Abstract
Obesity remains a common metabolic disorder and a threat to health as it is associated with numerous complications. Lifestyle modifications and caloric restriction can achieve limited weight loss. Bariatric surgery is an effective way of achieving substantial weight loss as well as glycemic control secondary to weight-related type 2 diabetes mellitus. It has been suggested that an anorexigenic gut hormone response following bariatric surgery contributes to weight loss. Understanding the changes in gut hormones and their contribution to weight loss physiology can lead to new therapeutic treatments for weight loss. Two distinct types of neurons in the arcuate hypothalamic nuclei control food intake: proopiomelanocortin neurons activated by the anorexigenic (satiety) hormones and neurons activated by the orexigenic peptides that release neuropeptide Y and agouti-related peptide (hunger centre). The arcuate nucleus of the hypothalamus integrates hormonal inputs from the gut and adipose tissue (the anorexigenic hormones cholecystokinin, polypeptide YY, glucagon-like peptide-1, oxyntomodulin, leptin, and others) and orexigeneic peptides (ghrelin). Replicating the endocrine response to bariatric surgery through pharmacological mimicry holds promise for medical treatment. Obesity has genetic and environmental factors. New advances in genetic testing have identified both monogenic and polygenic obesity-related genes. Understanding the function of genes contributing to obesity will increase insights into the biology of obesity. This review includes the physiology of appetite control, the influence of genetics on obesity, and the changes that occur following bariatric surgery. This has the potential to lead to the development of more subtle, individualised, treatments for obesity.
Keywords:
hunger centre; satiety centre; POMC; AgRP/NPY; leptin; incretin; bariatric surgery; obesity therapy; obesity genetics 1. Introduction
The gut endocrine system and the gut-central nervous system axis function to digest and absorb food and, consequently, to regulate appetite. The gut-pancreatic endocrine system is one of the largest endocrine systems in the body. Most of the gut hormones secreted (incretins, cholecystokinin, and pancreatic polypeptide), except ghrelin, act to increase satiety and decrease food intake [1]. The adipose tissue is widely distributed in the body; has distinct functions both in energy homeostasis and as an endocrine organ. Leptin, an adipokine secreted by the adipose tissue, has a satiety effect on the central nervous system [2]. Many gut hormones act on the hypothalamus and brain stem areas of appetite control. Within the arcuate nucleus of the hypothalamus are the proopiomelanocortin (POMC)-secreting neurons, which suppress appetite, and a second population of neurons, which increase food intake and co-express neuropeptide Y (NPY) and agouti-related protein (AgRP) [3].
Obesity has become a major contributor to the global burden of chronic disease. Obesity is associated with several disease states, which include, among others, type 2 diabetes (T2DM), hypertension, dyslipidemia, cardiovascular disease, non-alcoholic fatty liver disease, and obstructive sleep apnea [4]. This has led to the development of several obesity treatment methods, including bariatric surgery.
Appropriate response to changes in the homeostatic control of energy intake is obligatory for a stable energetic state and weight maintenance. Understanding the mechanisms controlling appetite and weight transformation may facilitate the understanding of appetite changes as well as weight loss and hormonal changes following bariatric surgery. Bariatric surgery is associated with side effects such as dumping syndrome, postprandial hypoglycaemia, partial T2DM remission or T2DM relapse, and weight regain. Knowledge of the mechanisms of appetite homeostasis may facilitate a greater understanding of changes following bariatric surgery, the development of therapeutic approaches for avoiding the side effects of bariatric surgery, and the advancement of less invasive weight loss strategies [5].
The main goal of this review is to summarise recent advances in the physiology of energy homeostasis, the genetics of obesity and appetite regulation, and the changes that arise following bariatric surgery. The analyses will have clinical relevance in identifying future developments in treatment for mitigating the risk of obesity and its comorbidities.
2. Method
This narrative review covered several topics, and individual terms, e.g., incretins and leptin, were used to identify reviews or articles in PubMed, MEDLINE, Google Scholar, and Web of Science over the past 15 years. From each article/review we extracted further references for the studies included in this review.
3. Neurons and the Control of Appetite
Several neuronal populations, which are distributed throughout the brain, affect the capacity for food intake. Specific areas of the hypothalamus are believed to control feeding behaviour. Two distinct neuronal populations are thought to be significant in regulating energy balance. These are found in the arcuate nucleus of the hypothalamus (ARH), the anorexigenic (appetite suppressing) proopiomelanocortin (POMC) neurons, and the orexigenic (appetite-increasing) neuropeptide Y (NPY)/agouti-related peptide (AgRP) co-expressing neurons. The neurons are positioned to receive signals from both peripheral organs due to the rich blood supply to the arcuate nucleus as well as input from various parts of the central nervous system [6]. POMC-expressing neurons are also located in the nucleus tractus solitaries (NTS) and have different behavioral energy homeostasis functions in the ARH and NTS [7]. POMC neurons have been investigated extensively because of their central role in energy balance. POMC neurones in the ARH extend fibres to other ARH neurones and other multiple brain regions, including the paraventricular nucleus of the hypothalamus, lateral hypothalamus, commissural nucleus of the solitary tract, bed nucleus of the stria terminalis, nucleus accumbens, septal nucleus, ventral tegmental area, central amygdala, periaqueductal grey, and dorsal raphe nucleus [8]. The ARH contains distinct populations of POMC neurons that are either GABAergic or glutamatergic neurotransmitters and are activated by serotonin 5-HT2Cs, insulin, and leptin receptors (among others) [8]. The evidence for leptin acting on a different population of POMC neurons than insulin is conflicting [9]. Other studies suggest a heterogeneous population for POMC and that POMC neurons form several molecularly distinct clusters [10]. POMC and AgRP neurons change neuronal activity in response to glucose fluctuations [11]. Specific responses may be segregated into distinct populations of POMC neurons (Figure 1a–c). POMC neurons suppress appetite, and following processing of POMC by proprotein convertase (coded by PCSK1), release α-melanocyte stimulating hormone (α-MSH). The hormone α-MSH is an agonist of the melanocortin-4-receptors (MC4R), found in the paraventricular nucleus. The MC4R receptor has anorectic activity [6,12]. AgRP inhibits the MC4R agonist activity of melanocortin peptides (Figure 1a–c). In addition, a functional role in energy homeostasis for central and peripheral MC3R has been suggested [8]. In this system, hormones of the fed state (insulin and leptin) released by the pancreas and adipocytes (respectively) bind to their receptors on the POMC neurons and support the processing of proopiomelanocortin to α-MSH, promoting a signal to decrease food intake. Leptin also binds to AgRP /NPY neurons to inhibit their orexigenic activity. It is likely that leptin binding to neuronal cells other than the POMC neurons contributes to energy homeostasis [13] and that AgRP neurons engage other circuits that coordinate feeding. Axon projections of AgRP neurons extend to neurons in the bed nucleus of the stria terminalis, lateral hypothalamus, and parabrachial nuclei that control insulin sensitivity in brown adipose tissue. The axon projections of the AgRP neurons may trigger the motivational and autonomic circuits that contribute to feeding behavior. It is likely that multiple pathways function in the brain, one for homeostatic control of energy balance and feeding and another for non-homeostatic, reward-driven, hedonic feeding [13]. One suggestion is that the hedonic circuitry, once activated, can override the homeostatic energy balance circuitry and the chronic inhibition of AgRP [14]. The effects of lifestyle on epigenetic modifications of genes in the hypothalamus and its role in the regulation of energy homeostasis require further investigation [15].
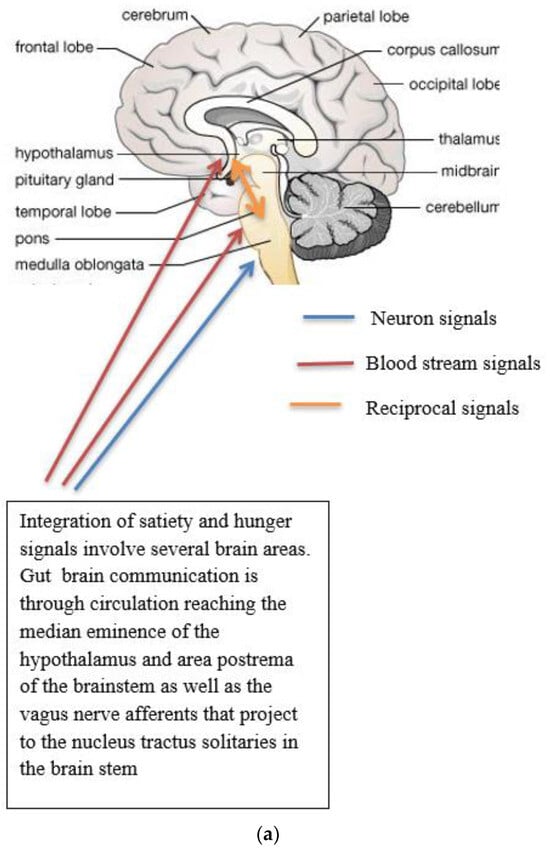
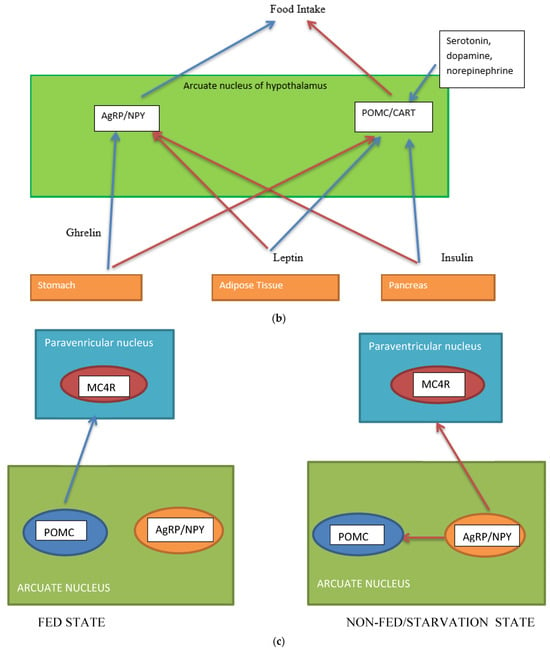
Figure 1.
(a) Gut-brain communication underlying appetite control; (b) neuron function in appetite control, AgRP/NPY (neuropeptide Y/agouti-related peptide neurons), POMC (proopiomelanocortin),  activation,
activation,  inhibition; (c) neuron interaction in appetite control, activation , inhibition . Populations of POMC neurons express different receptors. e.g., leptin and insulin. In the fed state, POMC neurons act to decrease food intake and increase energy expenditure. In the fasted state, AgRP/NPY neurons inhibit the activity of MC4R neurons and POMC neurons.
inhibition; (c) neuron interaction in appetite control, activation , inhibition . Populations of POMC neurons express different receptors. e.g., leptin and insulin. In the fed state, POMC neurons act to decrease food intake and increase energy expenditure. In the fasted state, AgRP/NPY neurons inhibit the activity of MC4R neurons and POMC neurons.
activation, inhibition; (c) neuron interaction in appetite control, activation , inhibition . Populations of POMC neurons express different receptors. e.g., leptin and insulin. In the fed state, POMC neurons act to decrease food intake and increase energy expenditure. In the fasted state, AgRP/NPY neurons inhibit the activity of MC4R neurons and POMC neurons.
Complex neurological and endocrine signals are relayed between the gut and the brain to regulate hunger and satiety. The gut neuroendocrine system and the gut-brain axis function to optimise digestion and absorption and regulate appetite. Numerous reciprocal connections exist between the brain stem (particularly the nucleus of the tractus solitaries) and the hypothalamus [16]. The brain stem receives vagus nerve afferents from the gastrointestinal tract and endocrine signals from the blood due to its closeness to the blood-brain barrier. It is positioned to act as a site of integration between endocrine and neuronal signals (Figure 1a). Afferent neurons of the vagus nerve targets for gut hormones [17].
4. The Endocrine System of the Gut and the Control of Appetite
Gut hormones are secreted by the enteroendocrine cells scattered throughout the epithelial cells of the gut. Gut hormones can act as both hormones and neurotransmitters. The incretin effect is described as the increase in insulin secretion by pancreatic β cells by exposure to intestinal absorption of glucose compared to isoglycemic levels by intravenous infusion. Gut derived hormones are the proglucagon-derived peptides, glucagon-like peptide 1 (GLP-1), GLP2, and gastric inhibitory peptide, also known as glucose-dependent insulinotropic polypeptide (GIP), as well as cholecystokinin (CCK) and peptide YY (PYY), released by the gastrointestinal tract into the general circulation, which mediate effects of feeding [18]. Both GIP and GLP-1, the real incretins, exert their effects by binding to their specific receptors, the GIP receptor (GIPR) and the GLP1 receptor (GLP1R), in pancreatic β cells and boosting the secretion of glucose-dependent insulin release [19,20]. Evidence points out that some enteroendocrine cells, which constitute approximately 1% of intestinal epithelial cells, are plurihormonal. In mice, GIP is secreted from K cells found predominantly in the duodenum, and GLP-1 is secreted from L cells located in the lower small intestine and colon. The presence of food (carbohydrates, protein, and fats) stimulates incretin hormone secretion. Similar to other gut peptides, GLP-1 is a neurotransmitter. Evidence for the involvement of GLP-1 in signalling in the CNS is the wide distribution of the GLP-1 receptor [21]. The GLP1R receptor is widely distributed in the body, for example, in the cardiovascular system, gastrointestinal tract, adipose tissue, brain, and bone [22]. Similarly, some studies report the GLP2R receptor to be widely expressed [23]. This highlights the diversity of incretin functions. A wide range of pharmacological drugs and pre-clinical studies have been used to clarify GLP1R/GIPR physiological function [24]. The suggested physiological roles for incretin hormones are summarised in Table 1.

Table 1.
Functions of incretin hormones.
4.1. Physiology of Incretin Secretion
Release of incretins (GLP-1 and GLP2 are cosecreted) requires the presence of nutrients, carbohydrates, proteins, and fat in the intestinal lumen and their absorption through the enterocytes [44]. To understand the process of altered incretin secretion following bariatric surgery, an understanding of the detailed molecular mechanism underlying its secretion is valuable and is included.
4.2. Secretion and Metabolism of Incretins
In the pancreas, the proglucagon molecule is processed by proconvertase 2 (PC2) to glucagon (Figure 2) and by proconvertase 1/3 (PC1/3) in the gut into GLP-1 (amino acids 1–37) and GLP2 (amino acids 1–33) [45]. Glucagon is secreted by the pancreatic α cell, stimulates glycogenolysis and gluconeogenesis in the liver, and opposes the hypoglycemic action of insulin. Bioactive GLP-1 (7–37) is generated from GLP-1 (1–37). Cleavage by PC1/3 leaves two basic residues at the C-terminus of GLP-1, which are removed by carboxypeptidase prior to carboxyamidation. Several immunoreactive forms of GLP-1 (including GLP-1 7–36 amide and GLP-1 7–37, which are thought to be equipotent), are released in vivo [46]. GLP-1 is inactivated by dipeptidyl peptidase IV (DPP-IV), which cleaves the N-terminal end. GLP-1 (7–36 amide) is thought to be the majority of circulating active GLP-1 in human plasma [47]. Degradation and inactivation of human GLP-2 (amino acids 1–33) with the enzyme DPP-IV resulted in the liberation of GLP-2 (amino acids 3–33) [48].
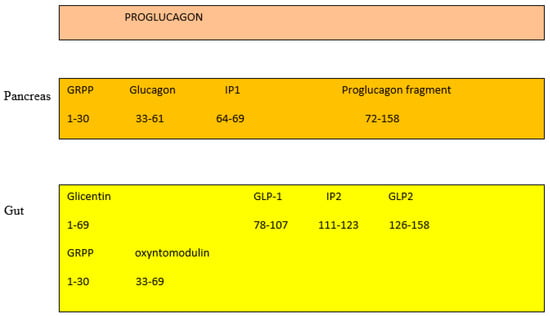
Figure 2.
Gut and pancreas processing of the proglucagon molecule.
GIP is derived by proteolytic processing of a 153-residue precursor, preproGIP [49]. GIP is a 42 amino acid, and as it contains an alanine in position 2, it is physiologically degraded and inactivated by DPP-IV to its bioinactive form (amino acid 3–42) within minutes of secretion [50]. Both inactive GIP and GLP-1 are cleared by the kidney [51]. In healthy humans, the half-lives of GLP-1 is 2–3 min, GIP 5 min [52], and GLP-2 7 min [53]. Which of the two, GLP-1 or GIP, is the most prominent is still a matter of debate. Depending on the method protocol, it has been suggested that either GIP or GLP-1 contributes equally or that either GIP or GLP-1 contribute the majority of the incretin effect in humans [54]. One suggestion is that the contribution to the incretin effect is GIP: approximately 44%, GLP-1: approximately 22%, Glucose: approximately 33%, and neural transmission: negligible [55].
4.3. Control of Incretin Secretion by Nutrients
It has been shown that the presence of macronutrients (carbohydrates, proteins, and fats) in the gut regulates incretin hormone secretion.
4.3.1. Carbohydrates
Glucose is a recognised stimulant of incretins and acts through the sodium glucose cotransporter 1 (SGLT1) with the influx of sodium ions, which depolarises the plasma membrane, opening the voltage-sensitive calcium channels, which is followed by exocytosis of GLP-1-containing secretory vesicles [56]. Facilitative glucose transport through the GLUT2 transporter appears to be of less importance in incretin secretion [57].
