
Hearing Loss in Baraitser–Winter Syndrome: Case Reports and Review of the Literature


Abstract
1. Introduction
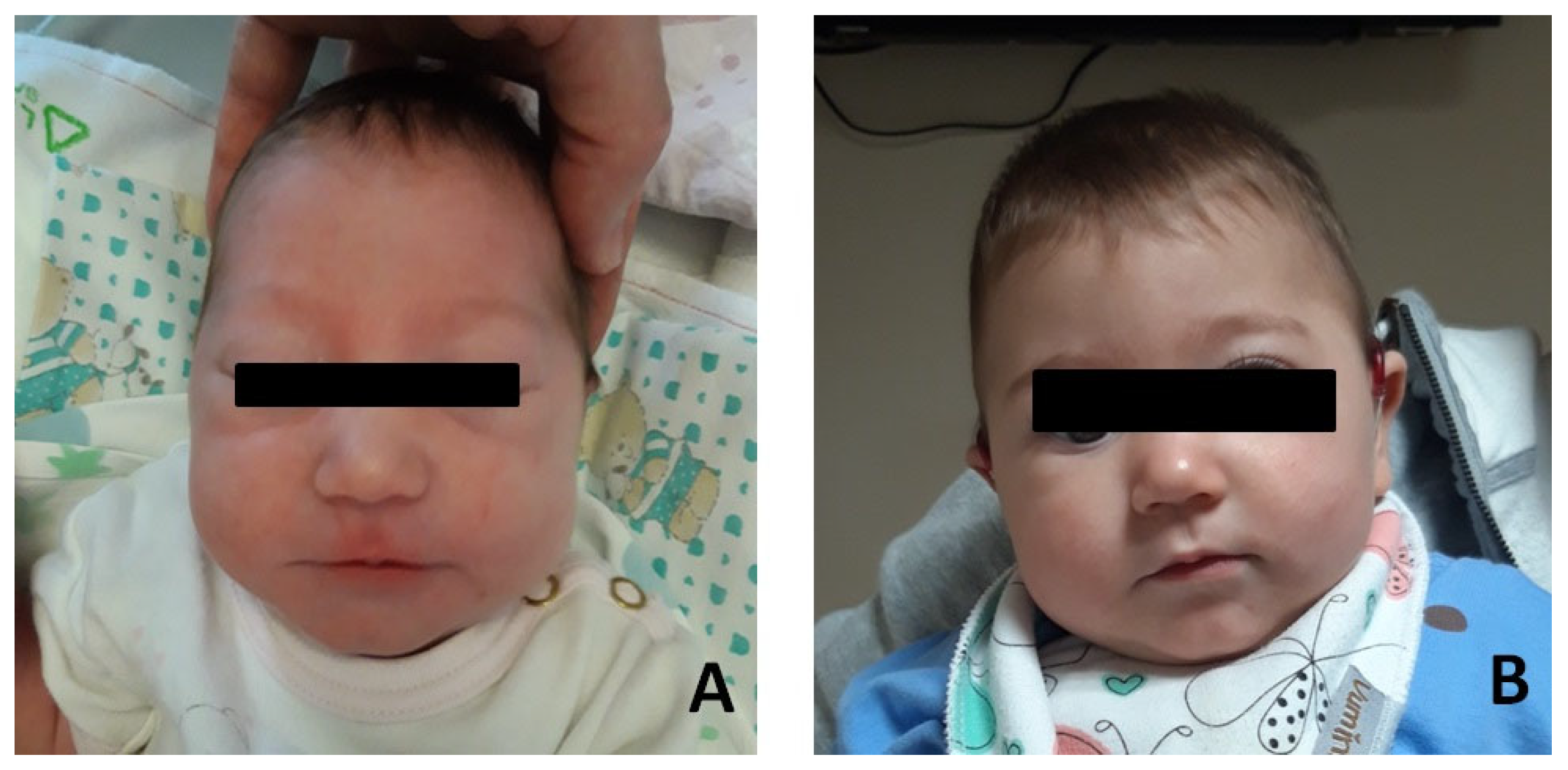
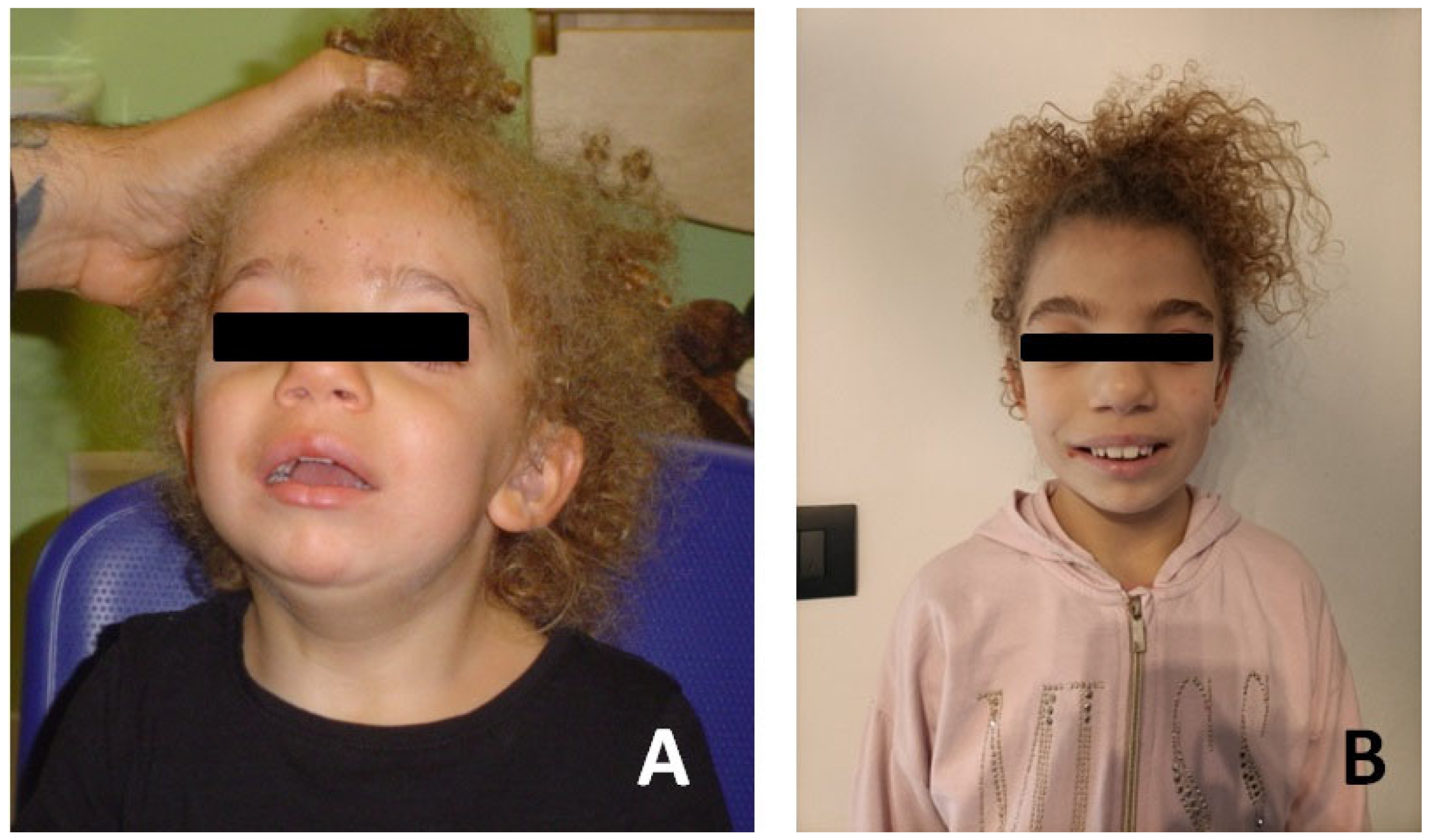
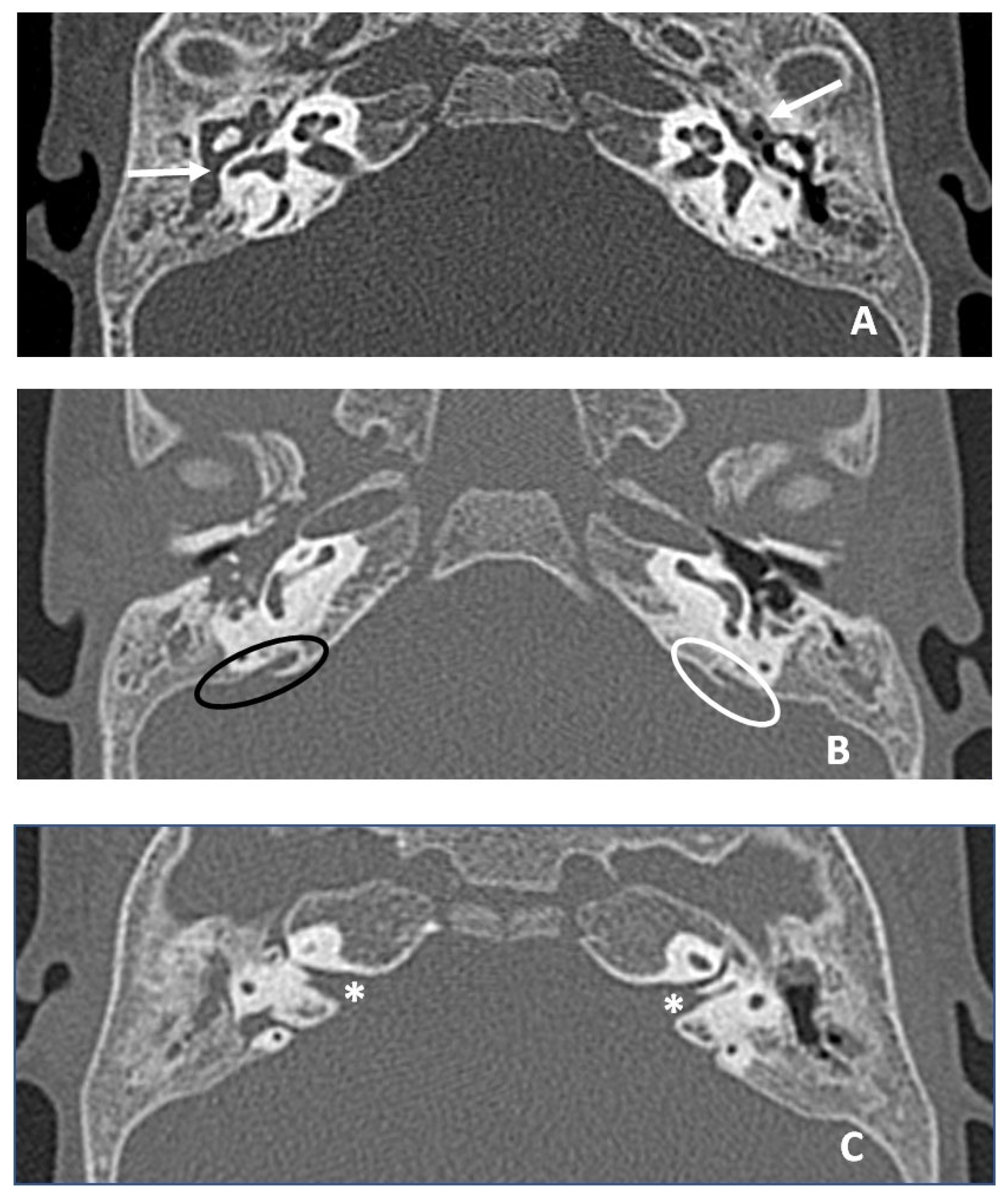
2. Case Reports
- ABR: V wave was detected at 60 dB on the left ear and 90 dB on the right ear;
- Impedenzometry: type A tympanogram bilaterally, acoustic reflex was present only on the left side.
