
Management of Hereditary Transthyretin Amyloidosis (ATTRv) Patients and Asymptomatic Carriers in Spain: The EMPATIa Study

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. Study Design
2.3. Clinical Management
2.4. ATTRv Amyloidosis Red Flags
2.5. Statistical Analysis
3. Results
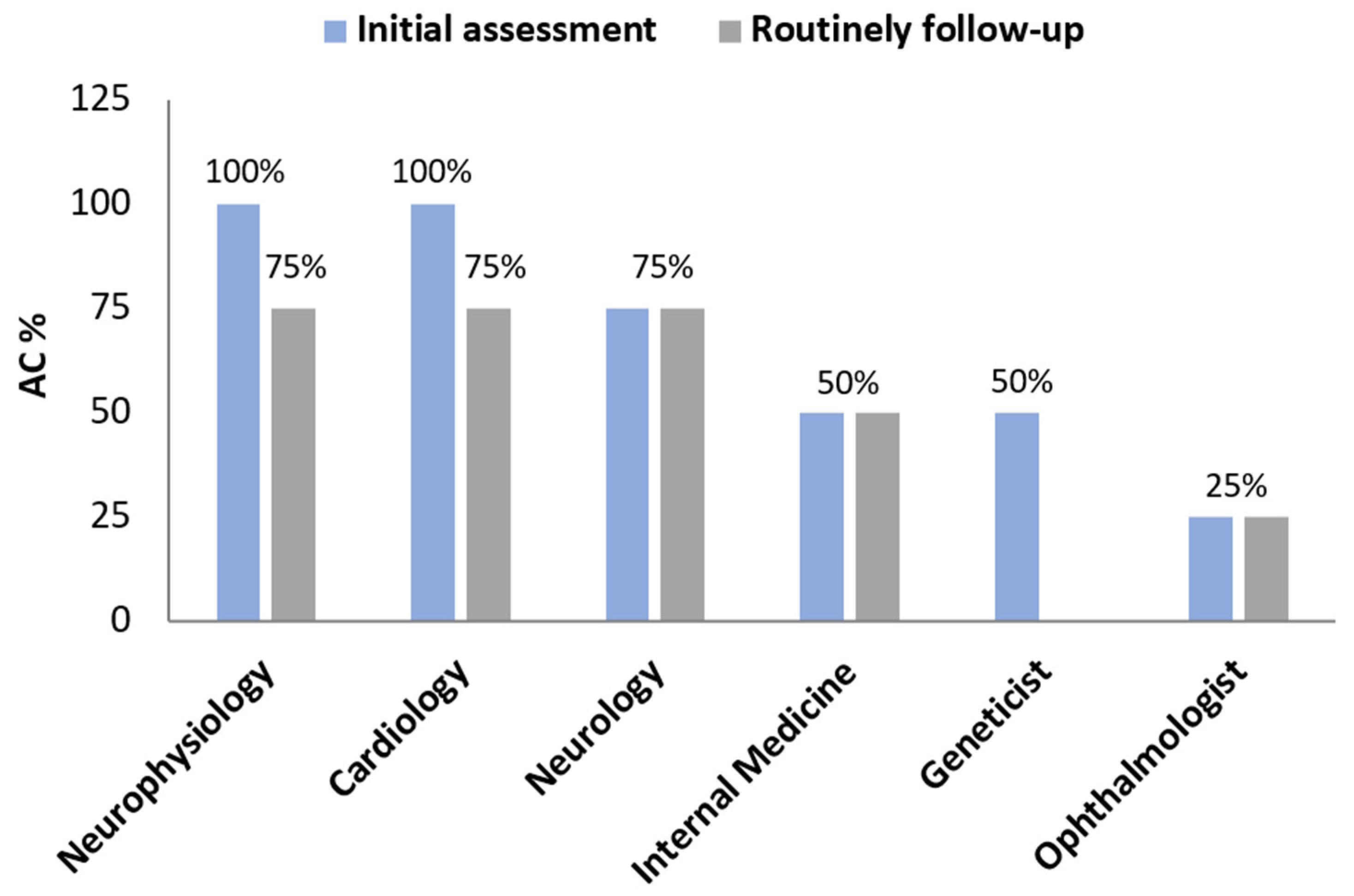
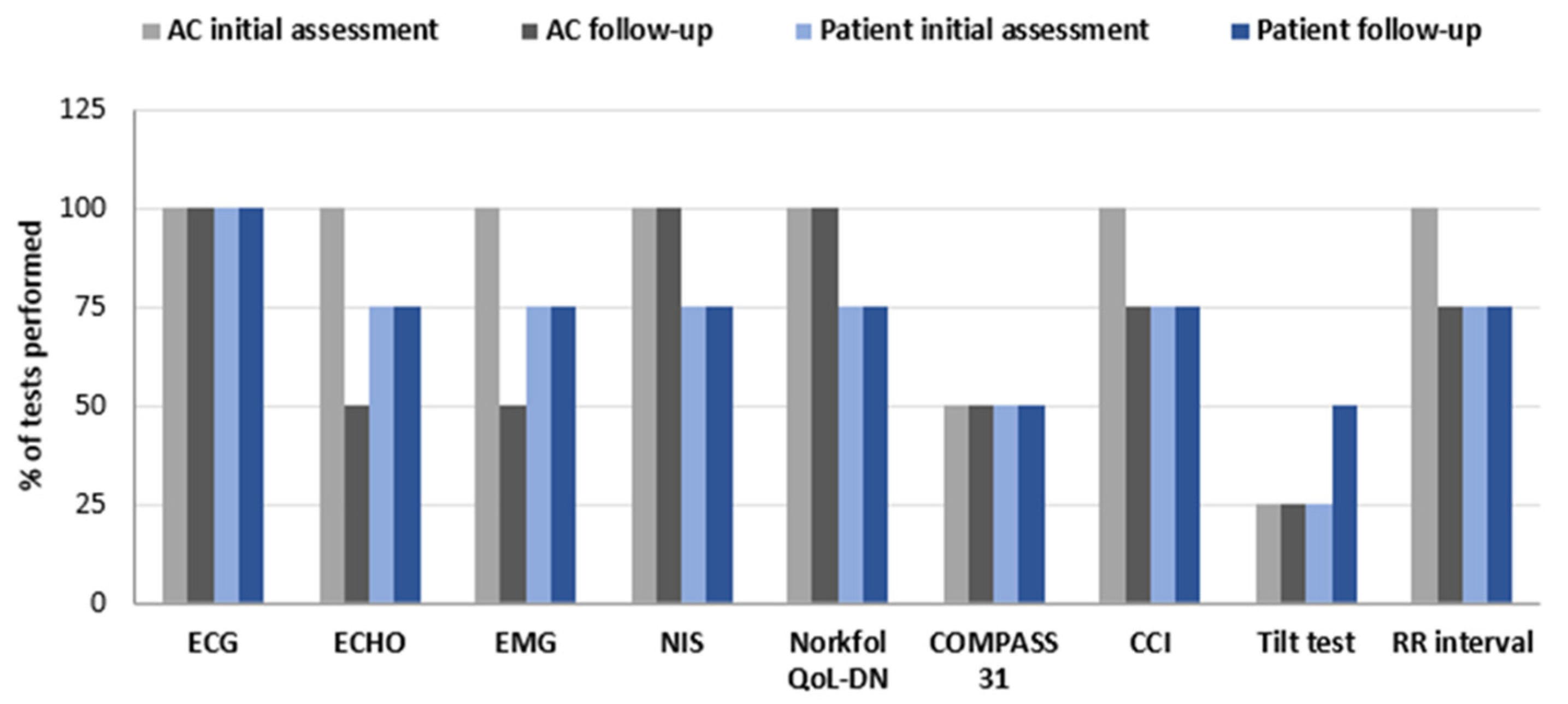
3.1. Clinical Management of Hereditary Transthyretin Amyloidosis
3.2. ACs Characteristics According to Follow-Up Frequency
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AC | asymptomatic carrier |
| ATTR | transthyretin amyloidosis |
| ATTRv | hereditary transthyretin amyloidosis |
| EMPATIa | Carrier Management Study in Transthyretin hereditary Amyloidosis (from the Spanish: “Estudio sobre el Manejo de los Portadores en Amiloidosis hereditaria por Transtiretina”) |
| ESC | electrochemical skin conductance |
| ECG | electrocardiogram |
| ECHO | echocardiogram |
| NORFOLK QoL-DN | Norfolk quality of life questionnaire for diabetic neuropathy |
| NIS | neuropathy impairment score |
| NT-proBNP | N-terminal prohormone of brain natriuretic peptide |
| QoL | quality of life |
| PN | polyneuropathy |
| TTR | transthyretin |
References
- Ueda, M. Transthyretin: Its function and amyloid formation. Neurochem. Int. 2022, 155, 105313. [Google Scholar] [CrossRef] [PubMed]
- Nativi-Nicolau, J.N.; Karam, C.; Khella, S.; Maurer, M.S. Screening for ATTR amyloidosis in the clinic: Overlapping disorders, misdiagnosis, and multiorgan awareness. Heart Fail. Rev. 2022, 27, 785–793. [Google Scholar] [CrossRef] [PubMed]
- Condoluci, A.; Theaudin, M.; Schwotzer, R.; Pazhenkottil, A.P.; Arosio, P.; Averaimo, M.; Bacher, U.; Bode, P.; Cavalli, A.; Dirnhofer, S.; et al. Management of transthyretin amyloidosis. Swiss Med. Wkly. 2021, 151, w30053. [Google Scholar] [CrossRef]