4.3.2. Proteins
The pathways underlying the mechanism of protein stimulation of gut hormones are still not clearly understood. At the molecular level, meat hydrolysate has been shown to recruit the G-protein coupled calcium sensing receptor (CaSR) to cause GLP-1 release from the rodent intestine [58], though it is likely that other sensory pathways are involved, e.g., an increase in the anorectic hormone PYY [58,59].
4.3.3. Fats
Fats also potentially stimulate CCK, PYY, and GLP-1 release. The release of CCK, PYY, and GLP-1 in plasma is attenuated by inhibitors of lipase. CCK, GLP-1, and PYY release were found to be dependent on fatty acid chain length, with only fatty acids greater than C10 being effective in stimulating hormone secretion [60,61]. Several G-protein-coupled receptors (GPCR) have been associated with the sensing of fatty acids and the release of gut hormones. GPR40 is a receptor for medium- and long-chain fatty acids. The distribution of GPR40 suggests it may act to regulate pancreatic islet and neurological cell function [62]. Close to the GPR40 locus are the GPR41 and GPR43 genes encoding receptors activated by short-chain fatty acids [63]. GPCR receptors linked to GIP secretion are GPR120 and GPR40, expressed in K cells in the upper small intestine [64,65]. GPR40, GPR41, GPR43, GPR119, and GP120 were identified as sensing receptors for fatty acids with a role in GLP-1 secretion [66,67,68,69,70].
4.4. Other Variables That Regulate Gut Hormones
- (i)
- Gut microbiome: Gut microbiota can contribute to incretin secretion either by fermentation of dietary fibre and production of short-chain fatty acids [71] or by increasing the basal level of GLP-1 and slowing intestinal movement [72].
- (ii)
- Bile acids: Using a pharmacological approach, the bile acid-responsive receptor, G protein-coupled bile acid receptor 1 (GPBAR1/TGR5), has been shown to promote GLP-1 and PYY secretion from intestinal L-cells [73,74]. While bile acids are recognised to signal through specific nuclear acid receptors [75], GPBAR1 is a cell surface receptor. GPBAR1 is a GPCR that is activated by bile acids, resulting in stimulation of Gαs proteins and downstream cAMP signalling pathways. Recent findings support the hypothesis that additional pathways such as elevation of intracellular Ca2+ concentrations [76] and closure of ATP-sensitive potassium (KATP) channels may play a role in GLP-1 release [77]. Available data suggest that bile acids were more effective if they were applied to the basolateral GPBAR1 (vascular side) on L-cells and that luminal-applied bile acids are effective after absorption across the intestinal epithelial layer [78].
- (iii)
- Proinflammatory cytokines TNFα, IL6, and Regulated on Activation, Normal T-cell Expressed and Secreted (RANTES) are increased in obesity in both rodents and humans. Acute treatment with TNFα increases GLP-1 release, and chronic elevation of TNFα decreases GLP-1 release. It has been suggested that TNFα alters cell signalling through the TNF receptor-NFκβ pathway [79]. IL6 regulates glucose homeostasis by stimulating L cells and pancreatic α cells to secrete GLP-1 and, as a result, increase insulin secretion. One suggestion is that acute effects of IL6 are caused by increased GLP-1 exocytosis in L cells and chronic effects by increased glucose responsiveness [80,81]. The chemokine RANTES reduced glucose and stimulated GLP-1 secretion in humans. One proposal is that RANTES acts through the CCR1 receptor to reduce cAMP levels and PKA activity [82]. The response of the L-cell is likely an integrated response to different cytokines using different signalling pathways.
- (iv)
- Other suspected stimulants of GLP-1 secretion are progesterone (studies on cell lines suggest stimulation through the extracellular signal-related kinase 1/2 (ERK1/2) [83], insulin (studies on cell lines suggest a role for phosphatidylinositol 3 kinase-Akt and MAPK kinase (MEK)-ERK1/2 pathways [84], and glucocorticoid (reduced GLP-1 secretion in rodents) [85].
The contribution of each of these additional effects to incretin secretion is unclear, and further studies are required to understand the mechanism underlying these effects.
5. Other Hormones Implicated in Appetite Regulation
The function of various hormones in regulating appetite and satiety to maintain energy homeostasis is included below.
5.1. Insulin
In the liver, insulin supports the transport of glucose from the blood to hepatocytes, where it is converted to glycogen, fatty acids, and triglycerides. In the skeletal muscle, insulin promotes the absorption of glucose. In adipose tissue, insulin supports the uptake of fatty acids, which are converted to triglycerides [86]. Insulin receptors are widely distributed throughout the CNS. Insulin has central anorexic effects. Insulin administration within the ARH can decrease the intake of food by reducing the activation of AgRP/NPY neurones and by increasing the activity of the POMC pathway. It has been proposed that insulin’s actions may be mediated additionally within non-hypothalamic regions [87].
5.2. Glucagon
Glucagon is a 29-amino acid peptide that is secreted from pancreatic α-cells in response to low levels of blood glucose. The glucagon receptor is a G-protein-coupled receptor that is mostly expressed in the liver with minor amounts in the brain and heart. Factors that regulate the secretion of glucagon are glucose, amino acids, gastrointestinally derived peptides, the autonomic nervous system, and possibly intra-islet regulation from beta cells and the inhibitory action of somatostatin. Glucagon secretion increases in hypoglycemic conditions with increased hepatic glucose production as a result of decreased glycogenesis and glycolysis and stimulation of glycogenolysis and gluconeogenesis. In the liver, glucagon can promote beta oxidation, decreased fatty acid synthesis, and decrease very low density lipoprotein release [88]. Early work suggests that glucagon contributes to satiety and that the satiety signal is transmitted through the vagus nerve [89]. A series of human studies confirmed the thermogenic effects of glucagon, an effect that was apparently dependent on the feeding state. One possibility is that the increase in energy expenditure is caused by the catabolic actions of glucagon and is not fully dependent on BAT [90]. Glucagon was thought to increase heart rate and contractility by acting through glucagon receptors and increasing cAMP, though recent results provide conflicting evidence [91,92].
5.3. Ghrelin
Ghrelin is an orexigenic hormone. The human ghrelin gene is localised on chromosome 3p25-26. Ghrelin mRNA is mostly expressed in gastric tissue, and it is also present at low levels in the intestine, pancreas, kidneys, and placenta. PC 1/3 processes proghrelin to ghrelin with acyl modification of Ser3 in ghrelin by the enzyme ghrelin O-acyltransferase. Preprandial rise and postprandial fall in plasma ghrelin levels support the hypothesis that ghrelin plays a physiological role in meal initiation in humans [93]. Ghrelin is an endogenous ligand of the growth hormone secretagogue receptor type 1a (GHS-R1a), which stimulates growth hormone (GH) secretion. GHS-R1a is a GPCR receptor. GHS-R1a is expressed in the hypothalamic and exra-hypothalamic regions and metabolic organs. GHS-R1a interacts with Gαq/11 and the phospholipase C and inositol triphosphate pathways. GHS-Rs are expressed in AgRP/NPY neurons and POMC neurons. The complexity of the role of ghrelin-mediated signalling in the arcuate nucleus and ventromedial nucleus of the hypothalamus are described in the review by Yanagi et al. [94]. However, studies with ghrelin KO mice models have shown that the mice have no decrease in appetite or appreciable difference in body weight, and it has been suggested that ghrelin may play a role during extreme nutritional challenges to protect bodyweight and has a further function in regulating blood glucose [95]. Ghrelin may act to relay meal-related information; ghrelin increases in preference for highly palatable food and may play a role in stress-induced food consumption, which results in hedonic feeding [96].
5.4. Secretin
Secretin is secreted by the enteroendocrine S-cells in the proximal duodenum in response to acidic chyme from the stomach. Both secretin and its receptor are ubiquitously distributed throughout the body, including the central nervous system. Secretin is associated with (i) stimulation of pancreatic water and bicarbonate secretion; (ii) body fluid homeostasis; and (iii) lipolytic actions in mouse adipocytes [97]. The half-life of secretin is approximately 2.5 min [98]. In mice, secretin and its receptor have been identified in ARH and the paraventricular nucleus, suggesting a central role for secretin in the control of appetite [99]. In humans (and mice), secretin activates (increased metabolism, thermogenesis) brown adipose tissue (BAT) and delays the central nervous system response to appetising food, and the impulse to refeed after a meal, and stimulates satiation [100,101]. However, secretin receptor KO mice on the same diet gained significantly less weight and had lower body fat content than their WT counterparts, suggesting further work is required on the role of the gut-BAT-brain axis in the role of chronic energy balance [102]. Secretin has been shown to have pleiotropic effects, and secretin receptors have been identified in the heart and kidney. Secretin has been shown to increase myocardial glucose uptake and increase renal filtration [103].
5.5. PYY, PP, and NPY
All three are members of the neuropeptide Y (NPY) family and have an amidated carboxyl terminus with a hairpin fold referred to as the PP fold, the latter required for the function of the NPY family. Peptide YY (PYY) hormone is primarily secreted by the L cells of the gut; there are two main forms of the hormone: PYY1–36 and PYY3–36, the latter created by DPP-IV by cleavage of the N-terminal amino acid residues. PYY immunoreactivity is also found within the central nervous system. There are 5Y receptors (Y1 to Y5, members of the G-protein-coupled receptor family), and PYY3–36 the major form in circulation, has a high affinity for Y2 and a lesser affinity for Y1 and Y5 receptors. In the rat, Y2 mRNA has been detected in the central nervous system [104]. The Y receptors are widely distributed in the central and peripheral tissues. Each Y receptor has a varying distribution across central and peripheral tissues [105,106]. Basal plasma PYY levels are low but rise in response to food, and postprandially they remain elevated for several hours. [107]. A high protein diet causes the greatest satiety and PYY release in humans [59]. Intravenous PYY3–36 decreased appetite in both healthy and obese subjects [108]. It has been suggested that PYY3–36 acts by modulating the activity of NPY and POMC neurons in the ARH of the hypothalamus [109], to decrease food intake. Other studies suggest that peripheral PYY3–36 may conduct satiety signals to the brain by the vagal afferent pathway [110].
Pancreatic polypeptide (PP) is a 36-amino acid peptide secreted by specialised pancreatic islet cells called F cells, and a small amount is found in the gut. Release of PP is postprandial and can remain elevated for up to 6 h following food intake [111]. PP, similar to PYY, is a satiety hormone, and though it binds all members of the Y receptor, it exhibits greater affinity for the Y4 receptor [112]. Investigations suggest possible mechanisms for PP-induced satiety are (i) delay in gastric emptying and increasing energy expenditure [113]; (ii) anorectic actions through increasing the activity of POMC neurons. The latter effect is thought to be a direct consequence of the activation of Y4 signalling [114]. Other data suggest that PP may regulate food intake by suppressing the orexigenic pathways by downregulating the orexigenic hormone orexin and simultaneously increasing the anorexigenic pathways by upregulating the anorexigenic brain-derived neurotrophic factor [115]. Following vagotomy, PP does not exert its anorectic effects, suggesting its function may depend on the vagus nerve [113].
The neuropeptide NPY, a 36 amino acid peptide, is a member of the NPY family and is abundantly expressed in the central and peripheral nervous systems. NPY is a member of the NPY family and is included in this section for completeness. NPY is found in the postganglionic sympathetic neurons, adrenal medulla, and peripheral tissues such as adipose tissue, pancreas, liver, skeletal muscle, and osteoblasts [116]. NPY is an orexigenic neuropeptide. NPY and PYY have identical affinities for the Y receptor [117]. In the central nervous system, the highest concentration of NPY is found in the ARH. The NPY receptors are found in many hypothalamic neurons, connecting the limbic and autonomic nervous systems with the hypothalamus [118]. It has been suggested that NPY has a role in decreasing the browning of white adipose tissue and thermogenesis in brown adipose tissue [119].
5.6. Cholecystokinin (CCK)
CCK producing cells are located primarily in the proximal small intestine in I cells. Studies suggest that the cells contained axon-like basal projections called neuropods. One hypothesis is that the axon-like processes drive hormone secretion and that hormone secretion is near enteric nerve cells [120]. CCK in plasma is a heterogeneous mixture of medium-sized peptides processed from the primary translational product preproCCK (115 aminoacids): CCK-58, CCK-33, CCK-22, CCK-8, and CCK-5, and the Y-77 site is mostly O-sulfated. In addition, CCK is a carboxyamidated peptide. CCK has been reported to be widely expressed in mammalian tissue, intestinal tissue, the cerebral and peripheral nervous system, extraintestinal endocrine glands, the urogenital tract, and the cardiovascular system [121]. In human plasma, using well characterised assays, Rehfeld [122] et al. showed that CCK-33 and CCK-22 were the most abundant and CCK-58 less abundant in human plasma, though there was significant species variation. Fasting levels of CCK averaged 1.0 pM and rose to 6 pM within 15 min following ingestion of a meal. Fat, protein, and amino acids were all stimulators of CCK secretion in contrast, glucose caused a smaller elevation in CCK levels [123]. In dogs, the half-life of CCK-58 was approximately 4 min and that of CCK-8 was 1.3 min, suggesting a short half-life for CCK [124]. The actions of CCK are mediated by two G-protein-coupled receptors, CCK1 and CCK2. CCK1 receptors are found in the gastrointestinal tract, myenteric plexus, and vagal afferents. CCK1 binds sulfated CCK with a higher affinity than non-sulfated CCK. CCK2 receptors are present in the stomach and brain and have similar affinities for sulfated and non-sulfated CCK [121].
Longer (CCK-58, CCK-33) as well as shorter forms are expressed in cerebral neurons and are neuronal peptides and potent neurotransmitters [125]. CCK interacts with CCK1 receptors localised in specialised regions of the hindbrain to induce satiety. In rat studies, CCK has been shown to inhibit the expression of orexigenic peptides (e.g., orexin [126]) in the hypothalamus and prevent stimulation by ghrelin [127]. When plasma CCK concentrations are low, vagal afferent neurons are associated with stimulation of food intake, while increased postprandial CCK vagal afferent neurons are linked to downregulation of food intake [128]. Other effects of CCK are to (i) stimulate exocrine pancreatic secretion, (ii) induce gall bladder contraction, and (iii) delay gastric emptying [129].
5.7. Somatostatin
Endocrine cell-secreting somatostatin (SST)-secreting cells are found in the stomach, intestine, and pancreas. SST is a cyclic oligonucleopeptide, with two forms of 14 aa and 28 aa. SST has five types of G-protein-coupled SST receptors (SSTR1 to SSTR5 and two isoforms SSTR2A and SSTR2B). About 5% of pancreatic cells are SST-secreting δ cells. SST is secreted in response to hypoglycaemia. Somatostatin secreted within the pancreas is a paracrine inhibitor of glucagon and insulin secretion [130].
About 90% of SST is located in the GIT D endocrinal cells, and 10% is located in the enteric nervous structures. One estimate is that GIT D cells contain 65% of the total body SST, the pancreatic cells 5%, and the rest in the CNS [130,131]. SST inhibits parietal cells, ghrelin cells, and gastrin through paracrine inhibition [131]. Removal of this restraint during ingestion of food increases acid and gastrin secretion [132]. SST has been shown to have a widespread distribution in the central nervous system of rodents and humans. Pleiotropic effects of brain-initiated SST have been described, and this includes GIT motility. In rodent studies, centrally injected SST stimulated food intake, though contradictory findings suggest that SST has an anorexigenic effect [133]. A summary of hormones and their functions in energy homeostasis are summarised in Table 2.
Table 2.
Hormonal functions in maintaining energy homeostasis.
6. Adipocyte Hormones
Adipocyte tissue is formed by mature adipocytes, fibroblasts, endothelial cells, adipocyte progenitors, and immune cells. Three main types of adipocyte exist in humans white, brown, and beige. White adipocytes can store energy, whereas mitochondria-rich brown adipocytes dissipate energy by thermogenesis. Both cell types are found in multiple fat depots forming the adipose organs. Adipocytes are also endocrine organs secreting adipokines, batokines (from brown adipose tissue), lipokines, microRNA, and inflammatory cytokines [153,154]. Leptin and adiponectin are the most known secretory products. Other adipose tissue secretory products have been reviewed extensively [153]. Beige adipocytes can also dissipate energy. White-to-brown conversion is in response to certain stimuli, such as chronic cold exposure or in response to β3 adrenergic stimulation [155]. In recent years, it has emerged that white, brown, and beige adipose tissue play a role in the regulation of metabolic health through the secretion of several adipokines. In addition, adipocytes respond to hormones to carry out lipogenesis and lipolysis. Due to these functions, adipose tissue is considered a vital regulator of metabolic homeostasis.
6.1. Leptin
Leptin is a 167 aa peptide that is primarily secreted by the adipose tissue. It is also found in several other tissues, including the lymphoid tissue [156]. Leptin is secreted in a circadian rhythm, with low levels at mid-afternoon and highest level at midnight. Levels increase when adipose tissue mass increases; decreasing food intake and weight loss lead to a decrease in leptin levels and consequently an increase in food intake. This serves to control the adipose tissue mass within a narrow range by linking the changes in energy stores and physiological responses. The principal site of action for leptin is the brain. Further sites established outside the brain for the direct action of leptin are the immune cells. Factors stated as controlling leptin secretion are insulin, glucocorticoids, and catecholamines [157]. Controversy exists over whether exercise has an effect on leptin levels [158,159]. In rodent studies, there is a cold-induced suppression of leptin gene expression in WAT, and it is likely that this is mediated by the sympathetic nervous system [160].
