A.; Scipioni, A.; Morisi, R.; Espinola, M.S.; Russo, D.; Durante, C.; Schlumberger, M.; Screpanti, I.; et al. Notch signaling is involved in expression of thyrocyte differentiation markers and is down-regulated in thyroid tumors. J. Clin. Endocrinol. Metab. 2008, 93, 4080–4087. [Google Scholar] [CrossRef] [PubMed]
- Bolós, V.; Grego-Bessa, J.; de la Pompa, J.L. Notch signaling in development and cancer. Endocr. Rev. 2007, 28, 339–363. [Google Scholar] [CrossRef] [PubMed]
- Miele, L. Notch signaling. Clin. Cancer Res. 2006, 12, 1074–1079. [Google Scholar] [CrossRef] [PubMed]
- Somnay, Y.R.; Yu, X.M.; Lloyd, R.V.; Leverson, G.; Aburjania, Z.; Jang, S.; Jaskula-Sztul, R.; Chen, H. Notch3 expression correlates with thyroid cancer differentiation, induces apoptosis, and predicts disease prognosis. Cancer 2017, 123, 769–782. [Google Scholar] [CrossRef] [PubMed]
- Talora, C.; Sgroi, D.C.; Crum, C.P.; Dotto, G.P. Specific down-modulation of Notch1 signaling in cervical cancer cells is required for sustained HPV-E6/E7 expression and late steps of malignant transformation. Genes Dev. 2002, 16, 2252–2263. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Ning, L.; Chen, H. Notch1 mediates growth suppression of papillary and follicular thyroid cancer cells by histone deacetylase inhibitors. Mol. Cancer Ther. 2009, 8, 350–356. [Google Scholar] [CrossRef]
- Zhang, X.; Li, D.; Li, M.; Ye, M.; Ding, L.; Cai, H.; Fu, D.; Lv, Z. MicroRNA-146a targets PRKCE to modulate papillary thyroid tumor development. Int. J. Cancer 2014, 134, 257–267. [Google Scholar] [CrossRef]
- Yang, S.J.; Wang, D.D.; Zhong, S.L.; Chen, W.Q.; Wang, F.L.; Zhang, J.; Xu, W.X.; Xu, D.; Zhang, Q.; Li, J.; et al. Tumor-derived exosomal circPSMA1 facilitates the tumorigenesis, metastasis, and migration in triple-negative breast cancer (TNBC) through miR-637/Akt1/β-catenin (cyclin D1) axis. Cell Death Dis. 2021, 12, 420. [Google Scholar] [CrossRef]
- Lakshmanan, A.; Wojcicka, A.; Kotlarek, M.; Zhang, X.; Jazdzewski, K.; Jhiang, S.M. microRNA-339-5p modulates Na+/I− symporter-mediated radioiodide uptake. Endocr. Relat. Cancer 2015, 22, 11–21. [Google Scholar] [CrossRef]
- Montero-Conde, C.; Graña-Castro, O.; Martín-Serrano, G.; Martínez-Montes, Á.M.; Zarzuela, E.; Muñoz, J.; Torres-Perez, R.; Pita, G.; Cordero-Barreal, A.; Leandro-García, L.J.; et al. Hsa-miR-139-5p is a prognostic thyroid cancer marker involved in HNRNPF-mediated alternative splicing. Int. J. Cancer 2020, 146, 521–530. [Google Scholar] [CrossRef]
- Riesco-Eizaguirre, G.; Wert-Lamas, L.; Perales-Patón, J.; Sastre-Perona, A.; Fernández, L.P.; Santisteban, P. The miR-146b-3p/PAX8/NIS Regulatory Circuit Modulates the Differentiation Phenotype and Function of Thyroid Cells during Carcinogenesis. Cancer Res. 2015, 75, 4119–4130. [Google Scholar] [CrossRef] [PubMed]
