
Retinal Imaging Findings in Inherited Retinal Diseases

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Panretinal Pigmentary Retinopathies
2.1. Cone Dystrophy and Cone–Rod Dystrophies (COD/CORD)
2.1.1. GUCA1A-Associated COD/CORD
2.1.2. GUCY2D-Associated COD/CORD
2.1.3. PRPH2-Associated CORD
2.1.4. ABCA4-Associated COD/CORD
2.1.5. RPGR-Associated COD/CORD
2.2. Rod–Cone Dystrophy (RCD) and Associated Diseases
2.2.1. Retinitis Pigmentosa (RP)
2.2.2. RPGR-Associated RP
2.2.3. RP2-Associated RP
2.2.4. USH2A-Associated RP
2.2.5. RLBP1-Associated RP
2.2.6. NR2E3-Associated RP
2.3. Leber Congenital Amaurosis (LCA)
2.3.1. RPE65-Associated LCA
2.3.2. CRB1-Associated LCA
2.3.3. GUCY2D-Associated LCA
2.3.4. CEP290-Associated LCA
2.3.5. RDH12-Associated LCA
2.4. Enhanced S-Cone Syndrome (ESCS, or Goldman-Favre Syndrome)
2.5. Choroideremia (CHM)
2.6. Gyrate Atrophy
3. Macular Dystrophies
3.1. Stargardt Disease (STGD)/Fundus Flavimaculatus
3.2. Bestrophinopathies
3.2.1. Best’s Disease (BD)/Best Vitelliform Macular Dystrophy (BVMD)
3.2.2. Autosomal Recessive Bestrophinopathy (ARB)
3.2.3. Adult-Onset Vitelliform Macular Dystrophy (AOVMD)
3.2.4. Pattern Dystrophy (PD)
3.2.5. Sorsby Fundus Dystrophy (SFD)
3.2.6. North Carolina Macular Dystrophy (NCMD)
3.2.7. Central Areolar Choroidal Dystrophy (CACD)
3.2.8. Autosomal Dominant Occult Macular Dystrophy (OCMD)
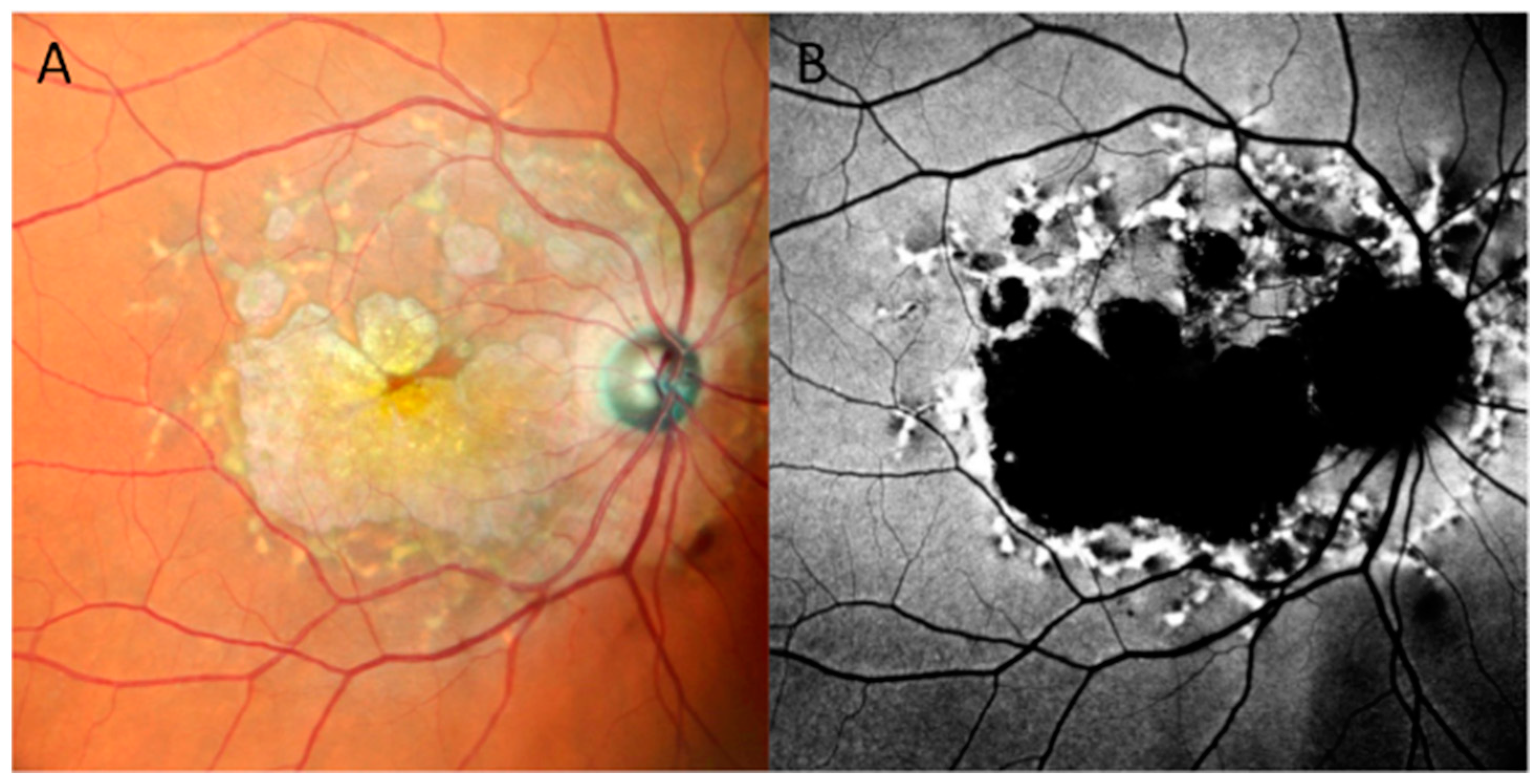
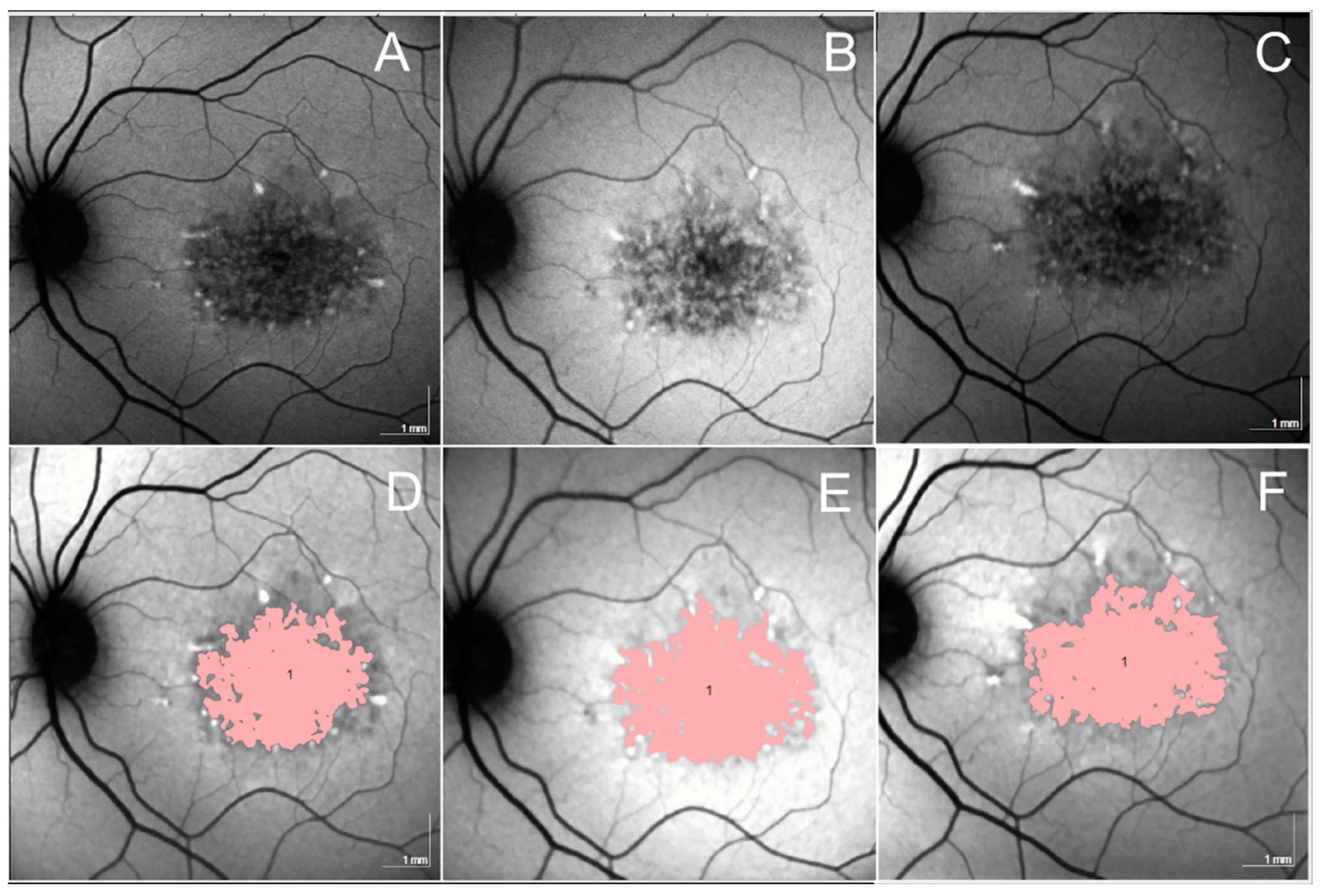
3.2.9. Doyne Honeycomb Retinal Dystrophy/Autosomal Dominant Drusen (ADD)
4. Stationary Conditions
4.1. Complete and Incomplete Congenital Stationary Night Blindness (CSNB)
4.2. Congenital Achromatopsia (ACHM)
5. Congenital Vitreoretinopathies
Congenital X-Linked Retinoschisis (XLRS)
6. Limitations, Strengths
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Krumpaszky, H.G.; Lüdtke, R.; Mickler, A.; Klauss, V.; Selbmann, H.K. Blindness incidence in Germany. A population-based study from Württemberg-Hohenzollern. Ophthalmologica 1999, 213, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Cremers, F.P.M.; Boon, C.J.F.; Bujakowska, K.; Zeitz, C. Special Issue Introduction: Inherited Retinal Disease: Novel Candidate Genes, Genotype-Phenotype Correlations, and Inheritance Models. Genes 2018, 9, 215. [Google Scholar] [CrossRef] [PubMed]
- Liew, G.; Michaelides, M.; Bunce, C. A comparison of the causes of blindness certifications in England and Wales in working age adults (16–64 years), 1999–2000 with 2009–2010. BMJ Open 2014, 4, e004015. [Google Scholar] [CrossRef]
- Duncan, J.L.; Pierce, E.A.; Laster, A.M.; Daiger, S.P.; Birch, D.G.; Ash, J.D.; Iannaccone, A.; Flannery, J.G.; Sahel, J.A.; Zack, D.J.; et al. Inherited Retinal Degenerations: Current Landscape and Knowledge Gaps. Transl. Vis. Sci. Technol. 2018, 7, 6. [Google Scholar] [CrossRef]
- Stone, E.M. Challenges in genetic testing for clinical trials of inherited and orphan retinal diseases. Retina 2005, 25, S72–S73. [Google Scholar] [CrossRef]
- Bighinati, A.; Adani, E.; Stanzani, A.; D’Alessandro, S.; Marigo, V. Molecular mechanisms underlying inherited photoreceptor degeneration as targets for therapeutic intervention. Front. Cell. Neurosci. 2024, 18, 1343544. [Google Scholar] [CrossRef] [PubMed]
- Campochiaro, P.A.; Strauss, R.W.; Lu, L.; Hafiz, G.; Wolfson, Y.; Shah, S.M.; Sophie, R.; Mir, T.A.; Scholl, H.P. Is There Excess Oxidative Stress and Damage in Eyes of Patients with Retinitis Pigmentosa? Antioxid. Redox Signal. 2015, 23, 643–648. [Google Scholar] [CrossRef] [PubMed]
- Bovi Dos Santos, G.; de Lima-Vasconcellos, T.H.; Móvio, M.I.; Birbrair, A.; Del Debbio, C.B.; Kihara, A.H. New Perspectives in Stem Cell Transplantation and Associated Therapies to Treat Retinal Diseases: From Gene Editing to 3D Bioprinting. Stem Cell Rev. Rep. 2024, 20, 722–737. [Google Scholar] [CrossRef] [PubMed]
- Brar, A.S.; Parameswarappa, D.C.; Takkar, B.; Narayanan, R.; Jalali, S.; Mandal, S.; Fujinami, K.; Padhy, S.K. Gene Therapy for Inherited Retinal Diseases: From Laboratory Bench to Patient Bedside and Beyond. Ophthalmol. Ther. 2024, 13, 21–50. [Google Scholar] [CrossRef]
- Fenner, B.J.; Tan, T.-E.; Barathi, A.V.; Tun, S.B.B.; Yeo, S.W.; Tsai, A.S.H.; Lee, S.Y.; Cheung, C.M.G.; Chan, C.M.; Mehta, J.S.; et al. Gene-Based Therapeutics for Inherited Retinal Diseases. Front. Genet. 2021, 12, 794805. [Google Scholar] [CrossRef]