- Garcia-Pavia, P.; Dominguez, F.; Gonzalez-Lopez, E. Transthyretin amyloid cardiomyopathy. Med. Clin. 2021, 156, 126–134. [Google Scholar] [CrossRef]
- Yee, A.W.; Aldeghi, M.; Blakeley, M.P.; Ostermann, A.; Mas, P.J.; Moulin, M.; de Sanctis, D.; Bowler, M.W.; Mueller-Dieckmann, C.; Mitchell, E.P.; et al. A molecular mechanism for transthyretin amyloidogenesis. Nat. Commun. 2019, 10, 925. [Google Scholar] [CrossRef] [PubMed]
- Dispenzieri, A.; Coelho, T.; Conceicao, I.; Waddington-Cruz, M.; Wixner, J.; Kristen, A.V.; Rapezzi, C.; Plante-Bordeneuve, V.; Gonzalez-Moreno, J.; Maurer, M.S.; et al. Clinical and genetic profile of patients enrolled in the Transthyretin Amyloidosis Outcomes Survey (THAOS): 14-year update. Orphanet. J. Rare Dis. 2022, 17, 236. [Google Scholar] [CrossRef]
- Conceição, I.; Damy, T.; Romero, M.; Galán, L.; Attarian, S.; Luigetti, M.; Sadeh, M.; Sarafov, S.; Tournev, I.; Ueda, M. Early diagnosis of ATTR amyloidosis through targeted follow-up of identified carriers of TTR gene mutations. Amyloid 2019, 26, 3–9. [Google Scholar] [CrossRef]
- Ibrahim, R.B.; Liu, Y.T.; Yeh, S.Y.; Tsai, J.W. Contributions of Animal Models to the Mechanisms and Therapies of Transthyretin Amyloidosis. Front. Physiol. 2019, 10, 338. [Google Scholar] [CrossRef]
- Galan Davila, L.; Martinez Valle, F.; Buades Reines, J.; Gonzalez-Moreno, J.; Losada Lopez, I.; Sevilla, T.; Munoz Beamud, F.; Barcena Llona, J.E.; Romero Acebal, M.; Setaro, F.; et al. A description of variant transthyretin amyloidosis (ATTRv) stage 1 patients and asymptomatic carriers in Spain: The EMPATIa study. Orphanet. J. Rare Dis. 2024, 19, 323. [Google Scholar] [CrossRef]