- Hou, S.; Xie, X.; Zhao, J.; Wu, C.; Li, N.; Meng, Z.; Cai, C.; Tan, J. Downregulation of miR-146b-3p Inhibits Proliferation and Migration and Modulates the Expression and Location of Sodium/Iodide Symporter in Dedifferentiated Thyroid Cancer by Potentially Targeting MUC20. Front. Oncol. 2021, 10, 566365. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.T.; Qiu, Z.L.; Song, H.J.; Wei, W.J.; Luo, Q.Y. miRNA-106a directly targeting RARB associates with the expression of Na(+)/I(−) symporter in thyroid cancer by regulating MAPK signaling pathway. J. Exp. Clin. Cancer Res. 2016, 35, 101. [Google Scholar] [CrossRef] [PubMed]
- Ricarte-Filho, J.C.; Fuziwara, C.S.; Yamashita, A.S.; Rezende, E.; da-Silva, M.J.; Kimura, E.T. Effects of let-7 microRNA on Cell Growth and Differentiation of Papillary Thyroid Cancer. Transl. Oncol. 2009, 2, 236–241. [Google Scholar] [CrossRef]
- Oh, J.M.; Ahn, B.C. Molecular mechanisms of radioactive iodine refractoriness in differentiated thyroid cancer: Impaired sodium iodide symporter (NIS) expression owing to altered signaling pathway activity and intracellular localization of NIS. Theranostics 2021, 11, 6251–6277. [Google Scholar] [CrossRef] [PubMed]
- Mu, Z.; Zhang, X.; Liang, D.; Fang, J.; Chen, G.; Guo, W.; Sun, D.; Sun, Y.; Kai, Z.; Huang, L.; et al. Risk stratification for radioactive iodine refractoriness using molecular alterations in distant metastatic differentiated thyroid cancer. Chin. J. Cancer Res. 2024, 36, 25–35. [Google Scholar] [CrossRef]
- Haugen, B.R.; Alexander, E.K.; Bible, K.C.; Doherty, G.M.; Mandel, S.J.; Nikiforov, Y.E.; Pacini, F.; Randolph, G.W.; Sawka, A.M.; Schlumberger, M.; et al. 2015 American Thyroid Association Management Guidelines for Adult Patients with Thyroid Nodules and Differentiated Thyroid Cancer: The American Thyroid Association Guidelines Task Force on Thyroid Nodules and Differentiated Thyroid Cancer. Thyroid 2016, 26, 1–133. [Google Scholar] [CrossRef] [PubMed]
- Filetti, S.; Durante, C.; Hartl, D.M.; Leboulleux, S.; Locati, L.D.; Newbold, K.; Papotti, M.G.; Berruti, A.; ESMO Guidelines Committee. ESMO Clinical Practice Guideline update on the use of systemic therapy in advanced thyroid cancer. Ann. Oncol. 2022, 33, 674–684. [Google Scholar] [CrossRef]
- Schlumberger, M.; Brose, M.; Elisei, R.; Leboulleux, S.; Luster, M.; Pitoia, F.; Pacini, F. Definition and management of radioactive iodine-refractory differentiated thyroid cancer. Lancet Diabetes Endocrinol. 2014, 2, 356–358. [Google Scholar] [CrossRef]
- Silaghi, H.; Lozovanu, V.; Georgescu, C.E.; Pop, C.; Nasui, B.A.; Cătoi, A.F.; Silaghi, C.A. State of the Art in the Current Management and Future Directions of Targeted Therapy for Differentiated Thyroid Cancer. Int. J. Mol. Sci. 2022, 23, 3470. [Google Scholar] [CrossRef]
- Nistor, C.; Ciuche, A.; Constantinescu, I. Emergency surgical tracheal decompression in a huge retrosternal goiter. Acta Endocrinol. 2017, 13, 370–374. [Google Scholar] [CrossRef]