- ABR: V wave was detected at 60 dB bilaterally;
- Distortion-product otoacoustic emission (DPOAE): partially present bilaterally;
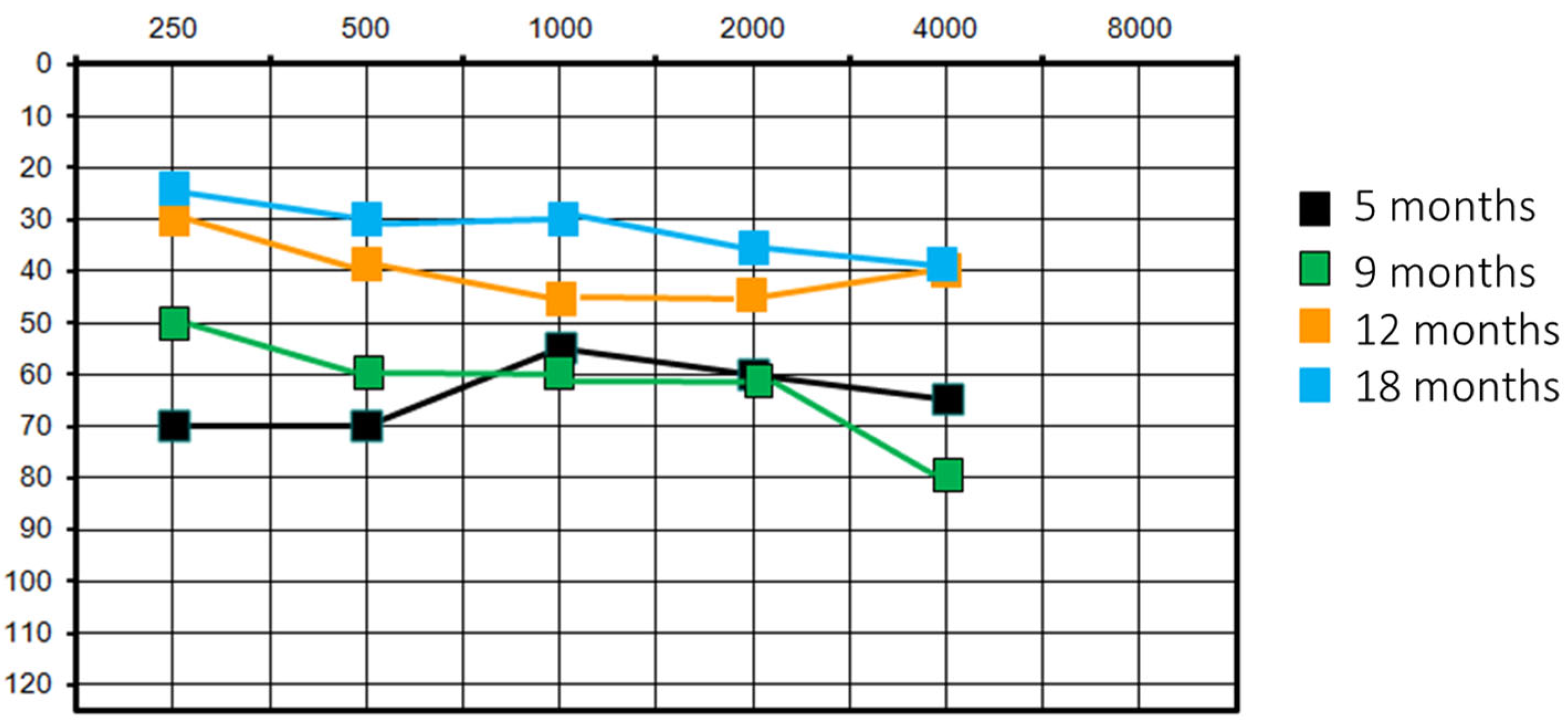
- Visual reinforcement audiometry (VRA): PTA (pure tone average) of 65 dB without HA and 40 dB with HA.
- Otoscopic examination: bilateral otitis media with effusion;
- Audiological test: mean PTA was 100 dB HL on the right ear and 90 dB HL on the left ear without HA; mean PTA was 55 dB with bilateral HA;
- Impedenzometry: type B tympanogram bilaterally; ABR: V wave was detected at 70 dB on the left ear and was absent on the right ear;
- Speech evaluation: poor perceptual-auditory skills, only inconstant voice detection. Language development was at the pre-verbal stage. A psychomotor delay was present.
- -
- Pure tone audiometry without hearing device: hearing residuals on the right, mean PTA of 75 dB on the left;
- -
- Speech perception without hearing device: 100% of intelligibility at 80 dB on the left, only detection threshold at 90 dB on the right;
- -
- Pure tone audiometry with right CI and left HA: mean free field PTA equal to 40 dB;
- -
- Speech perception with right CI and left HA: 100% of intelligibility at 50 dB;
- -
- Speech evaluation: phonetic skills were evolved with increased inventory. Poor vocal quality, presence of phonological processes of simplification of phonological structure and intelligibility of spontaneous production remains poor.
3. Literature Search
4. Hearing Loss in Baraitser–Winter Syndrome: Evidence Synthesis
5. Discussion
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene ACTB | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Change cDNA Amino Acid Change | N° Patients | Gender | HL | Age Onset HL | Progression | Hearing Intervention | Other Malformations | Reference | |
| 1 | c.208C > G, p.Pro70Ala | 1 | Male | Not reported | N/A | N/A | N/A | Craniofacial features, intellectual disability, motor and speech delay, eye problems | [43] |
| 2 | c.220 G4A p.Gly74Ser | 1 | Female | No | N/A | N/A | N/A | Heart defect, cleft lip and palate, motor delay, facial dysmorphism | [12] |
| 3 | c.359C4T p.Thr120Ile | 1 | Female | No | N/A | N/A | N/A | Eye problems, encephalic alterations, gastro-intestinal, renal alterations, facial dysmorphism, epilepsy, motor delay | [12] |
| 4 | c.478A > G (p.Thr160Ala) | 1 | Male | Not reported | N/A | N/A | N/A | Facial features, eye problems thrombocytopenia, intellectual developmental delay and attention deficit | [44] |
| 5 | c.484A > G, p.Thr162Ala | 1 | Male | Yes | Not reported | Not reported | Not reported | Development retardation, psycho-motor retardation, hypotonia, cerebral malformations, epilepsy. | [21] |
| 6 | c.511C > T, p.Leu171Phe | 1 | Female | Not reported | N/A | N/A | N/A | Cleft soft palate, ventricular arrhythmia, thrombocytopenia, dysmorphic features, development delay, eye problems | [43] |
| 7 | c.547C > T p.Arg183Trp | 1 | Male | Yes | 2.5 years | Not reported | CI | Facial dysmorphism | [15] |
| 1 | Male | Yes | 8 months | Not reported | Not reported | Dystonia, motor and language delays, mild intellectual disability | [16] | ||
| 1 | Male | Yes | Birth | Not reported | Not reported | Dystonia | [17] | ||
| 1 | Female | Yes | Congenital | Progressive | N/A | Facial dysmorphism, motor delay | [18] | ||
| 1 | Female | Yes | Birth | Not reported | Not reported | Dystonia Craniofacial dysmorphism | [19] | ||
| 1 | Female | Yes | Childhood | Progressive | Dystonia, epilepsy, scoliosis | [20] | |||
| 8 | c.586C > T (p.Arg196Cys) | 1 | Female | Not reported | N/A | N/A | N/A | Renal problem, facial dysmorphism, limb problem, heart anomalies, cerebral MRI alterations | [40] |
| 1 | Male | Yes | Not reported | Not reported | Not reported | Craniofacial anomalies, renal and genital anomalies | [12] | ||
| 9 | c.617G > A, p.Arg206Gl | 2 (one family) | 1 male 1 female | No | N/A | N/A | N/A | Intellectual disability, dysmorphic features, delayed motor and speech development, low muscle tone. | [29] |
| 10 | c.802G > T p.Gly268Arg | 1 | Male | Not reported | N/A | N/A | N/A | Hydrocephalus, cleft and lip palate, renal problems, dysmorphisms | [45] |
| 11 | c.826G > A (p.Glu276Lys) | 1 | Female | Yes | 2 | Not reported | Tube insertion | Facial features, brain abnormalities, ocular coloboma, cardiac defects, intellectual disabilities, short stature, developmental delay | [39] |
| 12 | c.938T > G p.(Met313Arg) | 2 | Males | Not reported | N/A | N/A | N/A | (1) Mild developmental delay, cleft lip, heart defect, microcephaly, leukocytosis and thrombocytopenia (2) Thrombocytopenia | [41] |