- Adams, D.; Ando, Y.; Beirão, J.M.; Coelho, T.; Gertz, M.A.; Gillmore, J.D.; Hawkins, P.N.; Lousada, I.; Suhr, O.B.; Merlini, G. Expert consensus recommendations to improve diagnosis of ATTR amyloidosis with polyneuropathy. J. Neurol. 2021, 268, 2109–2122. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.; Koike, H.; Slama, M.; Coelho, T. Hereditary transthyretin amyloidosis: A model of medical progress for a fatal disease. Nat. Rev. Neurol. 2019, 15, 387–404. [Google Scholar] [CrossRef] [PubMed]
- Conceição, I.; González-Duarte, A.; Obici, L.; Schmidt, H.H.J.; Simoneau, D.; Ong, M.-L.; Amass, L. “Red-flag” symptom clusters in transthyretin familial amyloid polyneuropathy. J. Peripher. Nerv. Syst. 2016, 21, 5–9. [Google Scholar] [CrossRef]
- Losada, I.; Gonzalez-Moreno, J.; Rodriguez, A.; Uson, M.; Ripoll-Vera, T.; Ferrer-Nadal, A.; Rigo, E.; Andreu, H.; Figuerola, A.; Montala, J.C.; et al. Multidisciplinary approach in the management of hATTR. Eur. J. Clin. Investig. 2020, 50, e13296. [Google Scholar] [CrossRef]
- Maurer, M.S.; Bokhari, S.; Damy, T.; Dorbala, S.; Drachman, B.M.; Fontana, M.; Grogan, M.; Kristen, A.V.; Lousada, I.; Nativi-Nicolau, J.; et al. Expert Consensus Recommendations for the Suspicion and Diagnosis of Transthyretin Cardiac Amyloidosis. Circ. Heart Fail. 2019, 12, e006075. [Google Scholar] [CrossRef]
- Lane, T.; Fontana, M.; Martinez-Naharro, A.; Quarta, C.C.; Whelan, C.J.; Petrie, A.; Rowczenio, D.M.; Gilbertson, J.A.; Hutt, D.F.; Rezk, T.; et al. Natural History, Quality of Life, and Outcome in Cardiac Transthyretin Amyloidosis. Circulation 2019, 140, 16–26. [Google Scholar] [CrossRef] [PubMed]
- López-Sainz, Á.; Hernandez-Hernandez, A.; Gonzalez-Lopez, E.; Domínguez, F.; Restrepo-Cordoba, M.A.; Cobo-Marcos, M.; Gómez-Bueno, M.; Hernandez-Perez, F.J.; Oteo, J.F.; Mirelis, J.G.; et al. Clinical profile and outcome of cardiac amyloidosis in a Spanish referral center. Rev. Esp. Cardiol. (Engl. Ed.) 2021, 74, 149–158. [Google Scholar] [CrossRef]