Leptin receptors (LepR) are in the class 1 cytokine receptor family with a single membrane spanning domain. Several isoforms of the receptor (LepRb is the longest) have been identified. These isoforms have homologous extracellular binding domains, but their intracellular domains vary in length due to alternative splicing. Leptin receptors activate several transduction pathways. The pathways include Janus kinase 2 (JAK2), signal transducer and activator of transcription 3 (STAT3), insulin receptor substrate (IRS)-phosphatidylinositol 3-kinase (PI3K), SH2-containing protein tyrosine phosphatase 2 (SHP2)-mitogen-activated protein kinase (MAPK), and 5’ adenosine monophosphate activated protein kinase (AMPK)/acetyl-CoA carboxylase (ACC), as well as other pathways [2]. LepR is widely distributed in several peripheral tissues. This raises questions about the role of leptin in these tissues. Leptin appears to have a range of roles in the peripheral tissues, which include the pancreas, liver, skeletal muscle, adipose tissues, immune cells, and cardiovascular system. Its effect may depend on whether leptin is acting centrally or directly on peripheral tissues [161]. Leptin is increased in obesity, and one of the major risk factors for obesity is leptin resistance, which leads to a decrease in satiety caused by disruption of signalling pathways of leptin. The changes in signalling pathways are triggered by inflammation [151].
In the ARH, leptin decreases food intake by activating POMC and inhibiting AgRP/NPY neurons. During fasting, the fall in leptin stimulates AgRP/NPY neurons and suppresses POMC to increase food intake and decrease energy expenditure [2]. In rats, leptin increased sympathetic nerve activity in the BAT and adrenal glands. Sympathetic activation of BAT would be expected to enhance thermogenesis [162]. One suggested mechanism is through increased expression of the uncoupling protein and increased thermogenesis [163]. Rodent studies suggest that leptin and insulin can act synergistically to promote WAT browning by acting on POMC [164]. The coupling of leptin levels to adaptations in food intake and energy expenditure enables long term weight maintenance. It has been stated that the organism’s sense of nutritional state (adipose tissue mass) is conveyed by leptin [165].
6.2. Adiponectin
Adiponectin is mainly synthesised in adipocytes. The monomeric protein is postranslationally modified into different multimers. The plasma adiponectin half-life was 2.5 h. Unlike other adipokines, adiponectin blood concentrations are inversely associated with fat mass. Weight loss due to caloric restriction in obese humans increases adiponectin tissue gene expression and plasma concentrations towards normal lean levels. Adiponectin increases insulin sensitivity and decreases hepatic glucose output. AdipoR1 and AdipoR2 were identified as receptors for adiponectin. Both receptors are expressed widely, though AdipoR1 is more highly expressed in skeletal muscle and AdipoR2 is more restricted to the liver. Adiponectin’s insulin sensitising actions are mainly in the liver and skeletal muscle [166]. In mice, adiponectin decreases glucose production in the liver [167] and increases fatty acid oxidation in rodent skeletal muscle [168]. Transgenic mice lacking leptin but overexpressing adiponectin had significantly higher levels of adipose tissue, suggesting that adiponectin promoted the storage of triglycerides in the adipose tissue [169]. AdipoR1 and AdipoR2 are expressed in human and rat pancreatic cells [170]. Studies on human pancreatic cells suggest that, in addition to its anti-apoptotic action, adiponectin has another effect on beta cells by potentiating insulin secretion [171]. Chronic cold exposure led to an elevated production of adiponectin in subcutaneous WAT. Adiponectin knockout mice showed blunted browning of subcutaneous adipose tissue in response to cold exposure, suggesting a role for adiponectin in cold-induced browning of subcutaneous WAT [172]. Studies on mice suggest that adiponectin is an orexigenic hormone and that it may stimulate the phosphorylation of AMP-activated protein kinase in the ARH [173]. Adiponectin is also an anti-inflammatory agent and, as a result, has protective effects on the vasculature, lung, heart, and colon [166]. The role of several hormones in energy homeostasis is summarised in Figure 3.
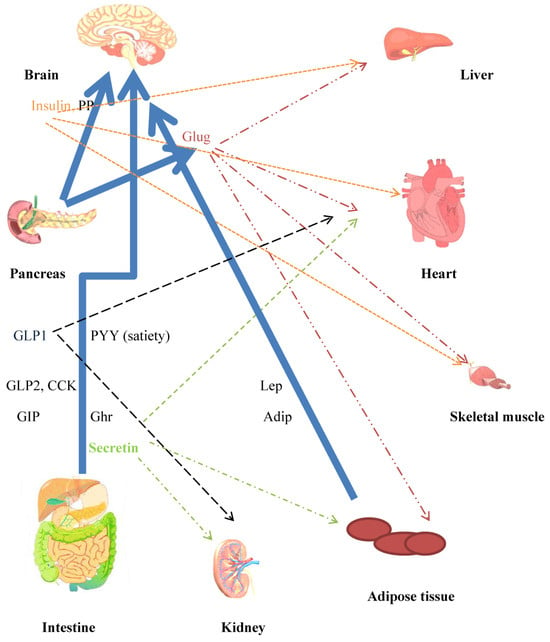
Figure 3.
Crosstalk between peripheral and central mechanisms that contribute towards whole body energy homeostasis. Major mediators of mechanisms for controlling energy homeostasis. Neuroendocrine hormones are released from the gut in response to nutrient intake. Incretins released from the gut have several effects, in addition to increasing insulin secretion. The CNS is a target for the anorexigenic/orexigenic and satiety effects of the hormones. Blue unbroken arrows show routes for hormone secretion. Effects on peripheral tissues, heart liver, skeletal muscle, and adipose tissue are indicated by broken arrow lines, color coded to agree with hormones. Hepatocytes and adipocytes are implicated in glucose/lipid and amino acid metabolism. Adipocytes secrete the adipokines leptin and adiponectin. Insulin, leptin, and secretin are hormones concerned in WAT browning and BAT thermogenesis. CCK (decreases food intake), Lep: leptin (anorexigenic), Adip: adiponectin (orexigenic), Ghr: ghrelin (orexigenic), Sec: secretin (satiety hormone), Glug: glucagon.
7. Genetic Mutations That Affect Energy Homeostasis
Individual contributions to mechanisms that control energy homeostasis are illustrated in mutations that affect metabolic function. Monogenic obesity has contributed to the understanding of the process of energy/metabolic homeostasis.
- (i)
- POMC mutations: patients with complete loss of POMC gene function were diagnosed on the basis of secondary hypocortisolism, red hair, and extreme obesity [174].
- (ii)
- Leptin and leptin receptor mutations: associated with morbidly obese phenotypes and disturbances in metabolic and immune functions [175].
- (iii)
- MC4R mutations are linked to severe obesity [176].
- (iv)
- Ghrelin receptor mutations: patients present with short stature and obesity [177].
- (v)
- Loss of function of PCSK1 variants (encoding prohormone convertase subtilisin/kexin type 1) with a deleterious effect on PC1/3 proconveratse activity contributes to obesity [178].
- (vi)
- NPY-variants within the gene are associated with obesity [179].
- (vii)
- Brain-derived neutrotropic factor (BDNF, a neurotropin) and its receptor tropomyosin receptor kinase B (Trkb) have been shown to act in the brain to regulate neuronal survival, growth, and plasticity. BDNF and TrkB regulate several physiological processes. BDNF has been suggested to play a role in weight loss, energy expenditure, and thermogenesis. Mutations in TrkB have been associated with obesity [180].
- (viii)
- Polygenic forms of obesity: genome wide association studies (GWAS) and next-generation sequencing (NGS) have increased awareness of the polygenic forms of obesity. GWAS studies have suggested that the FTO gene (widely expressed in tissues and the brain) is a significant genome contributing to obesity. A total of 127 sites in the human gene have been linked to obesity through GWAS studies. Some of the genes implicated in the development of obesity are: uncoupling proteins in the brown adipose tissue, the SLC6A14 gene, a transporter of amino acids [181].
- (ix)
- Syndromic forms of obesity have been reviewed [182]. Some examples are: Prader–Willi syndrome (PWS), Down syndrome, Bardet–Biedl syndrome, fragile X syndrome, Alstrom syndrome, and Cornelia de Lange syndrome.
8. Bariatric Surgery
Obesity is considered a chronic disease that increases morbidity and mortality. Bariatric surgery (surgical procedures for obesity treatment—referred to interchangeably as bariatric surgery, weight loss surgery, metabolic surgery, or metabolic/bariatric surgery) has been shown to be effective in achieving weight loss, and studies show a lower mortality compared to control subjects under non-surgical conventional treatment [183,184]. Three common forms of bariatric surgery are sleeve gastrectomy (SG), adjustable gastric band (AGB), and Roux-en-Y-gastric bypass (RYGB). The surgical procedures with advantages and disadvantages are summarised in Figure 4 [185]. Guidelines suggest that bariatric surgery should be considered in individuals with BMI ≥ 40 kg/m2 or BMI ≥ 35–40 kg/m2 with co-morbidities [185,186,187] that are anticipated to improve with weight loss. They further address optimised care before, during, and following surgery [185,186,187], https://www.nice.org.uk/guidance/cg189 (accessed on 23 February 2024). Patients following bariatric surgery are required to have nutritional surveillance and laboratory screening for nutritional deficiencies. In the Swedish Obese Subjects study, surgically treated patients had greater weight loss, more physical activity, and decreased energy intake compared to control subjects [188]. One guideline mentions a review for monogenic and syndromic forms of obesity if historical and physical findings require further investigations and decisions made on a case by case basis [185]. For polygenic obesity, common variants have been identified; each variant has a small effect on body BMI, with interaction with the environment playing a key role. Variants in or near genes that result in severe obesity may have a more subtle impact on individual BMI. Patient genotype may allow a more exact understanding of patient obesity and permit a more exact treatment [189].
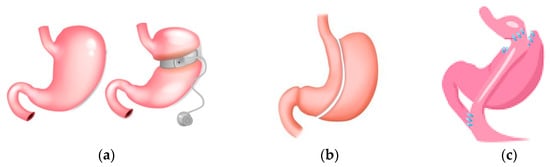
Figure 4.
Surgical procedures common in bariatic surgery. (a) Gastric banding: A silicone band that is adjustable is placed below the gastroesophageal junction; this suppresses hunger. The level of restriction can be adjusted by the amount of fluid in the band. It is a reversible method with the lowest morbidity and mortality. Disadvantages are gastric band leak, gastric band dilation, and the highest reoperation rate. (b) Gastric sleeve: Gastric volume is reduced by the removal of a large portion of the stomach. The disadvantages are staple line leaks, gastroesophageal reflux disease and dilation of the remaining gastric remnant. (c) Gastric bypass (RYGB): Gastric volume is reduced, and the small gastric pouch created is joined to the jejunum. Nutrients flow from the gastric pouch to the proximal jejunum, with bypassing of the lower stomach and duodenum. Disadvantages are staple line leaks, dumping syndrome, and gallstones.
Briefly, bariatric surgery addresses health and quality of life when lifestyle changes cannot decrease weight adequately to prevent complications associated with obesity. However, bariatric surgery has to be balanced alongside the risks associated with surgery and consequent mortality/morbidity, prospective nutritional deficiency, weight regain, and the need for lifelong support and medical monitoring. Nutrition surveillance includes iron, folate, vitamin B12, vitamin D, calcium, fat-soluble vitamins, and trace elements [190].
8.1. Bariatric Surgery and Absorption
Following surgery, the gastrointestinal physiology is altered. Mechanisms that lead to weight loss are still incompletely understood. Gastric pouch emptying is altered, though further studies are required, including the variables gastric pouch size and emptying and meal size and texture [191]. This may lead to changes in the postoperative patient driven by changes in diet, changes in anatomy and physiology of the GIT tract, which may be based on the type of procedure performed and may lead to changes in the digestion and absorption of food and changes in nutritional state. Changes in gastric microbiota are found in the obese state compared to lean microbiota composition. Alterations in the microbiota of obese patients following bariatric surgery have been observed. Accelerated gastric transit time and decreased gastric acid production can contribute to changes in gut microbiota. The extent to which gut microbiota affect the host BMI is difficult to determine [192]. Both RYGB and SG have been shown to increase systemic bile acid concentrations in the circulation. One hypothesis is that the bile acid receptor TGR5 shows an increased response to elevations of bile acid and may play a role in GLP-1 secretion [193].
Physiological changes following bariatric surgery are still to be investigated extensively. Studies have either investigated SG and RYGB patients separately during ongoing weight loss, during the weight stable phase, or studies lacked a control group. There are multifactorial mechanisms affecting weight loss following RYGB/SG surgery. The favourable effects are not caused by malabsorption and food restriction alone but also by changes in GIT hormones.
8.2. Gastrin
Gastrin levels were reported to fall following RYGB, increase during GS, and show no change during AGB. Histological changes associated with RYGB are atrophic gastritis and chronic gastritis, and patients are given proton pump inhibitors postoperatively [194].
8.3. Ghrelin
It has been shown that ghrelin levels fall following RYGB and GS, though studies suggest a long term increase in ghrelin levels following RYGB in some patients [194,195]. Peterli et al. [196] report that in patients following GS, ghrelin showed decreased levels at 1 year, though slightly higher than at 3 months.
8.4. Cholecytokinin
Studies have shown an increase in CCK levels postprandially after RYGB; in one study, GS showed a higher response in CCK than RYGB [194].
8.5. GLP-1
Studies suggest an increase in the satiety hormones GLP-1 and PYY, as well as oxyntomodulin, following RYGB. One suggestion is that the rise in gut hormones in RYGB can be related to the rate of pouch emptying and the direct delivery of nutrients into the proximal jejunum. Increased satiety hormones appear to play a role in optimal weight loss. Postprandial GLP-1 has been shown to be elevated compared to pre-operative levels at different time points post-surgery [195]. Increases in GLP-1 and PYY are also seen following SG [196].
8.6. GLP2
Postprandial GLP2 levels have been shown to increase following RYGB surgery, though no significant differences were demonstrated between fasting levels in preoperative and postoperative patients [195]. Cazzo et al. [197] suggest an increase in GLP2 following SG. Further investigations on the effects of bariatric surgery on GLP2 are required.
8.7. PYY
Postprandial plasma levels of PYY rise in patients following RYGB, though reports of increased PYY are not always consistent, suggesting further work is desirable [195]. An increase in PYY has been shown to occur following AGB, GS, and RYGB [194]. In a comparison of good versus poor responders in RYGB for weight loss, suboptimal PYY and GLP-1 postprandial responses were associated with poor responses [198].
8.8. GIP
In a meta-analysis, Gao et al. [199] state that fasting GIP levels are significantly reduced following RYGB surgery and that the decrease was more pronounced in diabetic subjects. They acknowledge the need for randomised prospective studies with larger sample sizes to confirm these findings.
8.9. PP
PP has been generally reported as unchanged after RYGB and SG, though some studies have reported lower fasting levels post-RYGB [200].
8.10. Adipokines
Following both RYGB and GS, adiponectin levels increased and leptin levels decreased [201]. This was paralleled by improved insulin sensitivity and normalisation of lipid profiles [202].
9. Gut Hormone Changes after Metabolic Bariatric Surgery and Their Contribution to Weight Loss
The alterations in gut hormone levels following bariatric surgery have been suggested as an important mediator of eating behaviours favouring weight loss in the long term. The main changes in gut hormones and adipokines are shown in Table 3. The increase in GLP-1 and PYY is thought to increase satiety and reduce hunger. Studies suggest that postprandial glucose and protein absorption and gut hormone secretions differ between SG and RYGB. RYGB was characterised by increased absorption of glucose and amino acids, whereas protein metabolism after SG did not differ very significantly from controls. In one study, there was a marked difference in gastric pouch emptying between SG and RYGB. The authors suggest that diverging rates of intestinal nutrient entry and absorption may explain the differences in the secretion of GIT hormones [203]. Other studies confirm the higher postprandial levels of GLP-1 following RYGB compared to SG [204], though other authors report a lack of difference in GLP-1 secretion between RYGB and SG [205]. The differences between studies can be attributed to various factors, including differences in surgical techniques and different limb lengths in RYGB. The role of post-surgical vagal nerve fibres in alterations to changes in food intake and GIT hormone secretion has not been completely investigated [206].
Table 3.
Summary of selected changes following metabolic bariatric surgery.
It has been suggested that each bariatric procedure has its own unique hormone profile. Differences in the rate of gastric pouch emptying, food intake restriction, and changes in the gut hormone profile may play a synergistic role in increasing satiety and reducing post-operative food intake. A few studies suggest that increased postprandial GLP-1 and PYY secretion 6 years after RYGB was associated with increased weight loss. The causality of the association has been queried, and it has been suggested that it has to be interpreted in the context of other hormone levels, e.g., a reduction in insulin and leptin levels. Studies that report an increase in fasting ghrelin following weight loss suggest that ghrelin changes are a consequence, not a cause of weight loss [207]. A study using the blockade of the actions of both GLP-1 and PYY suggests that both hormones are involved in inhibiting food intake following RYGB [208].
10. Postprandial Hypoglycaemia following Surgery
Postprandial hyperinsulinemic hypoglycaemia is a complication of RYGB and GS [209]. Hypoglycaemia may accompany dumping syndrome [210], though dumping syndrome can often be missed [211]. Studies further suggest that postprandial hypoglycaemia is not exclusively postprandial [212]. Symptoms of hypoglycaemia can vary from mild (sweating, weakness, and dizziness) to more severe neuroglycopenic symptoms, which include seizures, coma, and loss of consciousness [213].
Several hypotheses have been put forward to explain the pathophysiology of post-bariatric surgery hypoglycaemia
- (i)
- Rapid gastric emptying, which results in a steep rise in plasma glucose compared to non-surgical controls [214].