- Kiss, A.; Szili, B.; Bakos, B.; Ármós, R.; Putz, Z.; Árvai, K.; Kocsis-Deák, B.; Tobiás, B.; Balla, B.; Pikó, H.; et al. Comparison of surgical strategies in the treatment of low-risk differentiated thyroid cancer. BMC Endocr. Disord. 2023, 23, 23. [Google Scholar] [CrossRef] [PubMed]
- Nistor, C.E.; Găvan, C.S.; Ciritel, A.A.; Nemes, A.F.; Ciuche, A. The Association of Minimally Invasive Surgical Approaches and Mortality in Patients with Malignant Pleuropericarditis—A 10 Year Retrospective Observational Study. Medicina 2022, 58, 718. [Google Scholar] [CrossRef] [PubMed]
- Young, M.; John, S. Hepatic Chemoembolization. StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2018. [Google Scholar]
- Minocha, J.; Salem, R.; Lewandowski, R.J. Transarterial chemoembolization and yittrium-90 for liver cancer and other lesions. Clin. Liver Dis. 2014, 18, 877–890. [Google Scholar] [CrossRef] [PubMed]
- Choy, P.Y.; Koea, J.; McCall, J.; Holden, A.; Osbourne, M. The role of radiofrequency ablation in the treatment of primary and metastatic tumours of the liver: Initial lessons learned. N. Z. Med. J. 2002, 115, 1–7. [Google Scholar]
- Mazzeo, S.; Cervelli, R.; Elisei, R.; Tarantini, G.; Cappelli, C.; Molinaro, E.; Galleri, D.; De Napoli, L.; Comite, C.; Cioni, R.; et al. mRECIST criteria to assess recurrent thyroid carcinoma treatment response after radiofrequency ablation: A prospective study. J. Endocrinol. Investig. 2018, 41, 1389–1399. [Google Scholar] [CrossRef]
- Hay, I.D.; Lee, R.A.; Davidge-Pitts, C.; Reading, C.C.; Charboneau, J.W. Long-term outcome of ultrasound-guided percutaneous ethanol ablation of selected “recurrent” neck nodal metastases in 25 patients with TNM stages III or IVA papillary thyroid carcinoma previously treated by surgery and 131I therapy. Surgery 2013, 154, 1448–1455. [Google Scholar] [CrossRef] [PubMed]
- Deschamps, F.; de Baere, T. Cementoplasty of bone metastases. Diagn. Interv. Imaging 2012, 93, 685–689. [Google Scholar] [CrossRef]
- Karapanou, O.; Simeakis, G.; Vlassopoulou, B.; Alevizaki, M.; Saltiki, K. Advanced RAI-refractory thyroid cancer: An update on treatment perspectives. Endocr. Relat. Cancer 2022, 29, R57–R66. [Google Scholar] [CrossRef]
- Brose, M.S.; Nutting, C.M.; Jarzab, B.; Elisei, R.; Siena, S.; Bastholt, L.; De La Fouchardiere, C.; Pacini, F.; Paschke, R.; Shong, Y.K.; et al. Sorafenib in radioactive iodine-refractory, locally advanced or metastatic differentiated thyroid cancer: A randomised, double-blind, phase 3 trial. Lancet 2014, 384, 319–328. [Google Scholar] [CrossRef]
- Schlumberger, M.; Tahara, M.; Wirth, L.J.; Robinson, B.; Brose, M.S.; Elisei, R.; Habra, M.A.; Newbold, K.; Shah, M.H.; Hoff, A.O.; et al. Lenvatinib versus placebo in radioiodine-refractory thyroid cancer. N. Engl. J. Med. 2015, 372, 621–630. [Google Scholar] [CrossRef] [PubMed]