- Adams, D.; Suhr, O.B.; Hund, E.; Obici, L.; Tournev, I.; Campistol, J.M.; Slama, M.S.; Hazenberg, B.P.; Coelho, T.; European Network for, T.-F. First European consensus for diagnosis, management, and treatment of transthyretin familial amyloid polyneuropathy. Curr. Opin. Neurol. 2016, 29 (Suppl. S1), S14–S26. [Google Scholar] [CrossRef]
- Conceição, I.; Coelho, T.; Rapezzi, C.; Parman, Y.; Obici, L.; Galán, L.; Rousseau, A. Assessment of patients with hereditary transthyretin amyloidosis—Understanding the impact of management and disease progression. Amyloid 2019, 26, 103–111. [Google Scholar] [CrossRef]
- Ando, Y.; Adams, D.; Benson, M.D.; Berk, J.L.; Plante-Bordeneuve, V.; Coelho, T.; Conceicao, I.; Ericzon, B.G.; Obici, L.; Rapezzi, C.; et al. Guidelines and new directions in the therapy and monitoring of ATTRv amyloidosis. Amyloid 2022, 29, 143–155. [Google Scholar] [CrossRef]
- Garcia-Pavia, P.; Bengel, F.; Brito, D.; Damy, T.; Duca, F.; Dorbala, S.; Nativi-Nicolau, J.; Obici, L.; Rapezzi, C.; Sekijima, Y.; et al. Expert consensus on the monitoring of transthyretin amyloid cardiomyopathy. Eur. J. Heart Fail. 2021, 23, 895–905. [Google Scholar] [CrossRef]
- Formiga, F.; Garcia-Pavia, P.; Martin Sanchez, F.J.; Navarro-Ruiz, A.; Rubio-Terres, C.; Peral, C.; Tarilonte, P.; Lopez-Ibanez de Aldecoa, A.; Rubio-Rodriguez, D. Health and economic impact of the correct diagnosis of transthyretin cardiac amyloidosis in Spain. Expert Rev. Pharmacoecon Outcomes Res. 2021, 21, 1127–1133. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| >1 Visit per Year | ≤1 Visit per Year | p-Value | |||
|---|---|---|---|---|---|
| N | % | N | % | ||
| CNS alterations | 3 | 13.6 | 3 | 5.4 | 0.342 |
| Ophthalmic alterations | 0 | 0.0 | 3 | 5.4 | 0.555 |
| Cardiac alterations | 3 | 13.6 | 5 | 8.9 | 0.680 |