- Martinez Velazquez, L.A.; Ballios, B.G. The Next Generation of Molecular and Cellular Therapeutics for Inherited Retinal Disease. Int. J. Mol. Sci. 2021, 22, 11542. [Google Scholar] [CrossRef] [PubMed]
- Ren, D.; Fisson, S.; Dalkara, D.; Ail, D. Immune Responses to Gene Editing by Viral and Non-Viral Delivery Vectors Used in Retinal Gene Therapy. Pharmaceutics 2022, 14, 1973. [Google Scholar] [CrossRef]
- Chen, X.; Xu, N.; Li, J.; Zhao, M.; Huang, L. Stem cell therapy for inherited retinal diseases: A systematic review and meta-analysis. Stem Cell Res. Ther. 2023, 14, 286. [Google Scholar] [CrossRef] [PubMed]
- Thompson, D.A.; Iannaccone, A.; Ali, R.R.; Arshavsky, V.Y.; Audo, I.; Bainbridge, J.W.B.; Besirli, C.G.; Birch, D.G.; Branham, K.E.; Cideciyan, A.V.; et al. Advancing Clinical Trials for Inherited Retinal Diseases: Recommendations from the Second Monaciano Symposium. Transl. Vis. Sci. Technol. 2020, 9, 2. [Google Scholar] [CrossRef] [PubMed]
- Ashourizadeh, H.; Fakhri, M.; Hassanpour, K.; Masoudi, A.; Jalali, S.; Roshandel, D.; Chen, F.K. Pearls and Pitfalls of Adaptive Optics Ophthalmoscopy in Inherited Retinal Diseases. Diagnostics 2023, 13, 2413. [Google Scholar] [CrossRef] [PubMed]
- Schmetterer, L.; Scholl, H.; Garhöfer, G.; Janeschitz-Kriegl, L.; Corvi, F.; Sadda, S.R.; Medeiros, F.A. Endpoints for clinical trials in ophthalmology. Prog. Retin. Eye Res. 2023, 97, 101160. [Google Scholar] [CrossRef] [PubMed]
- Csaky, K.; Ferris, F., 3rd; Chew, E.Y.; Nair, P.; Cheetham, J.K.; Duncan, J.L. Report From the NEI/FDA Endpoints Workshop on Age-Related Macular Degeneration and Inherited Retinal Diseases. Investig. Ophthalmol. Vis. Sci. 2017, 58, 3456–3463. [Google Scholar] [CrossRef] [PubMed]
- Coco-Martin, R.M.; Diego-Alonso, M.; Orduz-Montaña, W.A.; Sanabria, M.R.; Sanchez-Tocino, H. Descriptive Study of a Cohort of 488 Patients with Inherited Retinal Dystrophies. Clin. Ophthalmol. 2021, 15, 1075–1084. [Google Scholar] [CrossRef]
- Hamel, C.P. Cone rod dystrophies. Orphanet J. Rare Dis. 2007, 2, 7. [Google Scholar] [CrossRef]
- Sparrow, J.R.; Yoon, K.D.; Wu, Y.; Yamamoto, K. Interpretations of fundus autofluorescence from studies of the bisretinoids of the retina. Investig. Ophthalmol. Vis. Sci. 2010, 51, 4351–4357. [Google Scholar] [CrossRef]
- Kominami, A.; Ueno, S.; Kominami, T.; Nakanishi, A.; Ito, Y.; Fujinami, K.; Tsunoda, K.; Hayashi, T.; Kikuchi, S.; Kameya, S.; et al. Case of cone dystrophy with normal fundus appearance associated with biallelic POC1B variants. Ophthalmic Genet. 2018, 39, 255–262. [Google Scholar] [CrossRef]
- Wolfing, J.I.; Chung, M.; Carroll, J.; Roorda, A.; Williams, D.R. High-resolution retinal imaging of cone-rod dystrophy. Ophthalmology 2006, 113, 1019.e1. [Google Scholar] [CrossRef]
- Choi, S.S.; Doble, N.; Hardy, J.L.; Jones, S.M.; Keltner, J.L.; Olivier, S.S.; Werner, J.S. In vivo imaging of the photoreceptor mosaic in retinal dystrophies and correlations with visual function. Investig. Ophthalmol. Vis. Sci. 2006, 47, 2080–2092. [Google Scholar] [CrossRef] [PubMed]
- Duncan, J.L.; Zhang, Y.; Gandhi, J.; Nakanishi, C.; Othman, M.; Branham, K.E.H.; Swaroop, A.; Roorda, A. High-resolution imaging with adaptive optics in patients with inherited retinal degeneration. Investig. Ophthalmol. Vis. Sci. 2007, 48, 3283–3291. [Google Scholar] [CrossRef]
- Vincent, A.; Wright, T.; Garcia-Sanchez, Y.; Kisilak, M.; Campbell, M.; Westall, C.; Héon, E. Phenotypic characteristics including in vivo cone photoreceptor mosaic in KCNV2-related “cone dystrophy with supernormal rod electroretinogram”. Investig. Ophthalmol. Vis. Sci. 2013, 54, 898–908. [Google Scholar] [CrossRef]
- Kikuchi, S.; Kameya, S.; Gocho, K.; El Shamieh, S.; Akeo, K.; Sugawara, Y.; Yamaki, K.; Zeitz, C.; Audo, I.; Takahashi, H. Cone dystrophy in patient with homozygous RP1L1 mutation. Biomed. Res. Int. 2015, 2015, 545243. [Google Scholar] [CrossRef]
- Roorda, A.; Zhang, Y.; Duncan, J.L. High-resolution in vivo imaging of the RPE mosaic in eyes with retinal disease. Investig. Ophthalmol. Vis. Sci. 2007, 48, 2297–2303. [Google Scholar] [CrossRef] [PubMed]