- Brose, M.S.; Worden, F.P.; Newbold, K.L.; Guo, M.; Hurria, A. Effect of Age on the Efficacy and Safety of Lenvatinib in Radioiodine-Refractory Differentiated Thyroid Cancer in the Phase III SELECT Trial. J. Clin. Oncol. 2017, 35, 2692–2699. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Xu, Z.; Ji, Q.; Ge, M.; Shi, F.; Qin, J.; Wang, F.; Chen, G.; Zhang, Y.; Huang, R.; et al. A Randomized, Phase III Study of Lenvatinib in Chinese Patients with Radioiodine-Refractory Differentiated Thyroid Cancer. Clin. Cancer Res. 2021, 27, 5502–5509. [Google Scholar] [CrossRef] [PubMed]
- Wirth, L.J.; Brose, M.S.; Sherman, E.J.; Licitra, L.; Schlumberger, M.; Sherman, S.I.; Bible, K.C.; Robinson, B.; Rodien, P.; Godbert, Y.; et al. Open-Label, Single-Arm, Multicenter, Phase II Trial of Lenvatinib for the Treatment of Patients with Anaplastic Thyroid Cancer. J. Clin. Oncol. 2021, 39, 2359–2366. [Google Scholar] [CrossRef]
- Tahara, M.; Kiyota, N.; Hoff, A.O.; Badiu, C.; Owonikoko, T.K.; Dutcus, C.E.; Suzuki, T.; Ren, M.; Wirth, L.J. Impact of lung metastases on overall survival in the phase 3 SELECT study of lenvatinib in patients with radioiodine-refractory differentiated thyroid cancer. Eur. J. Cancer 2021, 147, 51–57. [Google Scholar] [CrossRef]
- Tahara, M.; Brose, M.S.; Wirth, L.J.; Suzuki, T.; Miyagishi, H.; Fujino, K.; Dutcus, C.E.; Gianoukakis, A. Impact of dose interruption on the efficacy of lenvatinib in a phase 3 study in patients with radioiodine-refractory differentiated thyroid cancer. Eur. J. Cancer 2019, 106, 61–68. [Google Scholar] [CrossRef]
- Gild, M.L.; Tsang, V.H.M.; Clifton-Bligh, R.J.; Robinson, B.G. Multikinase inhibitors in thyroid cancer: Timing of targeted therapy. Nat. Rev. Endocrinol. 2021, 17, 225–234. [Google Scholar] [CrossRef]
- Cabanillas, M.E.; Brose, M.S.; Holland, J.; Ferguson, K.C.; Sherman, S.I. A phase I study of cabozantinib (XL184) in patients with differentiated thyroid cancer. Thyroid 2014, 24, 1508–1514. [Google Scholar] [CrossRef]
- Cabanillas, M.E.; De Souza, J.A.; Geyer, S.; Wirth, L.J.; Menefee, M.E.; Liu, S.V.; Shah, K.; Wright, J.; Shah, M.H. Cabozantinib as Salvage Therapy for Patients with Tyrosine Kinase Inhibitor-Refractory Differentiated Thyroid Cancer: Results of a Multicenter Phase II International Thyroid Oncology Group Trial. J. Clin. Oncol. 2017, 35, 3315–3321. [Google Scholar] [CrossRef]
- Brose, M.S.; Robinson, B.; Sherman, S.I.; Krajewska, J.; Lin, C.C.; Vaisman, F.; Hoff, A.O.; Hitre, E.; Bowles, D.W.; Hernando, J.; et al. Cabozantinib for radioiodine-refractory differentiated thyroid cancer (COSMIC-311): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2021, 22, 1126–1138. [Google Scholar] [CrossRef]
- Duke, E.S.; Barone, A.K.; Chatterjee, S.; Mishra-Kalyani, P.S.; Shen, Y.L.; Isikwei, E.; Zhao, H.; Bi, Y.; Liu, J.; Rahman, N.A.; et al. FDA Approval Summary: Cabozantinib for Differentiated Thyroid Cancer. Clin. Cancer Res. 2022, 28, 4173–4177. [Google Scholar] [CrossRef] [PubMed]
- Leboulleux, S.; Bastholt, L.; Krause, T.; de la Fouchardiere, C.; Tennvall, J.; Awada, A.; Gómez, J.M.; Bonichon, F.; Leenhardt, L.; Soufflet, C.; et al. Vandetanib in locally advanced or metastatic differentiated thyroid cancer: A randomised, double-blind, phase 2 trial. Lancet Oncol. 2012, 13, 897–905. [Google Scholar] [CrossRef] [PubMed]