| Carpal tunnel syndrome | 4 | 18.2 | 6 | 10.7 | 0.455 |
| Gastrointestinal alterations | 1 | 4.5 | 9 | 16.1 | 0.267 |
| Autonomic neuropathy | 5 | 22.7 | 6 | 10.7 | 0.276 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Losada López, I.; Gonzalez-Moreno, J.; Buades Reinés, J.; Sevilla, T.; Martinez Valle, F.; Galán Dávila, L.; Muñoz Beamud, F.; Bárcena Llona, J.E.; Romero Acebal, M.; Tarilonte, P.; et al. Management of Hereditary Transthyretin Amyloidosis (ATTRv) Patients and Asymptomatic Carriers in Spain: The EMPATIa Study. J. Clin. Med. 2024, 13, 7587. https://doi.org/10.3390/jcm13247587
Losada López I, Gonzalez-Moreno J, Buades Reinés J, Sevilla T, Martinez Valle F, Galán Dávila L, Muñoz Beamud F, Bárcena Llona JE, Romero Acebal M, Tarilonte P, et al. Management of Hereditary Transthyretin Amyloidosis (ATTRv) Patients and Asymptomatic Carriers in Spain: The EMPATIa Study. Journal of Clinical Medicine. 2024; 13(24):7587. https://doi.org/10.3390/jcm13247587
Chicago/Turabian StyleLosada López, Inés, Juan Gonzalez-Moreno, Juan Buades Reinés, Teresa Sevilla, Fernando Martinez Valle, Lucía Galán Dávila, Francisco Muñoz Beamud, José Eulalio Bárcena Llona, Manuel Romero Acebal, Patricia Tarilonte, and et al. 2024. "Management of Hereditary Transthyretin Amyloidosis (ATTRv) Patients and Asymptomatic Carriers in Spain: The EMPATIa Study" Journal of Clinical Medicine 13, no. 24: 7587. https://doi.org/10.3390/jcm13247587
APA StyleLosada López, I., Gonzalez-Moreno, J., Buades Reinés, J., Sevilla, T., Martinez Valle, F., Galán Dávila, L., Muñoz Beamud, F., Bárcena Llona, J. E., Romero Acebal, M., Tarilonte, P., & Setaro, F. (2024). Management of Hereditary Transthyretin Amyloidosis (ATTRv) Patients and Asymptomatic Carriers in Spain: The EMPATIa Study. Journal of Clinical Medicine, 13(24), 7587. https://doi.org/10.3390/jcm13247587