- Duncan, J.L.; Talcott, K.E.; Ratnam, K.; Sundquist, S.M.; Lucero, A.S.; Day, S.; Zhang, Y.; Roorda, A. Cone structure in retinal degeneration associated with mutations in the peripherin/RDS gene. Investig. Ophthalmol. Vis. Sci. 2011, 52, 1557–1566. [Google Scholar] [CrossRef] [PubMed]
- Duncan, J.L.; Roorda, A.; Navani, M.; Vishweswaraiah, S.; Syed, R.; Soudry, S.; Ratnam, K.; Gudiseva, H.V.; Lee, P.; Gaasterland, T.; et al. Identification of a novel mutation in the CDHR1 gene in a family with recessive retinal degeneration. Arch. Ophthalmol. 2012, 130, 1301–1308. [Google Scholar] [CrossRef]
- Scoles, D.; Flatter, J.A.; Cooper, R.F.; Langlo, C.S.; Robison, S.; Neitz, M.; Weinberg, D.V.; Pennesi, M.E.; Han, D.P.; Dubra, A.; et al. Assessing Photoreceptor Structure Associated with Ellipsoid Zone Disruptions Visualized with Optical Coherence Tomography. Retina 2016, 36, 91–103. [Google Scholar] [CrossRef]
- Kubota, D.; Gocho, K.; Kikuchi, S.; Akeo, K.; Miura, M.; Yamaki, K.; Takahashi, H.; Kameya, S. CEP250 mutations associated with mild cone-rod dystrophy and sensorineural hearing loss in a Japanese family. Ophthalmic Genet. 2018, 39, 500–507. [Google Scholar] [CrossRef] [PubMed]
- Ueda-Consolvo, T.; Ozaki, H.; Nakamura, T.; Oiwake, T.; Hayashi, A. The association between cone density and visual function in the macula of patients with retinitis pigmentosa. Graefes Arch. Clin. Exp. Ophthalmol. 2019, 257, 1841–1846. [Google Scholar] [CrossRef] [PubMed]
- Michaelides, M.; Hardcastle, A.J.; Hunt, D.M.; Moore, A.T. Progressive cone and cone-rod dystrophies: Phenotypes and underlying molecular genetic basis. Surv. Ophthalmol. 2006, 51, 232–258. [Google Scholar] [CrossRef]
- Michaelides, M.; Wilkie, S.E.; Jenkins, S.; Holder, G.E.; Hunt, D.M.; Moore, A.T.; Webster, A.R. Mutation in the gene GUCA1A, encoding guanylate cyclase-activating protein 1, causes cone, cone-rod, and macular dystrophy. Ophthalmology 2005, 112, 1442–1447. [Google Scholar] [CrossRef] [PubMed]
- Tsokolas, G.; Almuhtaseb, H.; Griffiths, H.; Shawkat, F.; Pengelly, R.J.; Sarah, E.; Lotery, A. Long term follow-up of a family with GUCY2D dominant cone dystrophy. Int. J. Ophthalmol. 2018, 11, 1945–1950. [Google Scholar] [CrossRef] [PubMed]
- Verma, A.; Velaga, S.; Nittala, M.G.; Baker, K.; Huang, X.; Chhablani, J.; Sadda, S.R. Relationship between Preserved Ellipsoid Zone Area and Choroidal Vascularity Index in Autosomal Dominant Retinitis Pigmentosa. J. Clin. Exp. Ophthalmol. 2021, 13, 1–6. [Google Scholar]
- Ba-Abbad, R.; Robson, A.G.; MacPhee, B.; Webster, A.R.; Michaelides, M. Rod-cone dystrophy associated with the Gly167Asp variant in PRPH2. Ophthalmic Genet. 2019, 40, 188–189. [Google Scholar] [CrossRef] [PubMed]
- Soucy, M.; Kolesnikova, M.; Kim, A.H.; Tsang, S.H. Phenotypic variability in PRPH2 as demonstrated by a family with incomplete penetrance of autosomal dominant cone-rod dystrophy. Doc. Ophthalmol. 2023, 146, 267–272. [Google Scholar] [CrossRef]
- Danciger, M.; Hendrickson, J.; Lyon, J.; Toomes, C.; McHale, J.C.; Fishman, G.A.; Inglehearn, C.F.; Jacobson, S.G.; Farber, D.B. CORD9 a new locus for arCRD: Mapping to 8p11, estimation of frequency, evaluation of a candidate gene. Investig. Ophthalmol. Vis. Sci. 2001, 42, 2458–2465. [Google Scholar]
- Klevering, B.J.; Blankenagel, A.; Maugeri, A.; Cremers, F.P.M.; Hoyng, C.B.; Rohrschneider, K. Phenotypic spectrum of autosomal recessive cone-rod dystrophies caused by mutations in the ABCA4 (ABCR) gene. Investig. Ophthalmol. Vis. Sci. 2002, 43, 1980–1985. [Google Scholar]
- Thiadens, A.A.H.J.; Soerjoesing, G.G.; Florijn, R.J.; Tjiam, A.G.; den Hollander, A.I.; van den Born, L.I.; Riemslag, F.C.; Bergen, A.A.B.; Klaver, C.C.W. Clinical course of cone dystrophy caused by mutations in the RPGR gene. Graefes Arch. Clin. Exp. Ophthalmol. 2011, 249, 1527–1535. [Google Scholar] [CrossRef]
- Georgiou, M.; Robson, A.G.; Fujinami, K.; de Guimarães, T.A.C.; Fujinami-Yokokawa, Y.; Daich Varela, M.; Pontikos, N.; Kalitzeos, A.; Mahroo, O.A.; Webster, A.R.; et al. Phenotyping and genotyping inherited retinal diseases: Molecular genetics, clinical and imaging features, and therapeutics of macular dystrophies, cone and cone-rod dystrophies, rod-cone dystrophies, leber congenital amaurosis, and cone dysfunction syndromes. Prog. Retin. Eye Res. 2024, 100, 101244. [Google Scholar] [CrossRef]