| 13 | c.992_1008del, p.(Ala331Val_fs*27) | 2 | 1 Male 1 Female | Not reported | N/A | N/A | N/A | (1) Speech delay, borderline intellectual impairment and microcephaly, pseudarthrosis, facial anomalies (2) Mild intellectual disability, microcephaly, thrombocytopenia, facial anomalies | [41] |
| 14 | c.1012_1023del p.(Ser338_Ile341del) | 1 | Male | Not reported | N/A | N/A | N/A | Microcephaly, facial anomalies, speech delays, haematological anomalies | [41] |
| 15 | c.1043C > T; p.Ser348Leu | 2 (one family, twin) | 2 females | Not reported | N/A | N/A | N/A | (1) Craniofacial features, bowel atresia, pulmonary hypoplasia She died at 1 h of age (2) Craniofacial features, laryngeal dysgenesis, malformed trachea, jejunal atresia, megakaryocytes, hyperglycaemia, patent ductus arteriosus, ventricular septal defect. She died on day 23 | [37] |
| 16 | c.1090G > A p.(Glu364Lys) | 1 | Female | Not reported | N/A | N/A | N/A | Developmental delay, microcephaly, thrombocytopenia, encephalic MRI alterations | [41] |
| 17 | c.a092_1105dup14 p.Ile369SerfsX18 | 1 | Female | Not reported | N/A | N/A | N/A | Short stature, developmental delay, eye alterations, minor facial dysmorphism. motor and speech delay | [46] |
| 18 | c.1101dup p.(Ser368Leu_fs*13) | 1 | Female | Not reported | N/A | N/A | N/A | Photo-sensitivity, stomatitis, keratoconjunctivitis, thrombocytopenia, leukopenia, recurrent otitis media, furunculosis, polyarthralgia, intellectual impairment, short stature | [47] |
| 19 | c.1117A > T; p.Lys373Ter | 5 | 3 male 2 female | Not reported | N/A | N/A | N/A | Grown retardation, speech and motor delay, heart problem, scoliosis, renal and genital anomalies | [48] |
| Gene ACTG1 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Change | N° Patients | Gender | HL | Age Onset HL (Years) | Progression | Hearing Intervention | Other Malformations | Reference | |
| 1 | c.94C > T | 2 (one family) | Male | Yes | Birth | Unknown | CI | Cleft-lip palate | [49] |
| Male | Yes | Prelingual | Unknown | None | No | [49] | |||
| 1 | Male | Yes | 3 | Unknown | Unknown | No | [50] | ||
| 2 | c.102 C > G p.I34M | 2 (one family) | Male | Yes | 18 | Progressive | HA | Not reported | [24] |
| Female | Yes | 15 | Progressive | None | Not reported | [24] | |||
| 3 | c.110 G > A p.R37H | 2 (one family) | Female | Yes | Unknown | Unknown | None | Not reported | [24] |
| Male | Yes | 10 | Progressive | Unknown | Not reported | [24] | |||
| 4 | c.142 G > C p.G48R | 4 (one family) | Female | Yes | 11 | Progressive | HA | Not reported | [24] |
| Female | Yes | Not reported | Progressive | None | Not reported | [24] | |||
| Male | Yes | 7 | Progressive | None | Not reported | [24] | |||
| Female | Yes | Precritical | Unknown | None | Not reported | [24] | |||
| 4 (one family) | Female | Yes | 11 | Progressive | Not reported | Not reported | [35] | ||
| Male | Yes | 6 | Stable | Not reported | Not reported | [35] | |||
| Female | No | Not reported | Not reported | Not reported | Not reported | [35] | |||
| Female | Yes | Not reported | Not reported | Not reported | Not reported | [35] | |||
| 5 | c.151G > A | 8 (one family) | Yes | From birth to 34 years | 5 of 8 progressive | HA (3 in selection for CI) | Not reported | [51] | |
| 6 | c.176A > G, p.Gln59Arg | 1 | Male | Not reported | Not reported | Not reported | Not reported | Ptosis, coloboma, dysmorphic facial features, psychomotor delay, pachygyria | [23] |
| 7 | c.246 G > A p.M82I | 1 | Male | Yes | Unknown | Unknown | None | Not reported | [24] |
| 8 | c.266 C > T p.T89I | 1 | Male | Yes | Unknown | Progressive | HA | Not reported | [24] |
| 9 | c.353 A > T p.K118M | 3 (2 families) | Female | Yes | 26 | Progressive | HA | Not reported | [24] |
| Male | Yes | 17 | Progressive | HA | Not reported | [24] | |||
| Male | Yes | 17 | Progressive | HA | Not reported | [24] | |||
| 2 (one family) | Male | Yes | 17 | Progressive | Not reported | Not reported | [35] | ||
| Male | Yes | 17 | Not reported | Not reported | Not reported | [35] | |||
| 1 | Female | Yes | 26 | Progressive | HA | Not reported | [35] | ||
| 10 | c.364A > G; p.I122V | 11 (one family) | 8 males, 3 females | Yes | Mean 6 years | Progressive | Not reported | No | [52] |
| 11 | c.493 A > G p.I165V | 1 | Male | Yes | 11 | Progressive | HA | Not reported | [24] |
| 12 | c.542C > T (NM_001614.5), p.Ala181Val (NP_001605.1) | 1 | Male | Yes | 4 months | Hearing improving | HA (subsequently abandoned) | Renal, genital, cardiological, encephalic dysmorphic features | [30] |