- (ii)
- Studies that used GLP-1R antagonists corrected the underlying hypoglycaemia in symptomatic patients, suggesting a role for GLP-1 in the underlying pathophysiology [213]. In other studies following RYGB individuals with post-RYGB hypoglycaemia had greater postprandial GLP-1 and insulin concentrations but not GIP when compared with individuals who had the same bariatric surgery but did not develop hypoglycaemia [215]. One hypothesis, supported by cross-sectional studies, is that postprandial hyperinsulinemia after RYGB can be attributed to the combined effects of more rapid nutrient transit from the gastric pouch to the gut coupled with an increased incretin effect [214,216]. One study reported decreased insulin clearance in the latter portion of postprandial hypoglycaemia contributed to relative hyperinsulinemia. A multifactorial model for the glucose dysregulation in postprandial hypoglycaemia has been suggested [217].
- (iii)
- The possibility of other factors that may influence glucose metabolism has been suggested: reduced counter-regulatory hormone response, increased insulin sensitivity following weight loss, altered bile acid metabolism [214].
Various treatment modalities have been proposed and are covered in detail elsewhere, and a summary will be included in this review: (i) dietary modification to include complex carbohydrates; (ii) medical treatment with acarbose, octreotide, diazoxide, and calcium channel blockers; (iii) surgical treatment, latter only for severe refractory hypoglycaemia. Although the use of GLP-1 receptor agonists is counterintuitive, authors have suggested its use if other alternatives are not successful, though further work is needed prior to the use of these drugs [218].
11. Weight Regain following Bariatric Surgery
Following bariatric surgery, it is now accepted that a proportion of patients incur weight gain during long term follow-up. The reported prevalence of weight gain will depend on the definition and time since surgery. One study reports that during time points from 2–5 years, the weight regain fluctuated from 2.53–94.18% depending on the definition of weight regain [219].
Weight loss due to a low calorie diet results in significant reductions in levels of leptin, PYY, CCK amylin, and insulin and increases in levels of ghrelin, GIP, and PP [220]. PYY, GLP-1, CCK, PP and amylin inhibit food intake, while ghrelin stimulates hunger. In contrast, following bariatric surgery, exclusion of the foregut leads to an increase in hormones that promote satiety as well as downregulation of ghrelin, which stimulates food intake [221]. However, there is controversy over the role of appetite related hormones in weight loss relapse [222]. Additional lifestyle modifications that change eating habits and physical activity can improve weight loss outcomes [223].
A case series with rare biallelic mutations in the leptin-melanocortin pathway (leptin, MC4R, and POMC genes) showed a limited benefit of bariatric surgery [224]. Cooiman et al., [225], suggest that heterozygous mutations in POMC and PCSK1 do not affect the effectiveness of bariatric surgery in the first two years of follow-up. Other authors confirm that carriers of a heterozygous variant in the leptin-melanocortin pathway have a progressive and significant weight regain in the mid- and long-term after RYGB [226]. Studies that combine several SNPs suggest that weight loss and long term weight regain after bariatric surgery may be influenced by multiple genetic variants [227]. A combination of several genes and the development of a genetic risk score may have a predictive value for an individual’s predisposition to obesity [228]. Bonetti et al. [229], suggest that a next-generation sequencing panel (NGS), which include diagnostic and candidate genes, could play a role in predicting the outcomes of bariatric surgery.
In a recent review, van der Meer et al. [230] suggest that there is limited evidence supporting a role for genetic variants in outcomes following bariatric surgery. They suggest that more evidence and studies are needed to replicate findings across sexes and ethnicities. Identifying carriers of deleterious genes that may lead to suboptimal short term weight loss or long term weight regain might help implement individualised therapies that take into account impaired genetic pathways that are less functional in these individuals. Early genetic evaluation can identify treatable obesity and establish more personalised management.
The recent development of therapeutic options for the treatment of genetic obesity has increased treatment alternatives for genetic obesity other than lifestyle management and bariatric surgery. Options available are (i) GLP-1 analogues, (ii) MC4R agonists, and (iii) recombinant leptin. These drugs are reviewed extensively elsewhere [12,231].
12. Type 2 Diabetes Mellitus (T2DM) and Bariatric Surgery
Obesity is a risk factor for T2DM as well as cardiovascular disease, musculoskeletal disease, and many cancers [232]. Bariatric surgery and weight loss can lead to T2DM remission [233]. Surgery has been shown to be more effective than medical treatment for long term control of T2DM in obese patients. The authors suggest continued monitoring of patients as there is a potential relapse of hyperglycemia [234]. Population-based data show that bariatric surgery, RYGB and GS, increases the chance of remission of T2DM, though the risks of surgery have to be balanced against its benefits [235]. Patient compliance with lifestyle modifications post-bariatric surgery is linked to weight loss outcomes and comorbidity resolution [236]. Following bariatric surgery, markedly enhanced insulin secretion is observed. This has been explained by a rapid transit of nutrients to the distal part of the small intestine and an increased postprandial secretion of GLP-1 [237]. Both weight loss-dependent [238] and weight loss-independent [239] changes have been hypothesised to yield improvements in glucose metabolism. Postprandial increases in GLP-1 [240] and (in rat studies) improved pancreatic β cell function following RYGB can contribute to T2D remission [241]. Mice studies suggest an improvement in hepatic and muscle insulin sensitivity following SG [242]. It has been hypothesised that gut microbiome changes and bile acid signalling post-bariatric surgery may lead to improved insulin sensitivity [243].
Relapse of diabetes mellitus can occur following bariatric surgery and treatment with anti-diabetic drugs; sodium-glucose cotransporter 2 inhibitors [244] and GLP-1 receptor agonists are options to prolong diabetes remission or extend treatment in patients with less effective glycemic control [245]. These medications have been reviewed in previous articles [244,245].
13. Conclusions
Several interconnected neural circuits in the brain control both energy needs and feeding behaviours. Neurons that respond to satiety are the POMC neurons that produce α-MSH, which binds to MCR4 receptors to decrease food intake. Conversely, decreased food intake triggers NPY/AgRP neurons to increase inhibition of POMC neurons, inhibition of α-MSH, and an increase in feeding response. The neurons are located in the ARH, which is considered an important centre for the integration of hunger and satiety [14]. The brain, mainly the hypothalamus, integrates the gut-brain axis signals, which are the many gut peptides that influence metabolic homeostasis. Neural signalling occurs through the vagus afferent neurons. Intestinal microbiota can further influence the gut-brain axis [246]. Obesity remains a complex, multifactorial disease that is now a major public health issue. Bariatric surgery remains an effective treatment with weight loss and metabolic benefits. A better understanding of the mechanisms and benefits associated with bariatric surgery is crucial for future pharmacological interventions in patients with morbid obesity. Replicating gut hormone changes through pharmacological means holds promise for future medical interventions in bariatric surgery. Further studies are needed to clarify the changes in pathophysiological pathways as a consequence of bariatric surgery. Knowledge of patients genotypes may allow for a more personalised treatment of obesity or the establishment of prevention strategies. Understanding the genetics of obesity may lead us towards new therapeutic targets and a more personalised and precise medicine for obesity. Multimodal treatments that include lifestyle management, bariatric surgery, and pharmacological treatments can be routes for obesity management. Combination co-agonist treatments and clinical trials are in progress [247]. The physiology of food consumption, weight control, and energy expenditure may be required for a more successful integrated understanding of obesity and its treatment in the future.
Funding
This research received no external funding.
Institutional Review Board Statement
This paper is a narrative review and did not require ethical approval.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available in the references cited and also on request from the corresponding author.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Gribble, F.M.; Reimann, F. Function and mechanisms of enteroendocrine cells and gut hormones in metabolism. Nat. Rev. Endocrinol. 2019, 15, 226–237. [Google Scholar] [CrossRef] [PubMed]
- Park, H.-K.; Ahima, R.S. Physiology of leptin: Energy homeostasis, neuroendocrine function and metabolism. Metabolism 2015, 64, 24–34. [Google Scholar] [CrossRef]
- Vohra, M.S.; Benchoula, K.; Serpell, C.J.; Hwa, W.E. AgRP/NPY and POMC neurons in the arcuate nucleus and their potential role in treatment of obesity. Eur. J. Pharmacol. 2022, 915, 174611. [Google Scholar] [CrossRef] [PubMed]
- Ward, Z.J.; Bleich, S.N.; Cradock, A.L.; Barrett, J.L.; Giles, C.M.; Flax, C.; Long, M.W.; Gortmaker, S.L. Projected U.S. state-level prevalence of adult obesity and severe obesity. N. Engl. J. Med. 2019, 381, 2440–2450. [Google Scholar] [CrossRef]
- Ruban, A.; Stoenchev, K.; Ashrafian, H.; Teare, J. Current treatments for obesity. Clin. Med. 2019, 19, 205–212. [Google Scholar] [CrossRef]
- Sohn, J.-W. Network of hypothalamic neurons that control appetite. BMB Rep. 2015, 48, 229–233. [Google Scholar] [CrossRef]
- Zhan, C.; Zhou, J.; Feng, Q.; Zhang, J.-E.; Lin, S.; Bao, J.; Wu, P.; Luo, M. Acute and Long-Term Suppression of Feeding Behavior by POMC Neurons in the Brainstem and Hypothalamus, Respectively. J. Neurosci. 2013, 33, 3624–3632. [Google Scholar] [CrossRef] [PubMed]
- Mountjoy, K.G. Pro-Opiomelanocortin (POMC) Neurones, POMC-Derived Peptides, Melanocortin Receptors and Obesity: How Understanding of this System has Changed Over the Last Decade. J. Neuroendocr. 2015, 27, 406–418. [Google Scholar] [CrossRef]
- Qiu, J.; Zhang, C.; Borgquist, A.; Nestor, C.C.; Smith, A.W.; Bosch, M.A.; Ku, S.; Wagner, E.J.; Rønnekleiv, O.K.; Kelly, M.J. Insulin Excites Anorexigenic Proopiomelanocortin Neurons via Activation of Canonical Transient Receptor Potential Channels. Cell Metab. 2014, 19, 682–693. [Google Scholar] [CrossRef]
- Quarta, C.; Claret, M.; Zeltser, L.M.; Williams, K.W.; Yeo, G.S.H.; Tschöp, M.H.; Diano, S.; Brüning, J.C.; Cota, D. POMC neuronal heterogeneity in energy balance and beyond: An integrated view. Nat. Metab. 2021, 3, 299–308. [Google Scholar] [CrossRef]
- Yoon, N.A.; Diano, S. Hypothalamic glucose-sensing mechanisms. Diabetologia 2021, 64, 985–993. [Google Scholar] [CrossRef] [PubMed]
- Sohn, Y.B. Genetic obesity: An update with emerging therapeutic approaches. Ann. Pediatr. Endocrinol. Metab. 2022, 27, 169–175. [Google Scholar] [CrossRef]
- Baldini, G.; Phelan, K.D. The melanocortin pathway and control of appetite-progress and therapeutic implications. J. Endocrinol. 2019, 241, R1–R33. [Google Scholar] [CrossRef] [PubMed]
- Denis, R.G.; Joly-Amado, A.; Webber, E.; Langlet, F.; Schaeffer, M.; Padilla, S.L.; Cansell, C.; Dehouck, B.; Castel, J.; Delbès, A.-S.; et al. Palatability Can Drive Feeding Independent of AgRP Neurons. Cell Metab. 2015, 22, 646–657, Erratum in Cell Metab. 2017, 25, 975. [Google Scholar] [CrossRef] [PubMed]
- Benite-Ribeiro, S.A.; Putt, D.A.; Soares-Filho, M.C.; Santos, J.M. The link between hypothalamic epigenetic modifications and long-term feeding control. Appetite 2016, 107, 445–453. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, M.W.; Woods, S.C.; Porte, D., Jr.; Seeley, R.J.; Baskin, D.G. Central nervous system control of food intake. Nature 2000, 404, 661–671. [Google Scholar] [CrossRef] [PubMed]
- Khandekar, N.; Berning, B.A.; Sainsbury, A.; Lin, S. The role of pancreatic polypeptide in the regulation of energy homeostasis. Mol. Cell. Endocrinol. 2015, 418 Pt 1, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Pais, R.; Gribble, F.M.; Reimann, F. Stimulation of incretin secreting cells. Ther. Adv. Endocrinol. Metab. 2016, 7, 24–42. [Google Scholar] [CrossRef]
- El, K.; Campbell, J.E. The role of GIP in α-cells and glucagon secretion. Peptides 2020, 125, 170213. [Google Scholar] [CrossRef]
- McLean, M.A.; Wong, C.K.; Campbell, J.E.; Hodson, D.J.; Trapp, S.; Drucker, D.J. Revisiting the Complexity of GLP-1 Action from Sites of Synthesis to Receptor Activation. Endocr. Rev. 2021, 42, 101–132. [Google Scholar] [CrossRef]
- Chaudhri, O.; Small, C.; Bloom, S. Gastrointestinal hormones regulating appetite. Philos. Trans. R. Soc. B Biol. Sci. 2006, 361, 1187–1209. [Google Scholar] [CrossRef]
- Nauck, M.A.; Quast, D.R.; Wefers, J.; Pfeiffer, A.F.H. The evolving story of incretins (GIP and GLP-1) in metabolic and cardiovascular disease: A pathophysiological update. Diabetes Obes. Metab. 2021, 23 (Suppl. S3), 5–29. [Google Scholar] [CrossRef]