- Lorusso, L.; Pieruzzi, L.; Biagini, A.; Sabini, E.; Valerio, L.; Giani, C.; Passannanti, P.; Pontillo-Contillo, B.; Battaglia, V.; Mazzeo, S.; et al. Lenvatinib and other tyrosine kinase inhibitors for the treatment of radioiodine refractory, advanced, and progressive thyroid cancer. OncoTargets Ther. 2016, 9, 6467–6477. [Google Scholar] [CrossRef] [PubMed]
- Dadu, R.; Devine, C.; Hernandez, M.; Waguespack, S.G.; Busaidy, N.L.; Hu, M.I.; Jimenez, C.; Habra, M.A.; Sellin, R.V.; Ying, A.K.; et al. Role of salvage targeted therapy in differentiated thyroid cancer patients who failed first-line sorafenib. J. Clin. Endocrinol. Metab. 2014, 99, 2086–2094. [Google Scholar] [CrossRef]
- Lamartina, L.; Anizan, N.; Dupuy, C.; Leboulleux, S.; Schlumberger, M. Redifferentiation-facilitated radioiodine therapy in thyroid cancer. Endocr. Relat. Cancer 2021, 28, T179–T191. [Google Scholar] [CrossRef]
- Ho, A.L.; Grewal, R.K.; Leboeuf, R.; Sherman, E.J.; Pfister, D.G.; Deandreis, D.; Pentlow, K.S.; Zanzonico, P.B.; Haque, S.; Gavane, S.; et al. Selumetinib-enhanced radioiodine uptake in advanced thyroid cancer. N. Engl. J. Med. 2013, 368, 623–632. [Google Scholar] [CrossRef]
- Rothenberg, S.M.; McFadden, D.G.; Palmer, E.L.; Daniels, G.H.; Wirth, L.J. Redifferentiation of iodine-refractory BRAF V600E-mutant metastatic papillary thyroid cancer with dabrafenib. Clin. Cancer Res. 2015, 21, 1028–1035. [Google Scholar] [CrossRef]
- Dunn, L.A.; Sherman, E.J.; Baxi, S.S.; Tchekmedyian, V.; Grewal, R.K.; Larson, S.M.; Pentlow, K.S.; Haque, S.; Tuttle, R.M.; Sabra, M.M.; et al. Vemurafenib Redifferentiation of BRAF Mutant, RAI-Refractory Thyroid Cancers. J. Clin. Endocrinol. Metab. 2019, 104, 1417–1428. [Google Scholar] [CrossRef]
- Pešorda, M.; Kuna, S.K.; Huić, D.; Herceg, D.; Despot, M.; Samardžić, T.; Gnjidić, M.; Belev, B. Kinase Inhibitors in the Treatment of Thyroid Cancer: Institutional Experience. Acta Clin. Croat. 2020, 59 (Suppl. S1), 73–80. [Google Scholar] [CrossRef]
- Jaber, T.; Waguespack, S.G.; Cabanillas, M.E.; Elbanan, M.; Vu, T.; Dadu, R.; Sherman, S.I.; Amit, M.; Santos, E.B.; Zafereo, M.; et al. Targeted Therapy in Advanced Thyroid Cancer to Resensitize Tumors to Radioactive Iodine. J. Clin. Endocrinol. Metab. 2018, 103, 3698–3705. [Google Scholar] [CrossRef]
- Iravani, A.; Solomon, B.; Pattison, D.A.; Jackson, P.; Ravi Kumar, A.; Kong, G.; Hofman, M.S.; Akhurst, T.; Hicks, R.J. Mitogen-Activated Protein Kinase Pathway Inhibition for Redifferentiation of Radioiodine Refractory Differentiated Thyroid Cancer: An Evolving Protocol. Thyroid 2019, 29, 1634–1645. [Google Scholar] [CrossRef] [PubMed]
- Leboulleux, S.; Dupuy, C.; Lacroix, L.; Attard, M.; Grimaldi, S.; Corre, R.; Ricard, M.; Nasr, S.; Berdelou, A.; Hadoux, J.; et al. Redifferentiation of a BRAFK601E-Mutated Poorly Differentiated Thyroid Cancer Patient with Dabrafenib and Trametinib Treatment. Thyroid 2019, 29, 735–742. [Google Scholar] [CrossRef] [PubMed]