| 13 | c.608C > T, p.Thr203Met | 1 | Male | Not reported | N/A | N/A | N/A | Strabismus, coloboma, facial dysmorphism, genital alterations, psychomotor delay, MRI: cortical dysplasia, pachygyria, short and thick corpus callosum with rostral agenesis, hypoplastic cerebellar vermis | [23] |
| 14 | c.617G > A p.(Arg206Gln) | 1 | Female | Yes | 4 | Progressive | Not reported | Psychomotor delay, microcephaly dysmorphic features, cardiac septal hypertrophy, abdominal swelling | [25] |
| 15 | c.625G > A (p. Val209Met) | 1 | Male | Yes | 6 | Progressive | Not reported | Myopia, astigmatism | [9] |
| 16 | c.638A > G (p.K213R) | 8 (one family) | 6 males 2 females | Yes | Second decade (7 cases) Birth (one case) | Progressive | Not reported | No | [53] |
| 17 | c.721 G > A p.E241K | 3 (one family) | Female | Yes | 14 | Progressive | HA | Not reported | [24] |
| Male | Yes | 3 | Progressive | HA | Not reported | [24] | |||
| Female | Yes | 3 | Unknown | None | Not reported | [24] | |||
| 3 (one family) | Female | Yes | 14 | Not reported | HA | Moyamoya disease | [35] | ||
| Male | Yes | 4 years, 11 months | Not reported | HA | Developmental disorder | [35] | |||
| Female | Yes | 3 | Not reported | Not reported | Not reported | [35] | |||
| 18 | c.791 C > T p.P264L | 1 | Female | Yes | 12 | Progressive | Unknown | Not reported | [24] |
| 19 | c.823 C > T p.H275Y | 1 | Female | Yes | 34 | Progressive | Unknown | Not reported | [24] |
| 20 | c.895 C > G p.L299V | 4 (one family) | Male | Yes | 46 | Progressive | HA | Not reported | [24] |
| Male | Yes | 6 | Progressive | EAS | Not reported | [24] | |||
| Male | Yes | 15 | Progressive | HA | Not reported | [24] | |||
| Male | Yes | Precritical | Progressive | None | Not reported | [24] | |||
| 4 (one family) | Male | Yes | 12 | Progressive | EAS | Not reported | [35] | ||
| Male | Yes | 15 | Progressive | Not reported | Not reported | [35] | |||
| Male | Yes | Not reported | Not reported | Not reported | Not reported | [35] | |||
| Male | Yes | Not reported | Not reported | Not reported | Not reported | [35] | |||
| 21 | c.914 T > C p.M305T | 1 | Male | Yes | 6 | Progressive | EAS | Not reported | [24] |
| 3 (one family) | Female | Yes | Postlingual (about 30) | Progressive | HA | No | [25] | ||
| Male | Yes | Postlingual (about 30) | Progressive | HA→ CI | No | [25] | |||
| Male | Yes | Postlingual (about 30) | Progressive | HA→ CI | No | [25] | |||
| 22 | c.994 C > T p.P332S | 2 (one family) | Male | Yes | 59 | Progressive | HA | Not reported | [24] |
| Male | Yes | 38 | Progressive | Unknown | Not reported | [24] | |||
| 1 | Male | Yes | 35 | Progressive | HA | Not reported | [24] | ||
| 23 | c.1003C > T; p.(Arg335Cys | 1 | Male | Yes | 6 | Not reported | Not reported | Craniofacial dysmorphia signs, cardiac and ophthalmological alterations. | [27] |
| c.1109T > C; p.V370A | 19 (one family) | 12 females 7 males | Yes | From 6 to 24 | Progressive | HA and 3 CI | Not reported | [8] | |
| 24 | p.Ala58Val | 3 (one family) | Male | Yes | 7 | Not reported | HA | Eye problems | [28] |
| Male | Yes | 4 | Not reported | Not reported | Eye problems | [28] | |||
| Female | Yes | Early childhood | Not reported | HA | Eye problems | [28] | |||
| 25 | Not reported | 4 families | Male | Yes | Not reported | Progressive (high frequencies) | Not reported | Not reported | [36] |
| Not reported | Yes | Not reported | Progressive | Not reported | Not reported | ||||
| Not reported | Yes | Not reported | Progressive | Not reported | Not reported | ||||
| Not reported | Yes | Not reported | Progressive | Not reported | Not reported | ||||
References
- Sonnemann, K.J.; Fitzsimons, D.P.; Patel, J.R.; Liu, Y.; Schneider, M.F.; Moss, R.L.; Ervasti, J.M. Cytoplasmic gamma-actin is not required for skeletal muscle development but its absence leads to a progressive myopathy. Dev. Cell 2006, 11, 387–397. [Google Scholar] [CrossRef]
- Perrin, B.J.; Sonnemann, K.J.; Ervasti, J.M. Beta-actin and gamma-actin are each dispensable for auditory hair cell development but required for Stereocilia maintenance. PLoS Genet. 2010, 6, e1001158. [Google Scholar] [CrossRef]
- Drummond, M.C.; Belyantseva, I.A.; Friderici, K.H.; Friedman, T.B. Actin in hair cells and hearing loss. Hear. Res. 2012, 288, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Clin Var. Available online: https://www.ncbi.nlm.nih.gov/clinvar/?term=ACTB+gene (accessed on 8 February 2024).
- Clin Var. Available online: https://www.ncbi.nlm.nih.gov/clinvar/?term=ACTG1+gene (accessed on 8 February 2024).