- Hartong, D.T.; Berson, E.L.; Dryja, T.P. Retinitis pigmentosa. Lancet 2006, 368, 1795–1809. [Google Scholar] [CrossRef]
- Berni, A.; Arrigo, A.; Bianco, L.; Antropoli, A.; Saladino, A.; Mansour, A.M.; Vilela, M.; Bandello, F.; Parodi, M.B. New insights in the multimodal imaging of retinitis pigmentosa. Eur. J. Ophthalmol. 2024, 34, 357–366. [Google Scholar] [CrossRef]
- Aizawa, S.; Mitamura, Y.; Baba, T.; Hagiwara, A.; Ogata, K.; Yamamoto, S. Correlation between visual function and photoreceptor inner/outer segment junction in patients with retinitis pigmentosa. Eye 2009, 23, 304–308. [Google Scholar] [CrossRef]
- Hariri, A.H.; Zhang, H.Y.; Ho, A.; Francis, P.; Weleber, R.G.; Birch, D.G.; Ferris, F.L.; Sadda, S.R.; Trial of Oral Valproic Acid for Retinitis Pigmentosa Group. Quantification of Ellipsoid Zone Changes in Retinitis Pigmentosa Using en Face Spectral Domain-Optical Coherence Tomography. JAMA Ophthalmol. 2016, 134, 628–635. [Google Scholar] [CrossRef]
- Campochiaro, P.A.; Iftikhar, M.; Hafiz, G.; Akhlaq, A.; Tsai, G.; Wehling, D.; Lu, L.; Wall, G.M.; Singh, M.S.; Kong, X. Oral N-acetylcysteine improves cone function in retinitis pigmentosa patients in phase I trial. J. Clin. Investig. 2020, 130, 1527–1541. [Google Scholar] [CrossRef]
- Huang, C.-H.; Yang, C.-H.; Lai, Y.-J.; Hsiao, C.K.; Hou, Y.-C.; Yang, C.-M.; Chen, T.-C. Hyperreflective foci as important prognostic indicators of progression of retinitis pigmentosa. Retina 2022, 42, 388–395. [Google Scholar] [CrossRef]
- Iovino, C.; Au, A.; Hilely, A.; Violanti, S.; Peiretti, E.; Gorin, M.B.; Sarraf, D. Evaluation of the Choroid in Eyes with Retinitis Pigmentosa and Cystoid Macular Edema. Investig. Ophthalmol. Vis. Sci. 2019, 60, 5000–5006. [Google Scholar] [CrossRef]
- Murakami, T.; Akimoto, M.; Ooto, S.; Suzuki, T.; Ikeda, H.; Kawagoe, N.; Takahashi, M.; Yoshimura, N. Association between abnormal autofluorescence and photoreceptor disorganization in retinitis pigmentosa. Am. J. Ophthalmol. 2008, 145, 687–694. [Google Scholar] [CrossRef]
- Hariri, A.H.; Gui, W.; Datoo O’Keefe, G.A.; Ip, M.S.; Sadda, S.R.; Gorin, M.B. Ultra-Widefield Fundus Autofluorescence Imaging of Patients with Retinitis Pigmentosa: A Standardized Grading System in Different Genotypes. Ophthalmol. Retin. 2018, 2, 735–745. [Google Scholar] [CrossRef]
- Cicinelli, M.V.; Marchese, A.; Bordato, A.; Manitto, M.P.; Bandello, F.; Battaglia Parodi, M. Reviewing the Role of Ultra-Widefield Imaging in Inherited Retinal Dystrophies. Ophthalmol. Ther. 2020, 9, 249–263. [Google Scholar] [CrossRef]
- Ling, L.; Gao, F.; Zhang, Q.; He, T.; Zhao, Y.; Xing, Y.; Yu, Y.; Ji, K. Optical Coherence Tomography Angiography Assessed Retinal and Choroidal Microvasculature Features in Patients with Retinitis Pigmentosa: A Meta-Analysis. Biomed. Res. Int. 2019, 2019, 6723917. [Google Scholar] [CrossRef]
- Iovino, C.; Iodice, C.M.; Pisani, D.; Damiano, L.; Di Iorio, V.; Testa, F.; Simonelli, F. Clinical Applications of Optical Coherence Tomography Angiography in Inherited Retinal Diseases: An Up-to-Date Review of the Literature. J. Clin. Med. Res. 2023, 12, 3170. [Google Scholar] [CrossRef]
- Liu, R.; Lu, J.; Liu, Q.; Wang, Y.; Cao, D.; Wang, J.; Wang, X.; Pan, J.; Ma, L.; Jin, C.; et al. Effect of Choroidal Vessel Density on the Ellipsoid Zone and Visual Function in Retinitis Pigmentosa Using Optical Coherence Tomography Angiography. Investig. Ophthalmol. Vis. Sci. 2019, 60, 4328–4335. [Google Scholar] [CrossRef]
- Georgiou, M.; Kalitzeos, A.; Patterson, E.J.; Dubra, A.; Carroll, J.; Michaelides, M. Adaptive optics imaging of inherited retinal diseases. Br. J. Ophthalmol. 2018, 102, 1028–1035. [Google Scholar] [CrossRef]
- Coussa, R.G.; Basali, D.; Maeda, A.; DeBenedictis, M.; Traboulsi, E.I. Sector retinitis pigmentosa: Report of ten cases and a review of the literature. Mol. Vis. 2019, 25, 869–889. [Google Scholar]
- Georgiou, M.; Awadh Hashem, S.; Daich Varela, M.; Michaelides, M. Gene Therapy in X-linked Retinitis Pigmentosa Due to Defects in RPGR. Int. Ophthalmol. Clin. 2021, 61, 97–108. [Google Scholar] [CrossRef]
- Duncan, J.L.; Liang, W.; Maguire, M.G.; Porco, T.C.; Wong, J.; Audo, I.; Cava, J.A.; Grieve, K.; Kalitzeos, A.; Kreis, J.; et al. Change in Cone Structure Over 24 Months in USH2A-Related Retinal Degeneration. Am. J. Ophthalmol. 2023, 252, 77–93. [Google Scholar] [CrossRef]