- El-Jamal, N.; Erdual, E.; Neunlist, M.; Koriche, D.; Dubuquoy, C.; Maggiotto, F.; Chevalier, J.; Berrebi, D.; Dubuquoy, L.; Boulanger, E.; et al. Glugacon-like peptide-2: Broad receptor expression, limited therapeutic effect on intestinal inflammation and novel role in liver regeneration. Am. J. Physiol. Liver Physiol. 2014, 307, G274–G285. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Wang, M.; Wen, Z.; Lu, Z.; Cui, L.; Fu, C.; Xue, H.; Liu, Y.; Zhang, Y. GLP-1 Receptor Agonists: Beyond Their Pancreatic Effects. Front. Endocrinol. 2021, 12, 721135. [Google Scholar] [CrossRef] [PubMed]
- Flint, A.; Raben, A.; Ersbøll, A.; Holst, J.; Astrup, A. The effect of physiological levels of glucagon-like peptide-1 on appetite, gastric emptying, energy and substrate metabolism in obesity. Int. J. Obes. 2001, 25, 781–792. [Google Scholar] [CrossRef]
- Heppner, K.M.; Kirigiti, M.; Secher, A.; Paulsen, S.J.; Buckingham, R.; Pyke, C.; Knudsen, L.B.; Vrang, N.; Grove, K.L. Expression and Distribution of Glucagon-Like Peptide-1 Receptor mRNA, Protein and Binding in the Male Nonhuman Primate (Macaca mulatta) Brain. Endocrinology 2015, 156, 255–267. [Google Scholar] [CrossRef]
- Vrang, N.; Larsen, P.J. Preproglucagon derived peptides GLP-1, GLP-2 and oxyntomodulin in the CNS: Role of peripherally secreted and centrally produced peptides. Prog. Neurobiol. 2010, 92, 442–462. [Google Scholar] [CrossRef] [PubMed]
- Adriaenssens, A.; Gribble, F.; Reimann, F. The glucose-dependent insulinotropic polypeptide signaling axis in the central nervous system. Peptides 2020, 125, 170194. [Google Scholar] [CrossRef]
- Vrang, N.; Hansen, M.; Larsen, P.J.; Tang-Christensen, M. Characterization of brainstem preproglucagon projections to the paraventricular and dorsomedial hypothalamic nuclei. Brain Res. 2007, 1149, 118–126. [Google Scholar] [CrossRef]
- Heimbürger, S.M.; Bergmann, N.C.; Augustin, R.; Gasbjerg, L.S.; Christensen, M.B.; Knop, F.K. Glucose-dependent insulinotropic polypeptide (GIP) and cardiovascular disease. Peptides 2020, 125, 170174. [Google Scholar] [CrossRef]
- Nauck, M.A.; Heimesaat, M.M.; Behle, K.; Holst, J.J.; Nauck, M.S.; Ritzel, R.; Hüfner, M.; Schmiegel, W.H. Effects of Glucagon-Like Peptide 1 on Counterregulatory Hormone Responses, Cognitive Functions, and Insulin Secretion during Hyperinsulinemic, Stepped Hypoglycemic Clamp Experiments in Healthy Volunteers. J. Clin. Endocrinol. Metab. 2002, 87, 1239–1246. [Google Scholar] [CrossRef] [PubMed]
- Brubaker, P.L.; Drucker, D.J. Minireview: Glucagon-Like Peptides Regulate Cell Proliferation and Apoptosis in the Pancreas, Gut, and Central Nervous System. Endocrinology 2004, 145, 2653–2659. [Google Scholar] [CrossRef] [PubMed]
- Meier, J.J.; Nauck, M.A.; Pott, A.; Heinze, K.; Goetze, O.; Bulut, K.; Schmidt, W.E.; Gallwitz, B.; Holst, J.J. Glucagon-Like Peptide 2 Stimulates Glucagon Secretion, Enhances Lipid Absorption, and Inhibits Gastric Acid Secretion in Humans. Gastroenterology 2006, 130, 44–54. [Google Scholar] [CrossRef] [PubMed]
- Meier, J.J.; Goetze, O.; Anstipp, J.; Hagemann, D.; Holst, J.J.; Schmidt, W.E.; Gallwitz, B.; Nauck, M.A. Gastric inhibitory polypeptide does not inhibit gastric emptying in humans. Am. J. Physiol. Endocrinol. Metab. 2004, 286, E621–E625. [Google Scholar] [CrossRef]
- Hellström, P.M.; Näslund, E.; Edholm, T.; Schmidt, P.T.; Kristensen, J.; Theodorsson, E.; Holst, J.J.; Efendic, S. GLP-1 suppresses gastrointestinal motility and inhibits the migrating motor complex in healthy subjects and patients with irritable bowel syndrome. Neurogastroenterol. Motil. 2008, 20, 649–659. [Google Scholar] [CrossRef]
- Brubaker, P.L. Glucagon-like Peptide-2 and the Regulation of Intestinal Growth and Function. Compr. Physiol. 2018, 8, 1185–1210. [Google Scholar] [CrossRef]
- Miyawaki, K.; Yamada, Y.; Ban, N.; Ihara, Y.; Tsukiyama, K.; Zhou, H.; Fujimoto, S.; Oku, A.; Tsuda, K.; Toyokuni, S.; et al. Inhibition of gastric inhibitory polypeptide signaling prevents obesity. Nat. Med. 2002, 8, 738–742. [Google Scholar] [CrossRef]
- Kim, S.-J.; Nian, C.; McIntosh, C.H.S. Activation of Lipoprotein Lipase by Glucose-dependent Insulinotropic Polypeptide in Adipocytes. J. Biol. Chem. 2007, 282, 8557–8567. [Google Scholar] [CrossRef]
- Kim, S.-J.; Nian, C.; McIntosh, C.H. GIP increases human adipocyte LPL expression through CREB and TORC2-mediated trans-activation of the LPL gene. J. Lipid Res. 2010, 51, 3145–3157. [Google Scholar] [CrossRef]
- McIntosh, C.H.; Widenmaier, S.; Kim, S. Glucose-dependent insulinotropic polypeptide signaling in pancreatic β-cells and adipocytes. J. Diabetes Investig. 2012, 3, 96–106. [Google Scholar] [CrossRef]
- El Bekay, R.; Coín-Aragüez, L.; Fernández-García, D.; Oliva-Olivera, W.; Bernal-López, R.; Clemente-Postigo, M.; Delgado-Lista, J.; Diaz-Ruiz, A.; Guzman-Ruiz, R.; Vázquez-Martínez, R.; et al. Effects of glucagon-like peptide-1 on the differentiation and metabolism of human adipocytes. Br. J. Pharmacol. 2016, 173, 1820–1834. [Google Scholar] [CrossRef] [PubMed]
- Muskiet, M.H.A.; Tonneijck, L.; Smits, M.M.; Van Baar, M.J.B.; Kramer, M.H.H.; Hoorn, E.J.; Joles, J.A.; Van Raalte, D.H. GLP-1 and the kidney: From physiology to pharmacology and outcomes in diabetes. Nat. Rev. Nephrol. 2017, 13, 605–628. [Google Scholar] [CrossRef] [PubMed]
- Sherk, V.D.; Schauer, I.; Shah, V.N. Update on the Acute Effects of Glucose, Insulin, and Incretins on Bone Turnover In Vivo. Curr. Osteoporos. Rep. 2020, 18, 371–377. [Google Scholar] [CrossRef]
- Seino, Y.; Fukushima, M.; Yabe, D. GIP and GLP-1, the two incretin hormones: Similarities and differences. J. Diabetes Investig. 2010, 1, 8–23. [Google Scholar] [CrossRef]
- Rouillé, Y.; Martin, S.; Steiner, D.F. Differential Processing of Proglucagon by the Subtilisin-like Prohormone Convertases PC2 and PC3 to Generate either Glucagon or Glucagon-like Peptide. J. Biol. Chem. 1995, 270, 26488–26496. [Google Scholar] [CrossRef] [PubMed]
- Friis-Hansen, L.; Lacourse, K.; Samuelson, L.; Holst, J. Attenuated processing of proglucagon and glucagon-like peptide-1 in carboxypeptidase E-deficient mice. J. Endocrinol. 2001, 169, 595–602. [Google Scholar] [CrossRef]
- Ørskov, C.; Rabenhøj, L.; Wettergren, A.; Kofod, H.; Holst, J.J. Tissue and Plasma Concentrations of Amidated and Glycine-Extended Glucagon-Like Peptide I in Humans. Diabetes 1994, 43, 535–539. [Google Scholar] [CrossRef]
- Brubaker, P.L.; Crivici, A.; Izzo, A.; Ehrlich, P.; Tsai, C.-H.; Drucker, D.J. Circulating and Tissue Forms of the Intestinal Growth Factor, Glucagon-Like Peptide-2*. Endocrinology 1997, 138, 4837–4843. [Google Scholar] [CrossRef] [PubMed]
- Takeda, J.; Seino, Y.; Tanaka, K.; Fukumoto, H.; Kayano, T.; Takahashi, H.; Mitani, T.; Kurono, M.; Suzuki, T.; Tobe, T. Sequence of an intestinal cDNA encoding human gastric inhibitory polypeptide precursor. Proc. Natl. Acad. Sci. USA 1987, 84, 7005–7008. [Google Scholar] [CrossRef]
- Kieffer, T.J.; McIntosh, C.H.; Pederson, R.A. Degradation of glucose-dependent insulinotropic polypeptide and truncated glucagon-like peptide 1 in vitro and in vivo by dipeptidyl peptidase IV. Endocrinology 1995, 136, 3585–3596. [Google Scholar] [CrossRef]
- Mentlein, R. Mechanisms underlying the rapid degradation and elimination of the incretin hormones GLP-1 and GIP. Best Pract. Res. Clin. Endocrinol. Metab. 2009, 23, 443–452. [Google Scholar] [CrossRef]
- Meier, J.J.; Nauck, M.A.; Kranz, D.; Holst, J.J.; Deacon, C.F.; Gaeckler, D.; Schmidt, W.E.; Gallwitz, B. Secretion, Degradation, and Elimination of Glucagon-Like Peptide 1 and Gastric Inhibitory Polypeptide in Patients with Chronic Renal Insufficiency and Healthy Control Subjects. Diabetes 2004, 53, 654–662. [Google Scholar] [CrossRef]
- Hartmann, B.; Harr, M.B.; Jeppesen, P.B.; Wojdemann, M.; Deacon, C.F.; Mortensen, P.B.; Holst, J.J. In Vivo and in Vitro Degradation of Glucagon-Like Peptide-2 in Humans1. J. Clin. Endocrinol. Metab. 2000, 85, 2884–2888. [Google Scholar] [CrossRef]
- Gasbjerg, L.S.; Helsted, M.M.; Hartmann, B.; Jensen, M.H.; Gabe, M.B.N.; Sparre-Ulrich, A.H.; Veedfald, S.; Stensen, S.; Lanng, A.R.; Bergmann, N.C.; et al. Separate and Combined Glucometabolic Effects of Endogenous Glucose-Dependent Insulinotropic Polypeptide and Glucagon-like Peptide 1 in Healthy Individuals. Diabetes 2019, 68, 906–917. [Google Scholar] [CrossRef]
- Nauck, M.A.; Meier, J.J. GIP and GLP-1: Stepsiblings Rather Than Monozygotic Twins Within the Incretin Family. Diabetes 2019, 68, 897–900. [Google Scholar] [CrossRef] [PubMed]
- Gribble, F.M.; Williams, L.; Simpson, A.K.; Reimann, F. A Novel Glucose-Sensing Mechanism Contributing to Glucagon-Like Peptide-1 Secretion from the GLUTag Cell Line. Diabetes 2003, 52, 1147–1154. [Google Scholar] [CrossRef] [PubMed]
- Röder, P.V.; Geillinger, K.E.; Zietek, T.S.; Thorens, B.; Koepsell, H.; Daniel, H. The Role of SGLT1 and GLUT2 in Intestinal Glucose Transport and Sensing. PLoS ONE 2014, 9, e89977. [Google Scholar] [CrossRef]
- Pais, R.; Gribble, F.M.; Reimann, F. Signalling pathways involved in the detection of peptones by murine small intestinal enteroendocrine L-cells. Peptides 2016, 77, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Batterham, R.L.; Heffron, H.; Kapoor, S.; Chivers, J.E.; Chandarana, K.; Herzog, H.; Le Roux, C.W.; Thomas, E.L.; Bell, J.D.; Withers, D.J. Critical role for peptide YY in protein-mediated satiation and body-weight regulation. Cell Metab. 2006, 4, 223–233. [Google Scholar] [CrossRef]
- Feltrin, K.L.; Little, T.J.; Meyer, J.H.; Horowitz, M.; Smout, A.J.P.M.; Wishart, J.; Pilichiewicz, A.N.; Rades, T.; Chapman, I.M.; Feinle-Bisset, C. Effects of intraduodenal fatty acids on appetite, antropyloroduodenal motility, and plasma CCK and GLP-1 in humans vary with their chain length. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2004, 287, R524–R533. [Google Scholar] [CrossRef]
- Feltrin, K.L.; Little, T.J.; Meyer, J.H.; Horowitz, M.; Rades, T.; Wishart, J.; Feinle-Bisset, C. Effects of lauric acid on upper gut motility, plasma cholecystokinin and peptide YY, and energy intake are load, but not concentration, dependent in humans. J. Physiol. 2007, 581 Pt 2, 767–777. [Google Scholar] [CrossRef] [PubMed]
- Briscoe, C.P.; Tadayyon, M.; Andrews, J.L.; Benson, W.G.; Chambers, J.K.; Eilert, M.M.; Ellis, C.; Elshourbagy, N.A.; Goetz, A.S.; Minnick, D.T.; et al. The Orphan G Protein-coupled Receptor GPR40 Is Activated by Medium and Long Chain Fatty Acids. J. Biol. Chem. 2003, 278, 11303–11311. [Google Scholar] [CrossRef] [PubMed]
- Ridner, G.; Bartoov-Shifman, R.; Zalogin, T.; Avnit-Sagi, T.; Bahar, K.; Sharivkin, R.; Kantorovich, L.; Weiss, S.; Walker, M.D. Regulation of the GPR40 locus: Towards a molecular understanding. Biochem. Soc. Trans. 2008, 36 Pt 3, 360–362. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, K.; Harada, N.; Sasaki, K.; Yamane, S.; Iida, K.; Suzuki, K.; Hamasaki, A.; Nasteska, D.; Shibue, K.; Joo, E.; et al. Free Fatty Acid Receptor GPR120 Is Highly Expressed in Enteroendocrine K Cells of the Upper Small Intestine and Has a Critical Role in GIP Secretion After Fat Ingestion. Endocrinology 2015, 156, 837–846. [Google Scholar] [CrossRef] [PubMed]
- Sankoda, A.; Harada, N.; Kato, T.; Ikeguchi, E.; Iwasaki, K.; Yamane, S.; Murata, Y.; Hirasawa, A.; Inagaki, N. Free fatty acid receptors, G protein-coupled receptor 120 and G protein-coupled receptor 40, are essential for oil-induced gastric inhibitory polypeptide secretion. J. Diabetes Investig. 2019, 10, 1430–1437. [Google Scholar] [CrossRef]
- Overton, H.A.; Babbs, A.J.; Doel, S.M.; Fyfe, M.C.; Gardner, L.S.; Griffin, G.; Jackson, H.C.; Procter, M.J.; Rasamison, C.M.; Tang-Christensen, M.; et al. Deorphanization of a G protein-coupled receptor for oleoylethanolamide and its use in the discovery of small-molecule hypophagic agents. Cell Metab. 2006, 3, 167–175. [Google Scholar] [CrossRef]
- Edfalk, S.; Steneberg, P.; Edlund, H. Gpr40 Is Expressed in Enteroendocrine Cells and Mediates Free Fatty Acid Stimulation of Incretin Secretion. Diabetes 2008, 57, 2280–2287. [Google Scholar] [CrossRef]
- Hirasawa, A.; Tsumaya, K.; Awaji, T.; Katsuma, S.; Adachi, T.; Yamada, M.; Sugimoto, Y.; Miyazaki, S.; Tsujimoto, G. Free fatty acids regulate gut incretin glucagon-like peptide-1 secretion through GPR120. Nat. Med. 2005, 11, 90–94. [Google Scholar] [CrossRef] [PubMed]
- Chu, Z.-L.; Carroll, C.; Alfonso, J.; Gutierrez, V.; He, H.; Lucman, A.; Pedraza, M.; Mondala, H.; Gao, H.; Bagnol, D.; et al. A Role for Intestinal Endocrine Cell-Expressed G Protein-Coupled Receptor 119 in Glycemic Control by Enhancing Glucagon-Like Peptide-1 and Glucose-Dependent Insulinotropic Peptide Release. Endocrinology 2008, 149, 2038–2047. [Google Scholar] [CrossRef] [PubMed]
- Tolhurst, G.; Heffron, H.; Lam, Y.S.; Parker, H.E.; Habib, A.M.; Diakogiannaki, E.; Cameron, J.; Grosse, J.; Reimann, F.; Gribble, F.M. Short-Chain Fatty Acids Stimulate Glucagon-Like Peptide-1 Secretion via the G-Protein-Coupled Receptor FFAR2. Diabetes 2012, 61, 364–371. [Google Scholar] [CrossRef]
- Tremaroli, V.; Bäckhed, F. Functional interactions between the gut microbiota and host metabolism. Nature 2012, 489, 242–249. [Google Scholar] [CrossRef]
- Wichmann, A.; Allahyar, A.; Greiner, T.U.; Plovier, H.; Lundén, G.; Larsson, T.; Drucker, D.J.; Delzenne, N.M.; Cani, P.D.; Bäckhed, F. Microbial Modulation of Energy Availability in the Colon Regulates Intestinal Transit. Cell Host Microbe 2013, 14, 582–590. [Google Scholar] [CrossRef]
- Katsuma, S.; Hirasawa, A.; Tsujimoto, G. Bile acids promote glucagon-like peptide-1 secretion through TGR5 in a murine enteroendocrine cell line STC-1. Biochem. Biophys. Res. Commun. 2005, 329, 386–390. [Google Scholar] [CrossRef]
- Ullmer, C.; Sanchez, R.A.; Sprecher, U.; Raab, S.; Mattei, P.; Dehmlow, H.; Sewing, S.; Iglesias, A.; Beauchamp, J.; Conde-Knape, K. Systemic bile acid sensing by G protein-coupled bile acid receptor 1 (GPBAR1) promotes PYY and GLP-1 release. Br. J. Pharmacol. 2013, 169, 671–684. [Google Scholar] [CrossRef] [PubMed]
- Hylemon, P.B.; Zhou, H.; Pandak, W.M.; Ren, S.; Gil, G.; Dent, P. Bile acids as regulatory molecules. J. Lipid Res. 2009, 50, 1509–1520. [Google Scholar] [CrossRef] [PubMed]