- Morell, R.J.; Friderici, K.H.; Wei, S.; Elfenbein, J.L.; Friedman, T.B.; Fisher, R.A. A new locus for late-onset, progressive, hereditary hearing loss DFNA20 maps to 17q25. Genomics 2000, 63, 1–6. [Google Scholar] [CrossRef] [PubMed]
- DeWan, A.T.; Parrado, A.R.; Leal, S.M. A second kindred linked to DFNA20 (17q25.3) reduces the genetic interval. Clin. Genet. 2003, 63, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Rendtorff, N.D.; Zhu, M.; Fagerheim, T.; Antal, T.L.; Jones, M.; Teslovich, T.M.; Gillanders, E.M.; Barmada, M.; Teig, E.; Trent, J.M.; et al. A novel missense mutation in ACTG1 causes dominant deafness in a Norwegian DFNA20/26 family, but ACTG1 mutations are not frequent among families with hereditary hearing impairment. Europ. J. Hum. Genet. 2006, 14, 1097–1105. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, U.; Piccolo, C.; Rigon, C.; Brasson, V.; Trevisson, E.; Boaretto, F.; Martini, A.; Cassina, M. DFNA20/26 and Other ACTG1-Associated Phenotypes: A Case Report and Review of the Literature. Audiol. Res. 2021, 11, 582–593. [Google Scholar] [CrossRef] [PubMed]
- Riviere, J.B.; van Bon, B.W.M.; Hoischen, A.; Kholmanskikh, S.S.; O’Roak, B.J.; Gilissen, C.; Gijsen, S.; Sullivan, C.T.; Christian, S.L.; Abdul-Rahman, O.A.; et al. De novo mutations in the actin genes ACTB and ACTG1 cause Baraitser-Winter syndrome. Nat. Genet. 2012, 44, 440–444. [Google Scholar] [CrossRef]
- Verloes, A.; Di Donato, N.; Masliah-Planchon, J.; Jongmans, M.; Abdul-Raman, O.A.; Albrecht, B.; Allanson, J.; Brunner, H.; Bertola, D.; Chassaing, N.; et al. Baraitser-Winter cerebrofrontofacial syndrome: Delineation of the spectrum in 42 cases. Eur. J. Hum. Genet. 2015, 23, 292–301. [Google Scholar] [CrossRef]
- Di Donato, N.; Rump, A.; Koenig, R.; Der Kaloustian, V.M.; Halal, F.; Sonntag, K.; Krause, C.; Hackmann, K.; Hahn, G.; Schrock, E.; et al. Severe forms of Baraitser-Winter syndrome are caused by ACTB mutations rather than ACTG1 mutations. Eur. J. Hum. Genet 2014, 22, 179–183. [Google Scholar] [CrossRef]
- Armstrong, E.C. The well-built clinical question: The key to finding the best evidence efficently. WMJ 1999, 98, 25–28. [Google Scholar]
- Moher, D.; Liberati, A.; Tetzlaff, J.; Altman, D.G.; PRISMA Group. Reprint–preferred reporting items for systematic reviews and meta-analyses: The PRISMA statement. Phys. Ther. 2009, 89, 873–880. [Google Scholar] [CrossRef] [PubMed]
- Skogseid, I.M.; Røsby, O.; Konglund, A.; Connelly, J.P.; Nedregaard, B.; Jablonski, G.E.; Kvernmo, N.; Stray-Pedersen, A.; Glover, J.C. Dystonia-deafness syndrome caused by ACTB p.Arg183Trp heterozygosity shows striatal dopaminergic dysfunction and response to pallidal stimulation. J. Neurodev. Disord. 2018, 10, 17. [Google Scholar] [CrossRef] [PubMed]
- Conboy, E.; Vairo, F.; Waggoner, D.; Ober, C.; Das, S.; Dhamija, R.; Klee, E.W.; Pichurin, P. Pathogenic Variant in ACTB, p.Arg183Trp, Causes Juvenile-Onset Dystonia, Hearing Loss, and Developmental Delay without Midline Malformation. Case Rep. Genet. 2017, 2017, 9184265. [Google Scholar] [CrossRef]
- Kola, S.; Kandadai, R.M.; Kashyap, M.; Deepak, S.; Prasad, V.; Alugolu, R.; Borgohain, R. Dystonia Deafness Syndrome: A Rare Deep Brain Stimulation Responsive Dystonia. Ann. Indian Acad. Neurol. 2023, 26, 766–768. [Google Scholar] [CrossRef]
- Straccia, G.; Reale, C.; Castellani, M.; Colangelo, I.; Orunesu, E.; Meoni, S.; Moro, E.; Krack, P.; Prokisch, H.; Zech, M.; et al. ACTB gene mutation in combined Dystonia-Deafness syndrome with parkinsonism: Expanding the phenotype and highlighting the long-term GPi DBS outcome. Park. Relat. Disord. 2022, 104, 3–6. [Google Scholar] [CrossRef] [PubMed]
- Zavala, L.; Ziegler, G.; Morón, D.G.; Garretto, N. Dystonia-Deafness Syndrome: ACTB Pathogenic Variant in an Argentinean Family. Mov. Disord. Clin. Pract. 2021, 9, 122–124. [Google Scholar] [CrossRef]
- Freitas, J.L.; Vale, T.C.; Barsottini, O.G.P.; Pedroso, J.L. Expanding the Phenotype of Dystonia-Deafness Syndrome Caused by ACTB Gene Mutation. Mov. Disord. Clin. Pract. 2019, 7, 86–87. [Google Scholar] [CrossRef]
- Sun, Y.; Shen, X.; Li, Q.; Kong, Q. Child with cerebral malformations and epilepsy. Int. J. Neurosci. 2018, 128, 881–885. [Google Scholar] [CrossRef]
- Der Kaloustiana, V.M.; Pelletiera, M.; Costab, T.; Blackstonc, D.R.; Oudjhane, K. A new syndrome with craniofacial and skeletal dysmorphisms and developmental delay. Clin. Dysmorphol. 2001, 10, 87–93. [Google Scholar] [CrossRef]