- Dessalces, E.; Bocquet, B.; Bourien, J.; Zanlonghi, X.; Verdet, R.; Meunier, I.; Hamel, C.P. Early-onset foveal involvement in retinitis punctata albescens with mutations in RLBP1. JAMA Ophthalmol. 2013, 131, 1314–1323. [Google Scholar] [CrossRef]
- Coppieters, F.; Leroy, B.P.; Beysen, D.; Hellemans, J.; De Bosscher, K.; Haegeman, G.; Robberecht, K.; Wuyts, W.; Coucke, P.J.; De Baere, E. Recurrent mutation in the first zinc finger of the orphan nuclear receptor NR2E3 causes autosomal dominant retinitis pigmentosa. Am. J. Hum. Genet. 2007, 81, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Hauswirth, W.W.; Aleman, T.S.; Kaushal, S.; Cideciyan, A.V.; Schwartz, S.B.; Wang, L.; Conlon, T.J.; Boye, S.L.; Flotte, T.R.; Byrne, B.J.; et al. Treatment of leber congenital amaurosis due to RPE65 mutations by ocular subretinal injection of adeno-associated virus gene vector: Short-term results of a phase I trial. Hum. Gene Ther. 2008, 19, 979–990. [Google Scholar] [CrossRef] [PubMed]
- Maguire, A.M.; Russell, S.; Wellman, J.A.; Chung, D.C.; Yu, Z.-F.; Tillman, A.; Wittes, J.; Pappas, J.; Elci, O.; Marshall, K.A.; et al. Efficacy, Safety, and Durability of Voretigene Neparvovec-rzyl in RPE65 Mutation-Associated Inherited Retinal Dystrophy: Results of Phase 1 and 3 Trials. Ophthalmology 2019, 126, 1273–1285. [Google Scholar] [CrossRef] [PubMed]
- Heckenlively, J.R. Preserved para-arteriole retinal pigment epithelium (PPRPE) in retinitis pigmentosa. Br. J. Ophthalmol. 1982, 66, 26–30. [Google Scholar] [CrossRef]
- Jacobson, S.G.; Cideciyan, A.V.; Aleman, T.S.; Pianta, M.J.; Sumaroka, A.; Schwartz, S.B.; Smilko, E.E.; Milam, A.H.; Sheffield, V.C.; Stone, E.M. Crumbs homolog 1 (CRB1) mutations result in a thick human retina with abnormal lamination. Hum. Mol. Genet. 2003, 12, 1073–1078. [Google Scholar] [CrossRef] [PubMed]
- Corvi, F.; Juhn, A.; Corradetti, G.; Nguyen, T.V.; Fawzi, A.A.; Sarraf, D.; Sadda, S.R. Multimodal imaging of CRB1 retinitis pigmentosa with a peripheral retinal tumor. Retin. Cases Brief. Rep. 2022, 16, 407–410. [Google Scholar] [CrossRef]
- Bouzia, Z.; Georgiou, M.; Hull, S.; Robson, A.G.; Fujinami, K.; Rotsos, T.; Pontikos, N.; Arno, G.; Webster, A.R.; Hardcastle, A.J.; et al. GUCY2D-Associated Leber Congenital Amaurosis: A Retrospective Natural History Study in Preparation for Trials of Novel Therapies. Am. J. Ophthalmol. 2020, 210, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, S.G.; Cideciyan, A.V.; Sumaroka, A.; Roman, A.J.; Wu, V.; Swider, M.; Sheplock, R.; Krishnan, A.K.; Garafalo, A.V. Leber Congenital Amaurosis Due to GUCY2D Mutations: Longitudinal Analysis of Retinal Structure and Visual Function. Int. J. Mol. Sci. 2021, 22, 2031. [Google Scholar] [CrossRef]
- Walia, S.; Fishman, G.A.; Jacobson, S.G.; Aleman, T.S.; Koenekoop, R.K.; Traboulsi, E.I.; Weleber, R.G.; Pennesi, M.E.; Heon, E.; Drack, A.; et al. Visual acuity in patients with Leber’s congenital amaurosis and early childhood-onset retinitis pigmentosa. Ophthalmology 2010, 117, 1190–1198. [Google Scholar] [CrossRef]
- Sheck, L.; Davies, W.I.L.; Moradi, P.; Robson, A.G.; Kumaran, N.; Liasis, A.C.; Webster, A.R.; Moore, A.T.; Michaelides, M. Leber Congenital Amaurosis Associated with Mutations in CEP290, Clinical Phenotype, and Natural History in Preparation for Trials of Novel Therapies. Ophthalmology 2018, 125, 894–903. [Google Scholar] [CrossRef]
- Cideciyan, A.V.; Jacobson, S.G.; Ho, A.C.; Krishnan, A.K.; Roman, A.J.; Garafalo, A.V.; Wu, V.; Swider, M.; Sumaroka, A.; Van Cauwenbergh, C.; et al. Restoration of Cone Sensitivity to Individuals with Congenital Photoreceptor Blindness within the Phase 1/2 Sepofarsen Trial. Ophthalmol. Sci. 2022, 2, 100133. [Google Scholar] [CrossRef] [PubMed]
- Janecke, A.R.; Thompson, D.A.; Utermann, G.; Becker, C.; Hübner, C.A.; Schmid, E.; McHenry, C.L.; Nair, A.R.; Rüschendorf, F.; Heckenlively, J.; et al. Mutations in RDH12 encoding a photoreceptor cell retinol dehydrogenase cause childhood-onset severe retinal dystrophy. Nat. Genet. 2004, 36, 850–854. [Google Scholar] [CrossRef]
- Scott, H.A.; Place, E.M.; Ferenchak, K.; Zampaglione, E.; Wagner, N.E.; Chao, K.R.; DiTroia, S.P.; Navarro-Gomez, D.; Mukai, S.; Huckfeldt, R.M.; et al. Expanding the phenotypic spectrum in RDH12-associated retinal disease. Cold Spring Harb. Mol. Case Stud. 2020, 6, a004754. [Google Scholar] [CrossRef]