- Parker, H.; Wallis, K.; le Roux, C.; Wong, K.; Reimann, F.; Gribble, F. Molecular mechanisms underlying bile acid-stimulated glucagon-like peptide-1 secretion. Br. J. Pharmacol. 2012, 165, 414–423. [Google Scholar] [CrossRef]
- Thomas, C.; Gioiello, A.; Noriega, L.; Strehle, A.; Oury, J.; Rizzo, G.; Macchiarulo, A.; Yamamoto, H.; Mataki, C.; Pruzanski, M.; et al. TGR5-Mediated Bile Acid Sensing Controls Glucose Homeostasis. Cell Metab. 2009, 10, 167–177. [Google Scholar] [CrossRef]
- Brighton, C.A.; Rievaj, J.; Kuhre, R.E.; Glass, L.L.; Schoonjans, K.; Holst, J.J.; Gribble, F.M.; Reimann, F. Bile Acids Trigger GLP-1 Release Predominantly by Accessing Basolaterally Located G Protein–Coupled Bile Acid Receptors. Endocrinology 2015, 156, 3961–3970. [Google Scholar] [CrossRef]
- Gagnon, J.; Sauvé, M.; Zhao, W.; Stacey, H.M.; Wiber, S.C.; Bolz, S.-S.; Brubaker, P.L. Chronic Exposure to TNFα Impairs Secretion of Glucagon-Like Peptide-1. Endocrinology 2015, 156, 3950–3960. [Google Scholar] [CrossRef]
- Allen, T.L.; Whitham, M.; Febbraio, M.A. IL-6 Muscles in on the Gut and Pancreas to Enhance Insulin Secretion. Cell Metab. 2012, 15, 8–9. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ellingsgaard, H.; Hauselmann, I.; Schuler, B.; Habib, A.M.; Baggio, L.L.; Meier, D.T.; Eppler, E.; Bouzakri, K.; Wueest, S.; Muller, Y.D.; et al. Interleukin-6 enhances insulin secretion by increasing glucagon-like peptide-1 secretion from L cells and alpha cells. Nat. Med. 2011, 17, 1481–1489. [Google Scholar] [CrossRef] [PubMed]
- Pais, R.; Zietek, T.; Hauner, H.; Daniel, H.; Skurk, T. RANTES (CCL5) reduces glucose-dependent secretion of glucagon-like peptides 1 and 2 and impairs glucose-induced insulin secretion in mice. Am. J. Physiol. Liver Physiol. 2014, 307, G330–G337. [Google Scholar] [CrossRef] [PubMed]
- Flock, G.B.; Cao, X.; Maziarz, M.; Drucker, D.J. Activation of Enteroendocrine Membrane Progesterone Receptors Promotes Incretin Secretion and Improves Glucose Tolerance in Mice. Diabetes 2012, 62, 283–290. [Google Scholar] [CrossRef]
- Lim, G.E.; Huang, G.J.; Flora, N.; LeRoith, D.; Rhodes, C.J.; Brubaker, P.L. Insulin Regulates Glucagon-Like Peptide-1 Secretion from the Enteroendocrine L Cell. Endocrinology 2009, 150, 580–591. [Google Scholar] [CrossRef] [PubMed]
- Kappe, C.; Fransson, L.; Wolbert, P.; Ortsäter, H. Glucocorticoids suppress GLP-1 secretion: Possible contribution to their diabetogenic effects. Clin. Sci. 2015, 129, 405–414. [Google Scholar] [CrossRef]
- Rahman, S.; Hossain, K.S.; Das, S.; Kundu, S.; Adegoke, E.O.; Rahman, A.; Hannan, A.; Uddin, J.; Pang, M.-G. Role of Insulin in Health and Disease: An Update. Int. J. Mol. Sci. 2021, 22, 6403. [Google Scholar] [CrossRef]
- Mitchell, C.S.; Begg, D.P. The regulation of food intake by insulin in the central nervous system. J. Neuroendocr. 2021, 33, e12952. [Google Scholar] [CrossRef]
- Janah, L.; Kjeldsen, S.; Galsgaard, K.D.; Winther-Sørensen, M.; Stojanovska, E.; Pedersen, J.; Knop, F.K.; Holst, J.J.; Albrechtsen, N.J.W. Glucagon Receptor Signaling and Glucagon Resistance. Int. J. Mol. Sci. 2019, 20, 3314. [Google Scholar] [CrossRef]
- Woods, S.C.; Lutz, T.A.; Geary, N.; Langhans, W. Pancreatic signals controlling food intake; insulin, glucagon and amylin. Philos. Trans. R. Soc. B Biol. Sci. 2006, 361, 1219–1235. [Google Scholar] [CrossRef]
- Kleinert, M.; Sachs, S.; Habegger, K.M.; Hofmann, S.M.; Müller, T.D. Glucagon Regulation of Energy Expenditure. Int. J. Mol. Sci. 2019, 20, 5407. [Google Scholar] [CrossRef]
- Aranda-Domene, R.; Orenes-Piñero, E.; Arribas-Leal, J.M.; Canovas-Lopez, S.; Hernández-Cascales, J. Evidence for a lack of inotropic and chronotropic effects of glucagon and glucagon receptors in the human heart. Cardiovasc. Diabetol. 2023, 22, 128. [Google Scholar] [CrossRef]
- Parmley, W.W.; Glick, G.; Sonnenblick, E.H. Cardiovascular Effects of Glucagon in Man. N. Engl. J. Med. 1968, 279, 12–17. [Google Scholar] [CrossRef]
- Cummings, D.E.; Purnell, J.Q.; Frayo, R.S.; Schmidova, K.; Wisse, B.E.; Weigle, D.S. A Preprandial Rise in Plasma Ghrelin Levels Suggests a Role in Meal Initiation in Humans. Diabetes 2001, 50, 1714–1719. [Google Scholar] [CrossRef] [PubMed]
- Yanagi, S.; Sato, T.; Kangawa, K.; Nakazato, M. The Homeostatic Force of Ghrelin. Cell Metab. 2018, 27, 786–804. [Google Scholar] [CrossRef] [PubMed]
- Mani, B.K.; Zigman, J.M. Ghrelin as a Survival Hormone. Trends Endocrinol. Metab. 2017, 28, 843–854. [Google Scholar] [CrossRef]
- Deschaine, S.L.; Leggio, L. From “Hunger Hormone” to “It’s Complicated”: Ghrelin Beyond Feeding Control. Physiology 2022, 37, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Sekar, R.; Chow, B.K.C. Lipolytic actions of secretin in mouse adipocytes. J. Lipid Res. 2014, 55, 190–200. [Google Scholar] [CrossRef] [PubMed]
- Schnabl, K.; Li, Y.; U-Din, M.; Klingenspor, M. Secretin as a Satiation Whisperer With the Potential to Turn into an Obesity-curbing Knight. Endocrinology 2021, 162, bqab113. [Google Scholar] [CrossRef]
- Cheng, C.Y.Y.; Chu, J.Y.S.; Chow, B.K.C. Central and Peripheral Administration of Secretin Inhibits Food Intake in Mice through the Activation of the Melanocortin System. Neuropsychopharmacology 2011, 36, 459–471. [Google Scholar] [CrossRef]
- Laurila, S.; Sun, L.; Lahesmaa, M.; Schnabl, K.; Laitinen, K.; Klén, R.; Li, Y.; Balaz, M.; Wolfrum, C.; Steiger, K.; et al. Secretin activates brown fat and induces satiation. Nat. Metab. 2021, 3, 798–809. [Google Scholar] [CrossRef]
- Li, Y.; Schnabl, K.; Gabler, S.-M.; Willershäuser, M.; Reber, J.; Karlas, A.; Laurila, S.; Lahesmaa, M.; Din, M.U.; Bast-Habersbrunner, A.; et al. Secretin-Activated Brown Fat Mediates Prandial Thermogenesis to Induce Satiation. Cell 2018, 175, 1561–1574.e12. [Google Scholar] [CrossRef]
- Sekar, R.; Chow, B.K.C. Secretin receptor-knockout mice are resistant to high-fat diet-induced obesity and exhibit impaired intestinal lipid absorption. FASEB J. 2014, 28, 3494–3505. [Google Scholar] [CrossRef] [PubMed]
- Laurila, S.; Rebelos, E.; Lahesmaa, M.; Sun, L.; Schnabl, K.; Peltomaa, T.-M.; Klén, R.; U-Din, M.; Honka, M.-J.; Eskola, O.; et al. Novel effects of the gastrointestinal hormone secretin on cardiac metabolism and renal function. Am. J. Physiol. Endocrinol. Metab. 2022, 322, E54–E62. [Google Scholar] [CrossRef] [PubMed]
- Gustafson, E.L.; E Smith, K.; Durkin, M.M.; Walker, M.W.; Gerald, C.; Weinshank, R.; A Branchek, T. Distribution of the neuropeptide Y Y2 receptor mRNA in rat central nervous system. Mol. Brain Res. 1997, 46, 223–235. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Macia, L.; Turner, N.; Enriquez, R.F.; Riepler, S.J.; Nguyen, A.D.; Lin, S.; Lee, N.J.; Shi, Y.C.; Yulyaningsih, E.; et al. Peripheral neuropeptide Y Y1 receptors regulate lipid oxidation and fat accretion. Int. J. Obes. 2010, 34, 357–373. [Google Scholar] [CrossRef]
- Shi, Y.; Lin, S.; Castillo, L.; Aljanova, A.; Enriquez, R.F.; Nguyen, A.D.; Baldock, P.A.; Zhang, L.; Bijker, M.S.; Macia, L.; et al. Peripheral-Specific Y2 Receptor Knockdown Protects Mice from High-Fat Diet-Induced Obesity. Obesity 2011, 19, 2137–2148. [Google Scholar] [CrossRef]
- Adrian, T.; Ferri, G.-L.; Bacarese-Hamilton, A.; Fuessl, H.; Polak, J.; Bloom, S. Human distribution and release of a putative new gut hormone, peptide YY. Gastroenterology 1985, 89, 1070–1077. [Google Scholar] [CrossRef]
- Batterham, R.L.; Cohen, M.A.; Ellis, S.M.; Le Roux, C.W.; Withers, D.J.; Frost, G.S.; Ghatei, M.A.; Bloom, S.R. Inhibition of Food Intake in Obese Subjects by Peptide YY3–36. N. Engl. J. Med. 2003, 349, 941–948. [Google Scholar] [CrossRef]
- Batterham, R.L.; Bloom, S.R. The Gut Hormone Peptide YY Regulates Appetite. Ann. N. Y. Acad. Sci. 2003, 994, 162–168. [Google Scholar] [CrossRef]
- Koda, S.; Date, Y.; Murakami, N.; Shimbara, T.; Hanada, T.; Toshinai, K.; Niijima, A.; Furuya, M.; Inomata, N.; Osuye, K.; et al. The Role of the Vagal Nerve in Peripheral PYY3–36-Induced Feeding Reduction in Rats. Endocrinology 2005, 146, 2369–2375. [Google Scholar] [CrossRef]
- Adrian, T.E.; Bloom, S.R.; Bryant, M.G.; Polak, J.M.; Heitz, P.H.; Barnes, A.J. Distribution and release of human pancreatic polypeptide. Gut 1976, 17, 940–944. [Google Scholar] [CrossRef]
- Blomqvist, A.G.; Herzog, H. Y-receptor subtypes—How many more? Trends Neurosci. 1997, 20, 294–298. [Google Scholar] [CrossRef] [PubMed]
- Asakawa, A.; Inui, A.; Yuzuriha, H.; Ueno, N.; Katsuura, G.; Fujimiya, M.; Fujino, M.A.; Niijima, A.; Meguid, M.M.; Kasuga, M. Characterization of the effects of pancreatic polypeptide in the regulation of energy balance. Gastroenterology 2003, 124, 1325–1336. [Google Scholar] [CrossRef]
- Lin, S.; Shi, Y.-C.; Yulyaningsih, E.; Aljanova, A.; Zhang, L.; Macia, L.; Nguyen, A.D.; Lin, E.-J.D.; During, M.J.; Herzog, H.; et al. Critical Role of Arcuate Y4 Receptors and the Melanocortin System in Pancreatic Polypeptide-Induced Reduction in Food Intake in Mice. PLoS ONE 2009, 4, e8488. [Google Scholar] [CrossRef] [PubMed]
- Sainsbury, A.; Shi, Y.-C.; Zhang, L.; Aljanova, A.; Lin, Z.; Nguyen, A.D.; Herzog, H.; Lin, S. Y4 receptors and pancreatic polypeptide regulate food intake via hypothalamic orexin and brain-derived neurotropic factor dependent pathways. Neuropeptides 2010, 44, 261–268. [Google Scholar] [CrossRef]
- Yan, C.; Zeng, T.; Lee, K.; Nobis, M.; Loh, K.; Gou, L.; Xia, Z.; Gao, Z.; Bensellam, M.; Hughes, W.; et al. Peripheral-specific Y1 receptor antagonism increases thermogenesis and protects against diet-induced obesity. Nat. Commun. 2021, 12, 2622. [Google Scholar] [CrossRef]
- Loh, K.; Herzog, H.; Shi, Y.-C. Regulation of energy homeostasis by the NPY system. Trends Endocrinol. Metab. 2015, 26, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Fetissov, S.O.; Kopp, J.; Hökfelt, T. Distribution of NPY receptors in the hypothalamus. Neuropeptides 2004, 38, 175–188. [Google Scholar] [CrossRef]
- Bi, S.; Li, L. Browning of white adipose tissue: Role of hypothalamic signaling. Ann. N. Y. Acad. Sci. 2013, 1302, 30–34. [Google Scholar] [CrossRef]
- Bohórquez, D.V.; Samsa, L.A.; Roholt, A.; Medicetty, S.; Chandra, R.; Liddle, R.A. An Enteroendocrine Cell—Enteric Glia Connection Revealed by 3D Electron Microscopy. PLoS ONE 2014, 9, e89881. [Google Scholar] [CrossRef]
- Rehfeld, J.F. Cholecystokinin—From Local Gut Hormone to Ubiquitous Messenger. Front. Endocrinol. 2017, 8, 47. [Google Scholar] [CrossRef]
- Rehfeld, J.F.; Sun, G.; Christensen, T.; Hillingsø, J.G. The Predominant Cholecystokinin in Human Plasma and Intestine Is Cholecystokinin-331. J. Clin. Endocrinol. Metab. 2001, 86, 251–258. [Google Scholar] [CrossRef][Green Version]
- Liddle, R.A.; Goldfine, I.D.; Rosen, M.S.; Taplitz, R.A.; Williams, J.A. Cholecystokinin bioactivity in human plasma. Molecular forms, responses to feeding, and relationship to gallbladder contraction. J. Clin. Investig. 1985, 75, 1144–1152. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, P.; Eberlein, G.A.; Reeve, J.R.; Bünte, R.H.; Grandt, D.; Goebell, H.; Eysselein, V.E. Comparison of clearance and metabolism of infused cholecystokinins 8 and 58 in dogs. Gastroenterology 1993, 105, 1732–1736. [Google Scholar] [CrossRef]
- Dodd, J.; Kelly, J. The actions of cholecystokinin and related peptides on pyramidal neurones of the mammalian hippocampus. Brain Res. 1981, 205, 337–350. [Google Scholar] [CrossRef]
- Gallmann, E.; Arsenijevic, D.; Spengler, M.; Williams, G.; Langhans, W. Effect of CCK-8 on insulin-induced hyperphagia and hypothalamic orexigenic neuropeptide expression in the rat. Peptides 2005, 26, 437–445. [Google Scholar] [CrossRef] [PubMed]
- Kobelt, P.; Tebbe, J.J.; Tjandra, I.; Stengel, A.; Bae, H.-G.; Andresen, V.; van der Voort, I.R.; Veh, R.W.; Werner, C.R.; Klapp, B.F.; et al. CCK inhibits the orexigenic effect of peripheral ghrelin. Am. J. Physiol. Integr. Comp. Physiol. 2005, 288, R751–R758. [Google Scholar] [CrossRef]
- De Lartigue, G.; Dimaline, R.; Varro, A.; Raybould, H.; De La Serre, C.B.; Dockray, G.J. Cocaine- and Amphetamine-Regulated Transcript Mediates the Actions of Cholecystokinin on Rat Vagal Afferent Neurons. Gastroenterology 2010, 138, 1479–1490. [Google Scholar] [CrossRef] [PubMed]
- Chandra, R.; Liddle, R.A. Cholecystokinin. Curr. Opin. Endocrinol. Diabetes 2007, 14, 63–67. [Google Scholar] [CrossRef]
- Rorsman, P.; Huising, M.O. The somatostatin-secreting pancreatic δ-cell in health and disease. Nat. Rev. Endocrinol. 2018, 14, 404–414. [Google Scholar] [CrossRef]
- Shamsi, B.H.; Chatoo, M.; Xu, X.K.; Xu, X.; Chen, X.Q. Versatile Functions of Somatostatin and Somatostatin Receptors in the Gastrointestinal System. Front. Endocrinol. 2021, 12, 652363. [Google Scholar] [CrossRef]
- Schubert, M.L.; Rehfeld, J.F. Gastric Peptides-Gastrin and Somatostatin. Compr. Physiol. 2019, 10, 197–228. [Google Scholar] [CrossRef] [PubMed]
- Stengel, A.; Taché, Y. Central somatostatin signaling and regulation of food intake. Ann. N. Y. Acad. Sci. 2019, 1455, 98–104. [Google Scholar] [CrossRef]
- Rehfeld, J.F. Gastrin and the Moderate Hypergastrinemias. Int. J. Mol. Sci. 2021, 22, 6977. [Google Scholar] [CrossRef] [PubMed]
- Raffort, J.; Lareyre, F.; Massalou, D.; Fénichel, P.; Panaïa-Ferrari, P.; Chinetti, G. Insights on glicentin, a promising peptide of the proglucagon family. Biochem. Medica 2017, 27, 308–324. [Google Scholar] [CrossRef] [PubMed]
- Holst, J.J.; Albrechtsen, N.J.; Gabe, M.B.N.; Rosenkilde, M.M. Oxyntomodulin: Actions and role in diabetes. Peptides 2018, 100, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Baggio, L.L.; Huang, Q.; Brown, T.J.; Drucker, D.J. Oxyntomodulin and glucagon-like peptide-1 differentially regulate murine food intake and energy expenditure. Gastroenterology 2004, 127, 546–558. [Google Scholar] [CrossRef] [PubMed]
- Kosinski, J.R.; Hubert, J.; Carrington, P.E.; Chicchi, G.G.; Mu, J.; Miller, C.; Cao, J.; Bianchi, E.; Pessi, A.; SinhaRoy, R.; et al. The Glucagon Receptor Is Involved in Mediating the Body Weight-Lowering Effects of Oxyntomodulin. Obesity 2012, 20, 1566–1571. [Google Scholar] [CrossRef]