- Chacon-Camacho, O.F.; Barragán-Arévalo, T.; Villarroel, C.E.; Almanza-Monterrubio, M.; Zenteno, J.C. Previously undescribed phenotypic findings and novel ACTG1 gene pathogenic variants in Baraitser-Winter cerebrofrontofacial syndrome. Eur. J. Med. Genet. 2020, 63, 103877. [Google Scholar] [CrossRef] [PubMed]
- Miyajima, H.; Moteki, H.; Day, T.; Nishio, S.Y.; Murata, T.; Ikezono, T.; Takeda, H.; Abe, S.; Iwasaki, S.; Takahashi, M.; et al. Novel ACTG1 mutations in patients identified by massively parallel DNA sequencing cause progressive hearing loss. Sci. Rep. 2020, 10, 7056. [Google Scholar] [CrossRef]
- Park, G.; Gim, J.; Kim, A.R.; Han, K.H.; Kim, H.S.; Oh, S.H.; Park, T.; Park, W.Y.; Choi, B.Y. Multiphasic analysis of whole exome sequencing data identifies a novel mutation of ACTG1 in a nonsyndromic hearing loss family. BMC Genom. 2013, 14, 191. [Google Scholar] [CrossRef] [PubMed]
- Graziani, L.; Cinnirella, G.; Ferradini, V.; Conte, C.; Bascio, F.L.; Bengala, M.; Sangiuolo, F.; Novelli, G. A likely pathogenic ACTG1 variant in a child showing partial phenotypic overlap with Baraitser-Winter syndrome. Am. J. Med. Genet. A 2023, 191, 1565–1569. [Google Scholar] [CrossRef] [PubMed]
- Göbe, T.; Berninger, L.; Schlump, A.; Feige, B.; Nickel, K.R.; Schiele, M.A.; van Elst, L.T.; Hotz, A.; Alter, S.; Domschke, K.; et al. Obsessive–compulsive symptoms in ACTG1 associated Baraitser Winter cerebrofrontofacial syndrome. J. Neural. Transm. 2022, 129, 1387–1391. [Google Scholar] [CrossRef]
- Kemerley, A.; Sloan, C.; Pfeifer, W.; Smith, R.; Drack, A. A novel mutation in ACTG1 causing Baraitser-Winter syndrome with extremely variable expressivity in three generations. Ophthalmic Genet. 2017, 38, 152–156. [Google Scholar] [CrossRef]
- Hampshire, K.; Martin, P.M.; Carlston, C.; Slavotinek, A. Baraitser-Winter cerebrofrontofacial syndrome: Report of two adult siblings. Am. J. Med. Genet. A 2020, 182, 1923–1932. [Google Scholar] [CrossRef]
- Dawidziuk, M.; Kutkowska-Kazmierczak, A.; Bukowska-Olech, E.; Jurek, M.; Kalka, E.; Guilbride, D.L.; Furmanek, M.I.; Bekiesinska-Figatowska, M.; Bal, J.; Gawlinski, P. De Novo ACTG1 Variant Expands the Phenotype and Genotype of Partial Deafness and Baraitser-Winter Syndrome. Int. J. Mol. Sci. 2022, 23, 692. [Google Scholar] [CrossRef]
- Yates, T.M.; Turner, C.L.; Firth, H.V.; Berg, J.; Pilz, D.T. Baraitser-Winter cerebrofrontofacial syndrome. Clin. Genet. 2017, 92, 3–9. [Google Scholar] [CrossRef]
- National Center for Biotechnology Information. ClinVar; [VCV000449191.7]. Available online: https://www.ncbi.nlm.nih.gov/clinvar/variation/VCV000449191.7 (accessed on 5 January 2024).
- Aldè, M.; Di Berardino, F.; Ambrosetti, U.; Barozzi, S.; Piatti, G.; Consonni, D.; Zanetti, D.; Pignataro, L.; Cantarella, G. Hearing outcomes in preterm infants with confirmed hearing loss. Int. J. Pediatr. Otorhinolaryngol. 2022, 161, 111262. [Google Scholar] [CrossRef]
- Bovo, R.; Trevisi, P.; Ghiselli, S.; Benatti, A.; Martini, A. Is very early hearing assessment always reliable in selecting patients for cochlear implants? A case series study. Int. J. Pediatr. Otorhinolaryngol. 2015, 79, 725–731. [Google Scholar] [CrossRef] [PubMed]
- Miyagawa, M.; Nishio, S.A.; Ichinose, A.; Iwasaki, S.; Murata, T.; Kitajiri, S.I.; Usami, S.I. Mutational spectrum and clinical features of patients with ACTG1 mutations identified by massively parallel DNA sequencing. Ann. Otol. Rhinol. Laryngol. 2015, 124 (Suppl. S1), 84S–93S. [Google Scholar] [CrossRef]
- Zhu, M.; Yang, T.; Wei, S.; DeWan, A.T.; Morell, R.J.; Elfenbein, J.L.; Fisher, R.A.; Leal, S.M.; Smith, R.J.; Friderici, K.H. Mutations in the gamma-actin gene (ACTG1) are associated with dominant progressive deafness (DFNA20/26). Am. J. Hum. Genet. 2003, 73, 1082–1091. [Google Scholar] [CrossRef] [PubMed]
- Sibbin, K.; Yap, P.; Nyaga, D.; Heller, R.; Evans, S.; Strachan, K.; Alburaiky, S.; Nguyen, H.M.A.; Hermann-Le Denmat, S.; Ganley, A.R.D.; et al. A de novo ACTB gene pathogenic variant in identical twins with phenotypic variation for hydrops and jejunal atresia. Am. J. Med. Genet. A 2022, 188, 1299–1306. [Google Scholar] [CrossRef] [PubMed]
- National Center for Biotechnology Information. ClinVar; [VCV000279997.28]. Available online: https://www.ncbi.nlm.nih.gov/clinvar/variation/VCV000279997.28 (accessed on 7 January 2024).