- Weber, M.; Kersting, D.; Riemann, B.; Brandenburg, T.; Führer-Sakel, D.; Grünwald, F.; Kreissl, M.C.; Dralle, H.; Weber, F.; Schmid, K.W.; et al. Enhancing Radioiodine Incorporation into Radio Iodine Refractory Thyroid Cancer with MAPK Inhibition (ERRITI): A Single-Center Prospective Two-Arm Study. Clin. Cancer Res. 2022, 28, 4194–4202. [Google Scholar] [CrossRef]
- Girolami, I.; Pantanowitz, L.; Mete, O.; Brunelli, M.; Marletta, S.; Colato, C.; Trimboli, P.; Crescenzi, A.; Bongiovanni, M.; Barbareschi, M.; et al. Programmed death-ligand 1 (PD-L1) is a potential biomarker of disease-free survival in papillary thyroid carcinoma: A systematic review and meta-analysis of PD-L1 immunoexpression in follicular epithelial derived thyroid carcinoma. Endocr. Pathol. 2020, 31, 291–300. [Google Scholar] [CrossRef]
- Shi, X.; Li, C.W.; Tan, L.C.; Wen, S.S.; Liao, T.; Zhang, Y.; Chen, T.Z.; Ma, B.; Yu, P.C.; Lu, Z.W.; et al. Immune co-inhibitory receptors PD-1, CTLA-4, TIM-3, LAG-3 and TIGIT in medullary thyroid cancers: A large cohort study. J. Clin. Endocrinol. Metab. 2020, 106, 120–132. [Google Scholar] [CrossRef]
- Chintakuntlawar, A.V.; Rumilla, K.M.; Smith, C.Y.; Jenkins, S.M.; Foote, R.L.; Kasperbauer, J.L.; Morris, J.C.; Ryder, M.; Alsidawi, S.; Hilger, C.; et al. Expression of PD-1 and PD-L1 in anaplastic thyroid cancer patients treated with multimodal therapy: Results from a retrospective study. J. Clin. Endocrinol. Metab. 2017, 102, 1943–1950. [Google Scholar] [CrossRef]
- Mehnert, J.M.; Varga, A.; Brose, M.S.; Aggarwal, R.R.; Lin, C.C.; Prawira, A.; De Braud, F.; Tamura, K.; Doi, T.; Piha-Paul, S.A.; et al. Safety and antitumor activity of the anti-PD-1 antibody pembrolizumab in patients with advanced, PD-L1-positive papillary or follicular thyroid cancer. BMC Cancer 2019, 19, 196. [Google Scholar] [CrossRef] [PubMed]
- Naing, A.; Gainor, J.F.; Gelderblom, H.; Forde, P.M.; Butler, M.O.; Lin, C.C.; Sharma, S.; De Olza, M.O.; Varga, A.; Taylor, M.; et al. A first-in-human phase 1 dose escalation study of spartalizumab (PDR001), an anti-PD-1 antibody, in patients with advanced solid tumors. J. Immunother. Cancer 2020, 8, e000530. [Google Scholar] [CrossRef] [PubMed]
- Capdevila, J.; Wirth, L.J.; Ernst, T.; Ponce Aix, S.; Lin, C.C.; Ramlau, R.; Butler, M.O.; Delord, J.P.; Gelderblom, H.; Ascierto, P.A.; et al. PD-1 blockade in anaplastic thyroid carcinoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2020, 38, 2620–2627. [Google Scholar] [CrossRef]
- Chintakuntlawar, A.V.; Yin, J.; Foote, R.L.; Kasperbauer, J.L.; Rivera, M.; Asmus, E.; Garces, N.I.; Janus, J.R.; Liu, M.; Ma, D.J.; et al. A phase 2 study of pembrolizumab combined with chemoradiotherapy as initial treatment for anaplastic thyroid cancer. Thyroid Off. J. Am. Thyroid Assoc. 2019, 29, 1615–1622. [Google Scholar] [CrossRef]