- de Carvalho, E.R.; Robson, A.G.; Arno, G.; Boon, C.J.F.; Webster, A.A.; Michaelides, M. Enhanced S-Cone Syndrome: Spectrum of Clinical, Imaging, Electrophysiologic, and Genetic Findings in a Retrospective Case Series of 56 Patients. Ophthalmol. Retin. 2021, 5, 195–214. [Google Scholar] [CrossRef] [PubMed]
- Talib, M.; van Schooneveld, M.J.; van Genderen, M.M.; Wijnholds, J.; Florijn, R.J.; Ten Brink, J.B.; Schalij-Delfos, N.E.; Dagnelie, G.; Cremers, F.P.; Wolterbeek, R.; et al. Genotypic and Phenotypic Characteristics of CRB1-Associated Retinal Dystrophies: A Long-Term Follow-up Study. Ophthalmology 2017, 124, 884–895. [Google Scholar] [CrossRef]
- Haider, N.B.; Naggert, J.K.; Nishina, P.M. Excess cone cell proliferation due to lack of a functional NR2E3 causes retinal dysplasia and degeneration in rd7/rd7 mice. Hum. Mol. Genet. 2001, 10, 1619–1626. [Google Scholar] [CrossRef] [PubMed]
- Yzer, S.; Barbazetto, I.; Allikmets, R.; van Schooneveld, M.J.; Bergen, A.; Tsang, S.H.; Jacobson, S.G.; Yannuzzi, L.A. Expanded clinical spectrum of enhanced S-cone syndrome. JAMA Ophthalmol. 2013, 131, 1324–1330. [Google Scholar] [CrossRef]
- Georgiou, M.; Fujinami, K.; Michaelides, M. Retinal imaging in inherited retinal diseases. Ann. Eye Sci. 2020, 5, 25. [Google Scholar] [CrossRef] [PubMed]
- McCulloch, C. Choroideremia: A clinical and pathologic review. Trans. Am. Ophthalmol. Soc. 1969, 67, 142–195. [Google Scholar]
- Hariri, A.H.; Velaga, S.B.; Girach, A.; Ip, M.S.; Le, P.V.; Lam, B.L.; Fischer, M.D.; Sankila, E.-M.; Pennesi, M.E.; Holz, F.G.; et al. Measurement and Reproducibility of Preserved Ellipsoid Zone Area and Preserved Retinal Pigment Epithelium Area in Eyes with Choroideremia. Am. J. Ophthalmol. 2017, 179, 110–117. [Google Scholar] [CrossRef]
- Foote, K.G.; Rinella, N.; Tang, J.; Bensaid, N.; Zhou, H.; Zhang, Q.; Wang, R.K.; Porco, T.C.; Roorda, A.; Duncan, J.L. Cone Structure Persists Beyond Margins of Short-Wavelength Autofluorescence in Choroideremia. Investig. Ophthalmol. Vis. Sci. 2019, 60, 4931–4942. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.W.; Johnson, R.D.; Williams, V.; Summerfelt, P.; Dubra, A.; Weinberg, D.V.; Stepien, K.E.; Fishman, G.A.; Carroll, J. Multimodal Imaging of Photoreceptor Structure in Choroideremia. PLoS ONE 2016, 11, e0167526. [Google Scholar] [CrossRef] [PubMed]
- Jain, N.; Jia, Y.; Gao, S.S.; Zhang, X.; Weleber, R.G.; Huang, D.; Pennesi, M.E. Optical Coherence Tomography Angiography in Choroideremia: Correlating Choriocapillaris Loss with Overlying Degeneration. JAMA Ophthalmol. 2016, 134, 697–702. [Google Scholar] [CrossRef] [PubMed]
- Tuten, W.S.; Vergilio, G.K.; Young, G.J.; Bennett, J.; Maguire, A.M.; Aleman, T.S.; Brainard, D.H.; Morgan, J.I. Visual Function at the Atrophic Border in Choroideremia Assessed with Adaptive Optics Microperimetry. Ophthalmol. Retin. 2019, 3, 888–899. [Google Scholar] [CrossRef]
- MacLaren, R.E.; Fischer, M.D.; Gow, J.A.; Lam, B.L.; Sankila, E.M.K.; Girach, A.; Panda, S.; Yoon, D.; Zhao, G.; Pennesi, M.E. Subretinal timrepigene emparvovec in adult men with choroideremia: A randomized phase 3 trial. Nat. Med. 2023, 29, 2464–2472. [Google Scholar] [CrossRef] [PubMed]
- Aleman, T.S.; Huckfeldt, R.M.; Serrano, L.W.; Pearson, D.J.; Vergilio, G.K.; McCague, S.; Marshall, K.A.; Ashtari, M.; Doan, T.M.; Weigel-DiFranco, C.A.; et al. Adeno-Associated Virus Serotype 2-hCHM Subretinal Delivery to the Macula in Choroideremia: Two-Year Interim Results of an Ongoing Phase I/II Gene Therapy Trial. Ophthalmology 2022, 129, 1177–1191. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M.D.; Ochakovski, G.A.; Beier, B.; Seitz, I.P.; Vaheb, Y.; Kortuem, C.; Reichel, F.F.L.; Kuehlewein, L.; Kahle, N.A.; Peters, T.; et al. Changes in retinal sensitivity after gene therapy in choroideremia. Retina 2020, 40, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Lam, B.L.; Davis, J.L.; Gregori, N.Z.; MacLaren, R.E.; Girach, A.; Verriotto, J.D.; Rodriguez, B.; Rosa, P.R.; Zhang, X.; Feuer, W.J. Choroideremia Gene Therapy Phase 2 Clinical Trial: 24-Month Results. Am. J. Ophthalmol. 2019, 197, 65–73. [Google Scholar] [CrossRef]