- Choi, G.J.; Kim, M.S.; Park, H.; Kim, J.Y.; Choi, J.M.; Lee, S.M.; Jang, J.H.; Cho, S.Y.; Jin, D.K. The First Korean Case of Baraitser-Winter Cerebro-Fronto-Facial Syndrome with a Novel Mutation in ACTB Diagnosed Via Targeted Gene Panel Sequencing and Literature Review. Ann. Clin. Lab. Sci. 2020, 50, 818–824. [Google Scholar] [PubMed]
- Eker, H.K.; Derinkuyu, B.E.; Ünal, S.; Masliah-Planchon, J.; Drunat, S.; Verloes, A. Cerebro-fronto-facial syndrome type 3 with polymicrogyria: A clinical presentation of Baraitser-Winter syndrome. Eur. J. Med. Genet. 2014, 57, 32–36. [Google Scholar] [CrossRef]
- Latham, S.L.; Ehmke, N.; Reinke, P.Y.A.; Taft, M.H.; Eicke, D.; Reindl, T.; Stenzel, W.; Lyons, M.J.; Friez, M.J.; Lee, J.A.; et al. Variants in exons 5 and 6 of ACTB cause syndromic thrombocytopenia. Nat. Commun. 2018, 9, 4250. [Google Scholar] [CrossRef]
- Ruthberg, J.S.; Kocharyan, A.; Farrokhian, N.; Stahl, M.C.; Hicks, K.; Scarborough, J.; Murray, G.S.; Wu, S.; Manzoor, N.; Otteson, T. Hearing loss patterns in enlarged vestibular aqueduct syndrome: Do fluctuations have clinical significance? Int. J. Pediatr. Otorhinolaryngol. 2022, 156, 111072. [Google Scholar] [CrossRef]
- Sandestig, A.; Green, A.; Jonasson, J.; Vogt, H.; Wahlström, J.; Pepler, A.; Ellnebo, K.; Biskup, S.; Stefanova, M. Could Dissimilar Phenotypic Effects of ACTB Missense Mutations Reflect the Actin Conformational Change? Two Novel Mutations and Literature Review. Mol. Syndromol. 2019, 9, 259–265. [Google Scholar] [CrossRef]
- Nie, M.; Liu, Q.; Yan, C. Skeletal Muscle Transcriptomic Comparison Between Men and Women in Response to Acute Sprint Exercise. Front. Genet. 2022, 13, 860815. [Google Scholar] [CrossRef]
- Weitensteiner, V.; Zhang, R.; Bungenberg, J.; Marks, M.; Gehlen, J.; Ralser, D.J.; Hilger, A.C.; Sharma, A.; Schumacher, J.; Gembruch, U.; et al. Exome sequencing in syndromic brain malformations identifies novel mutations in ACTB, and SLC9A6, and suggests BAZ1A as a new candidate gene. Birth Defects Res. 2018, 110, 587–597. [Google Scholar] [CrossRef]
- Rall, N.; Leon, A.; Gomez, R.; Daroca, J.; Lacassie, Y. New ocular finding in Baraitser-Winter syndrome (BWS). Eur. J. Med. Genet. 2018, 61, 21–23. [Google Scholar] [CrossRef] [PubMed]
- Nunoi, H.; Yamazaki, T.; Tsuchiya, H.; Kato, S.; Malech, H.L.; Matsuda, I.; Kanegasaki, S. A heterozygous mutation of beta-actin associated with neutrophil dysfunction and recurrent infection. Proc. Natl. Acad. Sci. USA 1999, 96, 8693–8698. [Google Scholar] [CrossRef] [PubMed]
- Cuvertino, S.; Stuart, H.M.; Chandler, K.E.; Roberts, N.A.; Armstrong, R.; Bernardini, L.; Bhaskar, S.; Callewaert, B.; Clayton-Smith, J.; Davalillo, C.H.; et al. ACTB Loss-of-Function Mutations Result in a Pleiotropic Developmental Disorder. Am. J. Hum. Genet. 2017, 101, 1021–1033. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.G.; Jang, J.; Jin, H.S. A novel missense mutation in the ACTG1 gene in a family with congenital autosomal dominant deafness: A case report. Mol. Med. Rep. 2018, 17, 7611–7617. [Google Scholar] [CrossRef]
- Wang, H.; Guan, J.; Lan, L.; Yu, L.; Xie, L.; Liu, X.; Yang, J.; Zhao, C.; Wang, D.; Wang, Q. A novel de novo mutation of ACTG1 in two sporadic non-syndromic hearing loss cases. Sci. China Life Sci. 2018, 61, 729–732. [Google Scholar] [CrossRef]
- De Heer, A.M.; Huygen, P.L.; Collin, R.W.; Oostrik, J.; Kremer, H.; Cremers, C.W. Audiometric and vestibular features in a second Dutch DFNA20/26 family with a novel mutation in ACTG1. Ann. Otol. Rhinol. Laryngol. 2009, 118, 382–390. [Google Scholar] [CrossRef]
- Liu, P.; Li, H.; Ren, X.; Mao, H.; Zhu, Q.; Zhu, Z.; Yang, R.; Yuan, W.; Liu, J.; Wang, Q.; et al. Novel ACTG1 mutation causing autosomal dominant non-syndromic hearing impairment in a Chinese family. J. Genet. Genom. 2008, 35, 553–558. [Google Scholar] [CrossRef]
- Yuan, Y.; Gao, X.; Huang, B.; Lu, J.; Wang, G.; Lin, X.; Qu, Y.; Dai, P. Phenotypic Heterogeneity in a DFNA20/26 family segregating a novel ACTG1 mutation. BMC Genet. 2016, 17, 33. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ghiselli, S.; Parmeggiani, G.; Zambonini, G.; Cuda, D. Hearing Loss in Baraitser–Winter Syndrome: Case Reports and Review of the Literature. J. Clin. Med. 2024, 13, 1500. https://doi.org/10.3390/jcm13051500
Ghiselli S, Parmeggiani G, Zambonini G, Cuda D. Hearing Loss in Baraitser–Winter Syndrome: Case Reports and Review of the Literature. Journal of Clinical Medicine. 2024; 13(5):1500. https://doi.org/10.3390/jcm13051500
Chicago/Turabian StyleGhiselli, Sara, Giulia Parmeggiani, Giulia Zambonini, and Domenico Cuda. 2024. "Hearing Loss in Baraitser–Winter Syndrome: Case Reports and Review of the Literature" Journal of Clinical Medicine 13, no. 5: 1500. https://doi.org/10.3390/jcm13051500
APA StyleGhiselli, S., Parmeggiani, G., Zambonini, G., & Cuda, D. (2024). Hearing Loss in Baraitser–Winter Syndrome: Case Reports and Review of the Literature. Journal of Clinical Medicine, 13(5), 1500. https://doi.org/10.3390/jcm13051500