- Laferrère, B.; Swerdlow, N.; Bawa, B.; Arias, S.; Bose, M.; Oliván, B.; Teixeira, J.; McGinty, J.; Rother, K.I. Rise of Oxyntomodulin in Response to Oral Glucose after Gastric Bypass Surgery in Patients with Type 2 Diabetes. J. Clin. Endocrinol. Metab. 2010, 95, 4072–4076. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Song, J.; Zaytseva, Y.Y.; Liu, Y.; Rychahou, P.; Jiang, K.; Starr, M.E.; Kim, J.T.; Harris, J.W.; Yiannikouris, F.B.; et al. An obligatory role for neurotensin in high-fat-diet-induced obesity. Nature 2016, 533, 411–415. [Google Scholar] [CrossRef]
- Schroeder, L.E.; Leinninger, G.M. Role of central neurotensin in regulating feeding: Implications for the development and treatment of body weight disorders. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 900–916. [Google Scholar] [CrossRef] [PubMed]
- Levin, B.E.; Lutz, T.A. Amylin and Leptin: Co-Regulators of Energy Homeostasis and Neuronal Development. Trends Endocrinol. Metab. 2017, 28, 153–164. [Google Scholar] [CrossRef] [PubMed]
- Lutz, T.A. Creating the amylin story. Appetite 2022, 172, 105965. [Google Scholar] [CrossRef] [PubMed]
- Boyle, C.N.; Zheng, Y.; Lutz, T.A. Mediators of Amylin Action in Metabolic Control. J. Clin. Med. 2022, 11, 2207. [Google Scholar] [CrossRef]
- Tomlinson, E.; Fu, L.; John, L.; Hultgren, B.; Huang, X.; Renz, M.; Stephan, J.P.; Tsai, S.P.; Powell-Braxton, L.; French, D.; et al. Transgenic Mice Expressing Human Fibroblast Growth Factor-19 Display Increased Metabolic Rate and Decreased Adiposity. Endocrinology 2002, 143, 1741–1747. [Google Scholar] [CrossRef] [PubMed]
- Wei, M.; Cao, W.-B.; Zhao, R.-D.; Sun, D.-P.; Liang, Y.-Z.; Huang, Y.-D.; Cheng, Z.-W.; Ouyang, J.; Yang, W.-S.; Yu, W.-B. Fibroblast growth factor 15, induced by elevated bile acids, mediates the improvement of hepatic glucose metabolism after sleeve gastrectomy. World J. Gastroenterol. 2023, 29, 3280–3291. [Google Scholar] [CrossRef] [PubMed]
- Mavanji, V.; Pomonis, B.; Kotz, C.M. Orexin, serotonin, and energy balance. WIREs Mech. Dis. 2022, 14, e1536. [Google Scholar] [CrossRef]
- Imperatore, R.; Palomba, L.; Cristino, L. Role of Orexin-A in Hypertension and Obesity. Curr. Hypertens. Rep. 2017, 19, 34. [Google Scholar] [CrossRef]
- Trovato, L.; Gallo, D.; Settanni, F.; Gesmundo, I.; Ghigo, E.; Granata, R. Obestatin: Is it really doing something? Front. Horm. Res. 2014, 42, 175–185. [Google Scholar] [CrossRef]
- van Galen, K.A.; ter Horst, K.W.; Serlie, M.J. Serotonin, food intake, and obesity. Obes. Rev. 2021, 22, e13210. [Google Scholar] [CrossRef]
- Kirichenko, T.V.; Markina, Y.V.; Bogatyreva, A.I.; Tolstik, T.V.; Varaeva, Y.R.; Starodubova, A.V. The Role of Adipokines in Inflammatory Mechanisms of Obesity. Int. J. Mol. Sci. 2022, 23, 14982. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, J.A.; Pothuraju, R.; Khan, P.; Sharma, G.; Muniyan, S.; Seshacharyulu, P.; Jain, M.; Nasser, M.W.; Batra, S.K. Pathophysiological role of growth differentiation factor 15 (GDF15) in obesity, cancer, and cachexia. Cytokine Growth Factor Rev. 2021, 64, 71–83. [Google Scholar] [CrossRef] [PubMed]
- Navarro-Perez, J.; Vidal-Puig, A.; Carobbio, S. Recent developments in adipose tissue-secreted factors and their target organs. Curr. Opin. Genet. Dev. 2023, 80, 102046. [Google Scholar] [CrossRef]
- Funcke, J.-B.; Scherer, P.E. Beyond adiponectin and leptin: Adipose tissue-derived mediators of inter-organ communication. J. Lipid Res. 2019, 60, 1648–1697. [Google Scholar] [CrossRef] [PubMed]
- Laiglesia, L.M.; Escoté, X.; Sáinz, N.; Felix-Soriano, E.; Santamaría, E.; Collantes, M.; Fernández-Galilea, M.; Colón-Mesa, I.; Martínez-Fernández, L.; Quesada-López, T.; et al. Maresin 1 activates brown adipose tissue and promotes browning of white adipose tissue in mice. Mol. Metab. 2023, 74, 101749. [Google Scholar] [CrossRef] [PubMed]
- Margetic, S.; Gazzola, C.; Pegg, G.G.; Hill, R.A. Leptin: A review of its peripheral actions and interactions. Int. J. Obes. Relat. Metab. Disord. 2002, 26, 1407–1433. [Google Scholar] [CrossRef] [PubMed]
- Moon, H.-S.; Dalamaga, M.; Kim, S.-Y.; Polyzos, S.A.; Hamnvik, O.-P.; Magkos, F.; Paruthi, J.; Mantzoros, C.S. Leptin’s Role in Lipodystrophic and Nonlipodystrophic Insulin-Resistant and Diabetic Individuals. Endocr. Rev. 2013, 34, 377–412. [Google Scholar] [CrossRef]
- Landt, M.; Lawson, G.M.; Helgeson, J.M.; Davila-Roman, V.G.; Ladenson, J.H.; Jaffe, A.S.; Hickner, R.C. Prolonged exercise decreases serum leptin concentrations. Metabolism 1997, 46, 1109–1112. [Google Scholar] [CrossRef]
- Bouassida, A.; Zalleg, D.; Bouassida, S.; Zaouali, M.; Feki, Y.; Zbidi, A.; Tabka, Z. Leptin, its implication in physical exercise and training: A short review. J. Sports Sci. Med. 2006, 5, 172–181. [Google Scholar]
- Trayhurn, P.; Duncan, J.S.; Rayner, D.V. Acute cold-induced suppression of ob (obese) gene expression in white adipose tissue of mice: Mediation by the sympathetic system. Biochem. J. 1995, 311 Pt 3, 729–733. [Google Scholar] [CrossRef]
- Pereira, S.; Cline, D.L.; Glavas, M.M.; Covey, S.D.; Kieffer, T.J. Tissue-Specific Effects of Leptin on Glucose and Lipid Metabolism. Endocr. Rev. 2021, 42, 1–28. [Google Scholar] [CrossRef]
- Haynes, W.G.; Morgan, D.A.; Walsh, S.A.; Mark, A.L.; Sivitz, W.I. Receptor-mediated regional sympathetic nerve activation by leptin. J. Clin. Investig. 1997, 100, 270–278. [Google Scholar] [CrossRef]
- Pollock, B.H.; Fischer, A.W.; Schlein, C.; Cannon, B.; Heeren, J.; Nedergaard, J.; Côté, I.; Sakarya, Y.; Green, S.M.; Carter, C.S.; et al. Leptin increases uncoupling protein expression and energy expenditure. Am. J. Physiol. Metab. 1997, 273 Pt 1, E226–E230. [Google Scholar] [CrossRef]
- Dodd, G.T.; Decherf, S.; Loh, K.; Simonds, S.E.; Wiede, F.; Balland, E.; Merry, T.L.; Münzberg, H.; Zhang, Z.-Y.; Kahn, B.B.; et al. Leptin and Insulin Act on POMC Neurons to Promote the Browning of White Fat. Cell 2015, 160, 88–104. [Google Scholar] [CrossRef]
- Friedman, J. 20 YEARS OF LEPTIN: Leptin at 20: An overview. J. Endocrinol. 2014, 223, T1–T8. [Google Scholar] [CrossRef] [PubMed]
- Fang, H.; Judd, R.L. Adiponectin Regulation and Function. Compr. Physiol. 2018, 8, 1031–1063. [Google Scholar] [CrossRef] [PubMed]
- Berg, A.H.; Combs, T.P.; Du, X.; Brownlee, M.; Scherer, P.E. The adipocyte-secreted protein Acrp30 enhances hepatic insulin action. Nat. Med. 2001, 7, 947–953. [Google Scholar] [CrossRef]
- Tomas, E.; Tsao, T.-S.; Saha, A.K.; Murrey, H.E.; Zhang, C.C.; Itani, S.I.; Lodish, H.F.; Ruderman, N.B. Enhanced muscle fat oxidation and glucose transport by ACRP30 globular domain: Acetyl–CoA carboxylase inhibition and AMP-activated protein kinase activation. Proc. Natl. Acad. Sci. USA 2002, 99, 16309–16313. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-Y.; van de Wall, E.; Laplante, M.; Azzara, A.; Trujillo, M.E.; Hofmann, S.M.; Schraw, T.; Durand, J.L.; Li, H.; Li, G.; et al. Obesity-associated improvements in metabolic profile through expansion of adipose tissue. J. Clin. Investig. 2007, 117, 2621–2637. [Google Scholar] [CrossRef] [PubMed]
- Kharroubi, I.; Rasschaert, J.; Eizirik, D.L.; Cnop, M. Expression of adiponectin receptors in pancreatic β cells. Biochem. Biophys. Res. Commun. 2003, 312, 1118–1122. [Google Scholar] [CrossRef] [PubMed]
- Patané, G.; Caporarello, N.; Marchetti, P.; Parrino, C.; Sudano, D.; Marselli, L.; Vigneri, R.; Frittitta, L. Adiponectin increases glucose-induced insulin secretion through the activation of lipid oxidation. Acta Diabetol. 2013, 50, 851–857. [Google Scholar] [CrossRef] [PubMed]
- Hui, X.; Gu, P.; Zhang, J.; Nie, T.; Pan, Y.; Wu, D.; Feng, T.; Zhong, C.; Wang, Y.; Lam, K.S.; et al. Adiponectin Enhances Cold-Induced Browning of Subcutaneous Adipose Tissue via Promoting M2 Macrophage Proliferation. Cell Metab. 2015, 22, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Kadowaki, T.; Yamauchi, T.; Kubota, N. The physiological and pathophysiological role of adiponectin and adiponectin receptors in the peripheral tissues and CNS. FEBS Lett. 2008, 582, 74–80. [Google Scholar] [CrossRef]
- Krude, H.; Biebermann, H.; Schnabel, D.; Tansek, M.Z.; Theunissen, P.; Mullis, P.E.; Grüters, A. Obesity Due to Proopiomelanocortin Deficiency: Three New Cases and Treatment Trials with Thyroid Hormone and ACTH4–10. J. Clin. Endocrinol. Metab. 2003, 88, 4633–4640. [Google Scholar] [CrossRef]
- Blüher, S.; Shah, S.; Mantzoros, C.S. Leptin Deficiency: Clinical Implications and Opportunities for Therapeutic Interventions. J. Investig. Med. 2009, 57, 784–788. [Google Scholar] [CrossRef]
- Lubrano-Berthelier, C.; Cavazos, M.; Dubern, B.; Shapiro, A.; LE Stunff, C.; Zhang, S.; Picart, F.; Govaerts, C.; Froguel, P.; Bougnères, P.; et al. Molecular Genetics of Human Obesity-Associated MC4R Mutations. Ann. N. Y. Acad. Sci. 2003, 994, 49–57. [Google Scholar] [CrossRef]
- Holst, B.; Schwartz, T.W. Ghrelin receptor mutations—Too little height and too much hunger. J. Clin. Investig. 2006, 116, 637–641. [Google Scholar] [CrossRef] [PubMed]
- Folon, L.; Baron, M.; Toussaint, B.; Vaillant, E.; Boissel, M.; Scherrer, V.; Loiselle, H.; Leloire, A.; Badreddine, A.; Balkau, B.; et al. Contribution of heterozygous PCSK1 variants to obesity and implications for precision medicine: A case-control study. Lancet Diabetes Endocrinol. 2023, 11, 182–190. [Google Scholar] [CrossRef]
- Bray, M.S.; Boerwinkle, E.; Hanis, C.L. Sequence Variation within the Neuropeptide Y Gene and Obesity in Mexican Americans. Obes. Res. 2000, 8, 219–226. [Google Scholar] [CrossRef]
- Podyma, B.; Parekh, K.; Güler, A.D.; Deppmann, C.D. Metabolic homeostasis via BDNF and its receptors. Trends Endocrinol. Metab. 2021, 32, 488–499. [Google Scholar] [CrossRef]
- Singh, R.K.; Kumar, P.; Mahalingam, K. Molecular genetics of human obesity: A comprehensive review. Comptes Rendus Biol. 2017, 340, 87–108. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, R.; Kimonis, V.; Butler, M.G. Genetics of Obesity in Humans: A Clinical Review. Int. J. Mol. Sci. 2022, 23, 11005. [Google Scholar] [CrossRef]
- Sjöström, L.; Narbro, K.; Sjöström, C.D.; Karason, K.; Larsson, B.; Wedel, H.; Lystig, T.; Sullivan, M.; Bouchard, C.; Carlsson, B.; et al. Effects of Bariatric Surgery on Mortality in Swedish Obese Subjects. N. Engl. J. Med. 2007, 357, 741–752. [Google Scholar] [CrossRef] [PubMed]
- O’brien, P.E.; Hindle, A.; Brennan, L.; Skinner, S.; Burton, P.; Smith, A.; Crosthwaite, G.; Brown, W. Long-Term Outcomes After Bariatric Surgery: A Systematic Review and Meta-analysis of Weight Loss at 10 or More Years for All Bariatric Procedures and a Single-Centre Review of 20-Year Outcomes After Adjustable Gastric Banding. Obes. Surg. 2019, 29, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Mechanick, J.I.; Apovian, C.; Brethauer, S.; Garvey, W.T.; Joffe, A.M.; Kim, J.; Kushner, R.F.; Lindquist, R.; Pessah-Pollack, R.; Seger, J.; et al. Clinical Practice Guidelines for the Perioperative Nutrition, Metabolic, and Nonsurgical Support of Patients Undergoing Bariatric Procedures—2019 Update: Cosponsored by American Association of Clinical Endocrinologists/American College of Endocrinology, The Obesity Society, American Society for Metabolic and Bariatric Surgery, Obesity Medicine Association, and American Society of Anesthesiologists. Obesity 2020, 28, O1–O58. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg, D.; Shikora, S.A.; Aarts, E.; Aminian, A.; Angrisani, L.; Cohen, R.V.; de Luca, M.; Faria, S.L.; Goodpaster, K.P.; Haddad, A.; et al. 2022 American Society of Metabolic and Bariatric Surgery (ASMBS) and International Federation for the Surgery of Obesity and Metabolic Disorders (IFSO) Indications for Metabolic and Bariatric Surgery. Obes. Surg. 2022, 18, 1345–1356, Erratum in Obes. Surg. 2023, 33, 3–14. [Google Scholar] [CrossRef]
- Lee, P.C.; Dixon, J. Bariatric-metabolic surgery: A guide for the primary care physician. Aust. Fam. Physician 2017, 46, 465–471. [Google Scholar]
- Sjöström, L.; Lindroos, A.-K.; Peltonen, M.; Torgerson, J.; Bouchard, C.; Carlsson, B.; Dahlgren, S.; Larsson, B.; Narbro, K.; Sjöström, C.D.; et al. Lifestyle, Diabetes, and Cardiovascular Risk Factors 10 Years after Bariatric Surgery. N. Engl. J. Med. 2004, 351, 2683–2693. [Google Scholar] [CrossRef]
- Loos, R.J.F.; Yeo, G.S.H. The genetics of obesity: From discovery to biology. Nat. Rev. Genet. 2022, 23, 120–133. [Google Scholar] [CrossRef] [PubMed]
- Di Lorenzo, N.; Antoniou, S.A.; Batterham, R.L.; Busetto, L.; Godoroja, D.; Iossa, A.; Carrano, F.M.; Agresta, F.; Alarçon, I.; Azran, C.; et al. Clinical practice guidelines of the European Association for Endoscopic Surgery (EAES) on bariatric surgery: Update 2020 endorsed by IFSO-EC, EASO and ESPCOP. Surg. Endosc. 2020, 34, 2332–2358. [Google Scholar] [CrossRef]
- Hedbäck, N.; Hindsø, M.; Bojsen-Møller, K.N.; Linddal, A.K.; Jørgensen, N.B.; Dirksen, C.; Møller, A.; Kristiansen, V.B.; Hartmann, B.; Holst, J.J.; et al. Effect of Meal Texture on Postprandial Glucose Excursions and Gut Hormones After Roux-en-Y Gastric Bypass and Sleeve Gastrectomy. Front. Nutr. 2022, 9, 889710. [Google Scholar] [CrossRef]
- Tedjo, D.I.; Wilbrink, J.A.; Boekhorst, J.; Timmerman, H.M.; Nienhuijs, S.W.; Stronkhorst, A.; Savelkoul, P.H.M.; Masclee, A.A.M.; Penders, J.; Jonkers, D.M.A.E. Impact of Sleeve Gastrectomy on Fecal Microbiota in Individuals with Morbid Obesity. Microorganisms 2023, 11, 2353. [Google Scholar] [CrossRef]
- Browning, M.G.; Pessoa, B.M.; Khoraki, J.; Campos, G.M. Changes in Bile Acid Metabolism, Transport, and Signaling as Central Drivers for Metabolic Improvements After Bariatric Surgery. Curr. Obes. Rep. 2019, 8, 175–184. [Google Scholar] [CrossRef]
- Meek, C.L.; Lewis, H.B.; Reimann, F.; Gribble, F.M.; Park, A.J. The effect of bariatric surgery on gastrointestinal and pancreatic peptide hormones. Peptides 2016, 77, 28–37. [Google Scholar] [CrossRef]