- Dierks, C.; Ruf, J.; Seufert, J.; Kreissl, M.; Klein, C.; Spitzweg, C.; Kroiss, M.; Thomusch, O.; Lorenz, K.; Zielke, A.; et al. 1646MO Phase II ATLEP trial: Final results for lenvatinib/pembrolizumab in metastasized anaplastic and poorly differentiated thyroid carcinoma. Ann. Oncol. 2022, 33, S1295. [Google Scholar] [CrossRef]
- French, J.D.; Haugen, B.R.; Worden, F.P.; Bowles, D.W.; Gianoukakis, A.G.; Konda, B.; Dadu, R.; Sherman, E.J.; McCue, S.; Foster, N.R.; et al. Combination Targeted Therapy with Pembrolizumab and Lenvatinib in Progressive, Radioiodine-Refractory Differentiated Thyroid Cancers. Clin. Cancer Res. 2024, 30, 3757–3767. [Google Scholar] [CrossRef] [PubMed]
- Volpe, D.F.; Nappi, C.; Zampella, E.; Di Donna, E.; Maurea, S.; Cuocolo, A.; Klain, M. Current Advances in Radioactive Iodine-Refractory Differentiated Thyroid Cancer. Curr. Oncol. 2024, 31, 3870–3884. [Google Scholar] [CrossRef]
- Jin, Y.; Van Nostrand, D.; Cheng, L.; Liu, M.; Chen, L. Radioiodine refractory differentiated thyroid cancer. Crit. Rev. Oncol. Hematol. 2018, 125, 111–120. [Google Scholar] [CrossRef] [PubMed]
| Inclusion Criteria |
|---|
| Original studies |
| Topic: gene data, radiodiodine-refractory |
| Published in PubMed |
| Timeframe of search: 1998–2024 |
| Exclusion criteria |
| Non-human data |
| Case report, case series |
| Editorial |
| Non-English paper |
| Pediatric data |
| Selective inhibitor of RET |
| Selective inhibitor of NTRK |
| RAI-avid |
| MTC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Voinea, I.-A.; Petrova, E.; Dumitru, N.; Cocoloș, A.; Ioachim, D.; Goldstein, A.L.; Ghemigian, A.M. Pathogenesis and Management Strategies in Radioiodine-Refractory Differentiated Thyroid Cancer: From Molecular Mechanisms Toward Therapeutic Approaches: A Comprehensive Review. J. Clin. Med. 2024, 13, 7161. https://doi.org/10.3390/jcm13237161
Voinea I-A, Petrova E, Dumitru N, Cocoloș A, Ioachim D, Goldstein AL, Ghemigian AM. Pathogenesis and Management Strategies in Radioiodine-Refractory Differentiated Thyroid Cancer: From Molecular Mechanisms Toward Therapeutic Approaches: A Comprehensive Review. Journal of Clinical Medicine. 2024; 13(23):7161. https://doi.org/10.3390/jcm13237161
Chicago/Turabian StyleVoinea, Iulia-Alexandra, Eugenia Petrova, Nicoleta Dumitru, Andra Cocoloș, Dumitru Ioachim, Andrei Liviu Goldstein, and Adina Mariana Ghemigian. 2024. "Pathogenesis and Management Strategies in Radioiodine-Refractory Differentiated Thyroid Cancer: From Molecular Mechanisms Toward Therapeutic Approaches: A Comprehensive Review" Journal of Clinical Medicine 13, no. 23: 7161. https://doi.org/10.3390/jcm13237161
APA StyleVoinea, I.-A., Petrova, E., Dumitru, N., Cocoloș, A., Ioachim, D., Goldstein, A. L., & Ghemigian, A. M. (2024). Pathogenesis and Management Strategies in Radioiodine-Refractory Differentiated Thyroid Cancer: From Molecular Mechanisms Toward Therapeutic Approaches: A Comprehensive Review. Journal of Clinical Medicine, 13(23), 7161. https://doi.org/10.3390/jcm13237161