- Dimopoulos, I.S.; Hoang, S.C.; Radziwon, A.; Binczyk, N.M.; Seabra, M.C.; MacLaren, R.E.; Somani, R.; Tennant, M.T.; MacDonald, I.M. Two-Year Results After AAV2-Mediated Gene Therapy for Choroideremia: The Alberta Experience. Am. J. Ophthalmol. 2018, 193, 130–142. [Google Scholar] [CrossRef]
- Palmer, E.; Stepien, K.M.; Campbell, C.; Barton, S.; Iosifidis, C.; Ghosh, A.; Broomfield, A.; Woodall, A.; Wilcox, G.; Sergouniotis, P.I.; et al. Clinical, biochemical and molecular analysis in a cohort of individuals with gyrate atrophy. Orphanet J. Rare Dis. 2023, 18, 265. [Google Scholar] [CrossRef]
- Simell, O.; Takki, K. Raised plasma-ornithine and gyrate atrophy of the choroid and retina. Lancet 1973, 1, 1031–1033. [Google Scholar] [CrossRef] [PubMed]
- Takki, K.K.; Milton, R.C. The natural history of gyrate atrophy of the choroid and retina. Ophthalmology 1981, 88, 292–301. [Google Scholar] [CrossRef] [PubMed]
- Elnahry, A.G.; Elnahry, G.A. Gyrate Atrophy of the Choroid and Retina: A Review. Eur. J. Ophthalmol. 2022, 32, 1314–1323. [Google Scholar] [CrossRef] [PubMed]
- Sergouniotis, P.I.; Davidson, A.E.; Lenassi, E.; Devery, S.R.; Moore, A.T.; Webster, A.R. Retinal structure, function, and molecular pathologic features in gyrate atrophy. Ophthalmology 2012, 119, 596–605. [Google Scholar] [CrossRef] [PubMed]
- Elnahry, A.G.; Hassan, F.K.; Abdel-Kader, A.A. Bevacizumab for the treatment of intraretinal cystic spaces in a patient with gyrate atrophy of the choroid and retina. Ophthalmic Genet. 2018, 39, 759–762. [Google Scholar] [CrossRef] [PubMed]
- Mansour, A.M.; Elnahry, A.G.; Tripathy, K.; Foster, R.E.; Mehanna, C.-J.; Vishal, R.; Çavdarlı, C.; Arrigo, A.; Parodi, M.B. Analysis of optical coherence angiography in cystoid macular oedema associated with gyrate atrophy. Eye 2021, 35, 1766–1774. [Google Scholar] [CrossRef] [PubMed]
- Khan, K.N.; Kasilian, M.; Mahroo, O.A.R.; Tanna, P.; Kalitzeos, A.; Robson, A.G.; Tsunoda, K.; Iwata, T.; Moore, A.T.; Fujinami, K.; et al. Early Patterns of Macular Degeneration in ABCA4-Associated Retinopathy. Ophthalmology 2018, 125, 735–746. [Google Scholar] [CrossRef] [PubMed]
- Fujinami, K.; Sergouniotis, P.I.; Davidson, A.E.; Wright, G.; Chana, R.K.; Tsunoda, K.; Tsubota, K.; Egan, C.A.; Robson, A.G.; Moore, A.T.; et al. Clinical and molecular analysis of Stargardt disease with preserved foveal structure and function. Am. J. Ophthalmol. 2013, 156, 487–501.e1. [Google Scholar] [CrossRef]
- Rahman, N.; Georgiou, M.; Khan, K.N.; Michaelides, M. Macular dystrophies: Clinical and imaging features, molecular genetics and therapeutic options. Br. J. Ophthalmol. 2020, 104, 451–460. [Google Scholar] [CrossRef]
- Strauss, R.W.; Kong, X.; Ho, A.; Jha, A.; West, S.; Ip, M.; Bernstein, P.S.; Birch, D.G.; Cideciyan, A.V.; Michaelides, M.; et al. Progression of Stargardt Disease as Determined by Fundus Autofluorescence Over a 12-Month Period: ProgStar Report No. 11. JAMA Ophthalmol. 2019, 137, 1134–1145. [Google Scholar] [CrossRef]
- Strauss, R.W.; Muñoz, B.; Ho, A.; Jha, A.; Michaelides, M.; Mohand-Said, S.; Cideciyan, A.V.; Birch, D.; Hariri, A.H.; Nittala, M.G.; et al. Incidence of Atrophic Lesions in Stargardt Disease in the Progression of Atrophy Secondary to Stargardt Disease (ProgStar) Study: Report No. 5. JAMA Ophthalmol. 2017, 135, 687–695. [Google Scholar] [CrossRef] [PubMed]
- Klufas, M.A.; Tsui, I.; Sadda, S.R.; Hosseini, H.; Schwartz, S.D. ULTRAWIDEFIELD AUTOFLUORESENCE IN ABCA4 STARGARDT DISEASE. Retina 2018, 38, 403–415. [Google Scholar] [CrossRef]
- Abalem, M.F.; Otte, B.; Andrews, C.; Joltikov, K.A.; Branham, K.; Fahim, A.T.; Schlegel, D.; Qian, C.X.; Heckenlively, J.R.; Jayasundera, T. Peripheral Visual Fields in ABCA4 Stargardt Disease and Correlation with Disease Extent on Ultra-widefield Fundus Autofluorescence. Am. J. Ophthalmol. 2017, 184, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Cai, C.X.; Light, J.G.; Handa, J.T. Quantifying the Rate of Ellipsoid Zone Loss in Stargardt Disease. Am. J. Ophthalmol. 2018, 186, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Velaga, S.B.; Nittala, M.G.; Jenkins, D.; Melendez, J.; Ho, A.; Strauss, R.W.; Scholl, H.P.; Sadda, S.R. Impact of segmentation density on spectral domain optical coherence tomography assessment in Stargardt disease. Graefes Arch. Clin. Exp. Ophthalmol. 2019, 257, 549–556. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Rossi, E.A.; Yang, Q.; Granger, C.E.; Latchney, L.R.; Chung, M.M. High-Resolution Adaptive Optics in Vivo Autofluorescence Imaging in Stargardt Disease. JAMA Ophthalmol. 2019, 137, 603–609. [Google Scholar] [CrossRef] [PubMed]