- Feris, F.; McRae, A.; Kellogg, T.A.; McKenzie, T.; Ghanem, O.; Acosta, A. Mucosal and hormonal adaptations after Roux-en-Y gastric bypass. Surg. Obes. Relat. Dis. 2023, 19, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Peterli, R.; Steinert, R.E.; Woelnerhanssen, B.; Peters, T.; Christoffel-Courtin, C.; Gass, M.; Kern, B.; von Fluee, M.; Beglinger, C. Metabolic and Hormonal Changes After Laparoscopic Roux-en-Y Gastric Bypass and Sleeve Gastrectomy: A Randomized, Prospective Trial. Obes. Surg. 2012, 22, 740–748. [Google Scholar] [CrossRef]
- Cazzo, E.; Gestic, M.A.; Utrini, M.P.; Chaim, F.D.; Geloneze, B.; Pareja, J.C.; Chaim, E.A.; Magro, D.O. GLP-2: A poorly understood mediator enrolled in various bariatric/metabolic surgery-related pathophysiologic mechanisms. Arq. Bras. Cir. Dig. 2016, 29, 272–275. [Google Scholar] [CrossRef] [PubMed]
- Pournaras, D.J.; le Roux, C.W. Obesity, gut hormones, and bariatric surgery. World J. Surg. 2009, 33, 1983–1988. [Google Scholar] [CrossRef]
- Gao, Z.; Yang, J.; Liang, Y.; Yang, S.; Zhang, T.; Gong, Z.; Li, M. Changes in Gastric Inhibitory Polypeptide (GIP) After Roux-en-Y Gastric Bypass in Obese Patients: A Meta-analysis. Obes. Surg. 2022, 32, 2706–2716. [Google Scholar] [CrossRef] [PubMed]
- Quercia, I.; Dutia, R.; Kotler, D.; Belsley, S.; Laferrère, B. Gastrointestinal changes after bariatric surgery. Diabetes Metab. 2014, 40, 87–94. [Google Scholar] [CrossRef]
- Šebunova, N.; Štšepetova, J.; Kullisaar, T.; Suija, K.; Rätsep, A.; Junkin, I.; Soeorg, H.; Lember, M.; Sillakivi, T.; Mändar, R. Changes in adipokine levels and metabolic profiles following bariatric surgery. BMC Endocr. Disord. 2022, 22, 33. [Google Scholar] [CrossRef]
- Woelnerhanssen, B.; Peterli, R.; Steinert, R.E.; Peters, T.; Borbély, Y.; Beglinger, C. Effects of postbariatric surgery weight loss on adipokines and metabolic parameters: Comparison of laparoscopic Roux-en-Y gastric bypass and laparoscopic sleeve gastrectomy—A prospective randomized trial. Surg. Obes. Relat. Dis. 2011, 7, 561–568. [Google Scholar] [CrossRef] [PubMed]
- Svane, M.S.; Bojsen-Møller, K.N.; Martinussen, C.; Dirksen, C.; Madsen, J.L.; Reitelseder, S.; Holm, L.; Rehfeld, J.F.; Kristiansen, V.B.; van Hall, G.; et al. Postprandial Nutrient Handling and Gastrointestinal Hormone Secretion After Roux-en-Y Gastric Bypass vs Sleeve Gastrectomy. Gastroenterology 2019, 156, 1627–1641.e1. [Google Scholar] [CrossRef]
- Fatima, F.; Hjelmesæth, J.; Birkeland, K.I.; Gulseth, H.L.; Hertel, J.K.; Svanevik, M.; Sandbu, R.; Småstuen, M.C.; Hartmann, B.; Holst, J.J.; et al. Gastrointestinal Hormones and β-Cell Function After Gastric Bypass and Sleeve Gastrectomy: A Randomized Controlled Trial (Oseberg). J. Clin. Endocrinol. Metab. 2021, 107, e756–e766. [Google Scholar] [CrossRef]
- Nannipieri, M.; Baldi, S.; Mari, A.; Colligiani, D.; Guarino, D.; Camastra, S.; Barsotti, E.; Berta, R.; Moriconi, D.; Bellini, R.; et al. Roux-en-Y Gastric Bypass and Sleeve Gastrectomy: Mechanisms of Diabetes Remission and Role of Gut Hormones. J. Clin. Endocrinol. Metab. 2013, 98, 4391–4399. [Google Scholar] [CrossRef] [PubMed]
- Berthoud, H.-R.; Shin, A.C.; Zheng, H. Obesity surgery and gut–brain communication. Physiol. Behav. 2011, 105, 106–119. [Google Scholar] [CrossRef]
- Lampropoulos, C.; Alexandrides, T.; Tsochatzis, S.; Kehagias, D.; Kehagias, I. Are the Changes in Gastrointestinal Hormone Secretion Necessary for the Success of Bariatric Surgery? A Critical Review of the Literature. Obes. Surg. 2021, 31, 4575–4584. [Google Scholar] [CrossRef] [PubMed]
- Svane, M.S.; Jørgensen, N.B.; Bojsen-Møller, K.N.; Dirksen, C.; Nielsen, S.; Kristiansen, V.B.; Toräng, S.; Albrechtsen, N.J.W.; Rehfeld, J.F.; Hartmann, B.; et al. Peptide YY and glucagon-like peptide-1 contribute to decreased food intake after Roux-en-Y gastric bypass surgery. Int. J. Obes. 2016, 40, 1699–1706. [Google Scholar] [CrossRef]
- Capristo, E.; Panunzi, S.; De Gaetano, A.; Spuntarelli, V.; Bellantone, R.; Giustacchini, P.; Birkenfeld, A.L.; Amiel, S.; Bornstein, S.R.; Raffaelli, M.; et al. Incidence of Hypoglycemia After Gastric Bypass vs Sleeve Gastrectomy: A Randomized Trial. J. Clin. Endocrinol. Metab. 2018, 103, 2136–2146. [Google Scholar] [CrossRef]
- Papamargaritis, D.; Koukoulis, G.; Sioka, E.; Zachari, E.; Bargiota, A.; Zacharoulis, D.; Tzovaras, G. Dumping Symptoms and Incidence of Hypoglycaemia After Provocation Test at 6 and 12 Months After Laparoscopic Sleeve Gastrectomy. Obes. Surg. 2012, 22, 1600–1606. [Google Scholar] [CrossRef]
- Masclee, G.M.C.; Masclee, A.A. Dumping Syndrome: Pragmatic Treatment Options and Experimental Approaches for Improving Clinical Outcomes. Clin. Exp. Gastroenterol. 2023, 16, 197–211. [Google Scholar] [CrossRef] [PubMed]
- Lupoli, R.; Lembo, E.; Ciciola, P.; Schiavo, L.; Pilone, V.; Capaldo, B. Continuous glucose monitoring in subjects undergoing bariatric surgery: Diurnal and nocturnal glycemic patterns. Nutr. Metab. Cardiovasc. Dis. 2020, 30, 1954–1960. [Google Scholar] [CrossRef]
- Hasan, M.A.; Schwartz, S.; McKenna, V.; Ing, R. An Imbalance of Pathophysiologic Factors in Late Postprandial Hypoglycemia Post Bariatric Surgery: A Narrative Review. Obes. Surg. 2023, 33, 2927–2937. [Google Scholar] [CrossRef]
- Salehi, M.; Vella, A.; McLaughlin, T.; Patti, M.-E. Hypoglycemia After Gastric Bypass Surgery: Current Concepts and Controversies. J. Clin. Endocrinol. Metab. 2018, 103, 2815–2826. [Google Scholar] [CrossRef] [PubMed]
- Jalleh, R.J.; Umapathysivam, M.M.; Plummer, M.P.; Deane, A.; Jones, K.L.; Horowitz, M. Postprandial plasma GLP-1 levels are elevated in individuals with postprandial hypoglycaemia following Roux-en-Y gastric bypass—A systematic review. Rev. Endocr. Metab. Disord. 2023, 24, 1075–1088. [Google Scholar] [CrossRef]
- Goldfine, A.B.; Mun, E.C.; Devine, E.; Bernier, R.; Baz-Hecht, M.; Jones, D.B.; Schneider, B.E.; Holst, J.J.; Patti, M.E. Patients with Neuroglycopenia after Gastric Bypass Surgery Have Exaggerated Incretin and Insulin Secretory Responses to a Mixed Meal. J. Clin. Endocrinol. Metab. 2007, 92, 4678–4685. [Google Scholar] [CrossRef] [PubMed]
- Salehi, M.; Gastaldelli, A.; D’Alessio, D.A. Altered islet function and insulin clearance cause hyperinsulinemia in gastric bypass patients with symptoms of postprandial hypoglycemia. J. Clin. Endocrinol. Metab. 2014, 99, 2008–2017. [Google Scholar] [CrossRef]
- Llewellyn, D.C.; Ellis, H.L.; Aylwin, S.J.B.; Oštarijaš, E.; Green, S.; Sheridan, W.; Chew, N.W.S.; le Roux, C.W.; Miras, A.D.; Patel, A.G.; et al. The efficacy of GLP-1RAs for the management of postprandial hypoglycemia following bariatric surgery: A systematic review. Obesity 2023, 31, 20–30. [Google Scholar] [CrossRef]
- Elhag, W.; Lock, M.; El Ansari, W. When Definitions Differ, are Comparisons Meaningful? Definitions of Weight Regain After Bariatric Surgery and Their Associations with Patient Characteristics and Clinical Outcomes—A Need for a Revisit? Obes. Surg. 2023, 33, 1390–1400. [Google Scholar] [CrossRef]
- Sumithran, P.; Prendergast, L.A.; Delbridge, E.; Purcell, K.; Shulkes, A.; Kriketos, A.; Proietto, J. Long-Term Persistence of Hormonal Adaptations to Weight Loss. N. Engl. J. Med. 2011, 365, 1597–1604. [Google Scholar] [CrossRef]
- El Ansari, W.; Elhag, W. Weight Regain and Insufficient Weight Loss After Bariatric Surgery: Definitions, Prevalence, Mechanisms, Predictors, Prevention and Management Strategies, and Knowledge Gaps—A Scoping Review. Obes. Surg. 2021, 31, 1755–1766. [Google Scholar] [CrossRef] [PubMed]
- Martins, C. Do we really know what drives relapse in obesity management? Eur. J. Intern. Med. 2021, 95, 113–114. [Google Scholar] [CrossRef] [PubMed]
- Vaccaro, S.; Itani, L.; Scazzina, F.; Bonilauri, S.; Cartelli, C.M.; El Ghoch, M.; Pellegrini, M. Do Lifestyle Interventions before Gastric Bypass Prevent Weight Regain after Surgery? A Five-Year Longitudinal Study. Nutrients 2022, 14, 3609. [Google Scholar] [CrossRef]
- Poitou, C.; Puder, L.; Dubern, B.; Krabusch, P.; Genser, L.; Wiegand, S.; Verkindt, H.; Köhn, A.; von Schwartzenberg, R.J.; Flück, C.; et al. Long-term outcomes of bariatric surgery in patients with bi-allelic mutations in the POMC, LEPR, and MC4R genes. Surg. Obes. Relat. Dis. 2021, 17, 1449–1456. [Google Scholar] [CrossRef]
- Cooiman, M.; Kleinendorst, L.; Aarts, E.; Janssen, I.; van Amstel, H.P.; Blakemore, A.; Hazebroek, E.; Meijers-Heijboer, H.; van der Zwaag, B.; Berends, F.; et al. Genetic Obesity and Bariatric Surgery Outcome in 1014 Patients with Morbid Obesity. Obes. Surg. 2020, 30, 470–477. [Google Scholar] [CrossRef]
- Campos, A.; Cifuentes, L.; Hashem, A.; Busebee, B.; Hurtado-Andrade, M.D.; Ricardo-Silgado, M.L.; McRae, A.; De la Rosa, A.; Feris, F.; Bublitz, J.T.; et al. Effects of Heterozygous Variants in the Leptin-Melanocortin Pathway on Roux-en-Y Gastric Bypass Outcomes: A 15-Year Case–Control Study. Obes. Surg. 2022, 32, 2632–2640. [Google Scholar] [CrossRef]
- Torrego-Ellacuría, M.; Barabash, A.; Matía-Martín, P.; Sánchez-Pernaute, A.; Torres, A.J.; Calle-Pascual, A.L.; Rubio-Herrera, M.A. Combined Effect of Genetic Variants on Long-Term Weight Response after Bariatric Surgery. J. Clin. Med. 2023, 12, 4288. [Google Scholar] [CrossRef]
- Goni, L.; Cuervo, M.; Milagro, F.I.; Martínez, J.A. A genetic risk tool for obesity predisposition assessment and personalized nutrition implementation based on macronutrient intake. Genes Nutr. 2015, 10, 445. [Google Scholar] [CrossRef]
- Bonetti, G.; Dhuli, K.; Ceccarini, M.R.; Kaftalli, J.; Samaja, M.; Precone, V.; Cecchin, S.; Maltese, P.E.; Guerri, G.; Marceddu, G.; et al. Next-Generation Sequencing of a Large Gene Panel for Outcome Prediction of Bariatric Surgery in Patients with Severe Obesity. J. Clin. Med. 2022, 11, 7531. [Google Scholar] [CrossRef] [PubMed]
- van der Meer, R.; Mohamed, S.A.; Monpellier, V.M.; Liem, R.S.L.; Hazebroek, E.J.; Franks, P.W.; Frayling, T.M.; Janssen, I.M.C.; Serlie, M.J. Genetic variants associated with weight loss and metabolic outcomes after bariatric surgery: A systematic review. Obes. Rev. 2023, 24, e13626. [Google Scholar] [CrossRef] [PubMed]
- Faccioli, N.; Poitou, C.; Clément, K.; Dubern, B. Current Treatments for Patients with Genetic Obesity. J. Clin. Res. Pediatr. Endocrinol. 2023, 15, 108–119. [Google Scholar] [CrossRef]
- Updated June, 2021. Available online: www.who.int/newsroom/fact-sheets/detail/obesity-and-overweight (accessed on 27 January 2024).
- American Diabetes Association Professional Practice Committee. Obesity and Weight Management for the Prevention and Treatment of Type 2 Diabetes: Standards of Care in Diabetes–2024. Diabetes Care 2024, 47 (Suppl. S1), S145–S157. [CrossRef]
- Mingrone, G.; Panunzi, S.; De Gaetano, A.; Guidone, C.; Iaconelli, A.; Nanni, G.; Castagneto, M.; Bornstein, S.; Rubino, F. Bariatric–metabolic surgery versus conventional medical treatment in obese patients with type 2 diabetes: 5 year follow-up of an open-label, single-centre, randomised controlled trial. Lancet 2015, 386, 964–973. [Google Scholar] [CrossRef] [PubMed]
- Yska, J.P.; van Roon, E.N.; de Boer, A.; Leufkens, H.G.M.; Wilffert, B.; de Heide, L.J.M.; de Vries, F.; Lalmohamed, A. Remission of Type 2 Diabetes Mellitus in Patients After Different Types of Bariatric Surgery. JAMA Surg. 2015, 150, 1126–1133. [Google Scholar] [CrossRef] [PubMed]
- Jackson, H.T.; Anekwe, C.; Chang, J.; Haskins, I.N.; Stanford, F.C. The Role of Bariatric Surgery on Diabetes and Diabetic Care Compliance. Curr. Diabetes Rep. 2019, 19, 125. [Google Scholar] [CrossRef] [PubMed]
- Svane, M.S.; Bojsen-Møller, K.N.; Nielsen, S.; Jørgensen, N.B.; Dirksen, C.; Bendtsen, F.; Kristiansen, V.B.; Hartmann, B.; Holst, J.J.; Madsbad, S. Effects of endogenous GLP-1 and GIP on glucose tolerance after Roux-en-Y gastric bypass surgery. Am. J. Physiol. Metab. 2016, 310, E505–E514. [Google Scholar] [CrossRef]
- Ferrannini, E.; Mingrone, G. Impact of Different Bariatric Surgical Procedures on Insulin Action and β-Cell Function in Type 2 Diabetes. Diabetes Care 2009, 32, 514–520. [Google Scholar] [CrossRef]
- Sandoval, D.A.; Patti, M.E. Glucose metabolism after bariatric surgery: Implications for T2DM remission and hypoglycaemia. Nat. Rev. Endocrinol. 2023, 19, 164–176. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.S.; Sandoval, D.A. Endocrine Function after Bariatric Surgery. Compr. Physiol. 2017, 7, 783–798. [Google Scholar] [CrossRef]
- Mosinski, J.D.; Aminian, A.; Axelrod, C.L.; Batayyah, E.; Romero-Talamas, H.; Daigle, C.; Mulya, A.; Scelsi, A.; Schauer, P.R.; Brethauer, S.A.; et al. Roux-en-Y gastric bypass restores islet function and morphology independent of body weight in ZDF rats. Am. J. Physiol. Metab. 2021, 320, E392–E398. [Google Scholar] [CrossRef]
- Abu-Gazala, S.; Horwitz, E.; Schyr, R.B.-H.; Bardugo, A.; Israeli, H.; Hija, A.; Schug, J.; Shin, S.; Dor, Y.; Kaestner, K.H.; et al. Sleeve Gastrectomy Improves Glycemia Independent of Weight Loss by Restoring Hepatic Insulin Sensitivity. Diabetes 2018, 67, 1079–1085. [Google Scholar] [CrossRef]
- Kirwan, J.P.; Münzberg, H.; Berthoud, H.-R. Mechanisms Responsible for Metabolic Improvements of Bariatric Surgeries. Diabetes 2018, 67, 1043–1044. [Google Scholar] [CrossRef] [PubMed]
- Scheen, A.J. Use of SGLT2 inhibitors after bariatric/metabolic surgery: Risk/benefit balance. Diabetes Metab. 2023, 49, 101453. [Google Scholar] [CrossRef] [PubMed]
- Alabduljabbar, K.; le Roux, C.W. Pharmacotherapy before and after bariatric surgery. Metabolism 2023, 148, 155692. [Google Scholar] [CrossRef] [PubMed]
- Wachsmuth, H.R.; Weninger, S.N.; Duca, F.A. Role of the gut–brain axis in energy and glucose metabolism. Exp. Mol. Med. 2022, 54, 377–392. [Google Scholar] [CrossRef]
- Frias, J.P.; Deenadayalan, S.; Erichsen, L.; Knop, F.K.; Lingvay, I.; Macura, S.; Mathieu, C.; Pedersen, S.D.; Davies, M. Efficacy and safety of co-administered once-weekly cagrilintide 2·4 mg with once-weekly semaglutide 2·4 mg in type 2 diabetes: A multicentre, randomised, double-blind, active-controlled, phase 2 trial. Lancet 2023, 402, 720–730. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).