- Mishra, Z.; Wang, Z.; Sadda, S.R.; Hu, Z. Automatic Segmentation in Multiple OCT Layers for Stargardt Disease Characterization Via Deep Learning. Transl. Vis. Sci. Technol. 2021, 10, 24. [Google Scholar] [CrossRef]
- Wang, Z.; Hu, Z.J. Artificial intelligence for assessment of Stargardt macular atrophy. Neural Regen. Res. 2022, 17, 2632–2636. [Google Scholar] [CrossRef] [PubMed]
- Sunness, J.S.; Ifrah, A.; Wolf, R.; Applegate, C.A.; Sparrow, J.R. Abnormal Visual Function Outside the Area of Atrophy Defined by Short-Wavelength Fundus Autofluorescence in Stargardt Disease. Investig. Ophthalmol. Vis. Sci. 2020, 61, 36. [Google Scholar] [CrossRef]
- Schönbach, E.M.; Strauss, R.W.; Muñoz, B.; Wolfson, Y.; Ibrahim, M.A.; Birch, D.G.; Zrenner, E.; Sunness, J.S.; Ip, M.S.; Sadda, S.R.; et al. Longitudinal Microperimetric Changes of Macular Sensitivity in Stargardt Disease After 12 Months: ProgStar Report No. 13. JAMA Ophthalmol. 2020, 138, 772–779. [Google Scholar] [CrossRef]
- Kong, X.; Ibrahim-Ahmed, M.; Bittencourt, M.G.; Strauss, R.W.; Birch, D.G.; Cideciyan, A.V.; Ervin, A.-M.; Ho, A.; Sunness, J.S.; Audo, I.S.; et al. Longitudinal Changes in Scotopic and Mesopic Macular Function as Assessed with Microperimetry in Patients with Stargardt Disease: SMART Study Report No. 2. Am. J. Ophthalmol. 2022, 236, 32–44. [Google Scholar] [CrossRef] [PubMed]
- Schönbach, E.M.; Strauss, R.W.; Cattaneo, M.E.G.V.; Fujinami, K.; Birch, D.G.; Cideciyan, A.V.; Sunness, J.S.; Zrenner, E.; Sadda, S.R.; Scholl, H.P. Longitudinal Changes of Fixation Stability and Location within 24 Months in Stargardt Disease: ProgStar Report No. 16. Am. J. Ophthalmol. 2022, 233, 78–89. [Google Scholar] [CrossRef] [PubMed]
- Mastropasqua, R.; Senatore, A.; Di Antonio, L.; Di Nicola, M.; Marchioni, M.; Perna, F.; Amore, F.; Borrelli, E.; De Nicola, C.; Carpineto, P.; et al. Correlation between Choriocapillaris Density and Retinal Sensitivity in Stargardt Disease. J. Clin. Med. Res. 2019, 8, 1432. [Google Scholar] [CrossRef] [PubMed]
- Kassa, E.; Ciulla, T.A.; Hussain, R.M.; Dugel, P.U. Complement inhibition as a therapeutic strategy in retinal disorders. Expert. Opin. Biol. Ther. 2019, 19, 335–342. [Google Scholar] [CrossRef]
- Csaky, K.G.; Bok, D.; Radu, R.A.; Sadda, S.R. Complement C5 Inhibition as a Potential Treatment for Autosomal Recessive Stargardt Disease (STGD1): Design of a Clinical Trial Assessing a Novel Treatment and Primary Outcome Measure. Investig. Ophthalmol. Vis. Sci. 2018, 59, 1569. [Google Scholar]
- Georgiou, M.; Kane, T.; Tanna, P.; Bouzia, Z.; Singh, N.; Kalitzeos, A.; Strauss, R.W.; Fujinami, K.; Michaelides, M. Prospective Cohort Study of Childhood-Onset Stargardt Disease: Fundus Autofluorescence Imaging, Progression, Comparison with Adult-Onset Disease, and Disease Symmetry. Am. J. Ophthalmol. 2020, 211, 159–175. [Google Scholar] [CrossRef] [PubMed]
- Marquardt, A.; Stöhr, H.; Passmore, L.A.; Krämer, F.; Rivera, A.; Weber, B.H. Mutations in a novel gene, VMD2, encoding a protein of unknown properties cause juvenile-onset vitelliform macular dystrophy (Best’s disease). Hum. Mol. Genet. 1998, 7, 1517–1525. [Google Scholar] [CrossRef] [PubMed]
- Gass, J.D.M. Best’s Disease. Stereoscopic atlas of macular diseases. In Diagnosis and Treatment; Mosby: St. Louis, MO, USA, 1997. [Google Scholar]
- Querques, G.; Zerbib, J.; Santacroce, R.; Margaglione, M.; Delphin, N.; Rozet, J.-M.; Kaplan, J.; Martinelli, D.; Noci, N.D.; Soubrane, G.; et al. Functional and clinical data of Best vitelliform macular dystrophy patients with mutations in the BEST1 gene. Mol. Vis. 2009, 15, 2960–2972. [Google Scholar] [PubMed]
- Querques, G.; Zerbib, J.; Georges, A.; Massamba, N.; Forte, R.; Querques, L.; Rozet, J.-M.; Kaplan, J.; Souied, E.H. Multimodal analysis of the progression of Best vitelliform macular dystrophy. Mol. Vis. 2014, 20, 575–592. [Google Scholar]
- Lima de Carvalho, J.R., Jr.; Paavo, M.; Chen, L.; Chiang, J.; Tsang, S.H.; Sparrow, J.R. Multimodal Imaging in Best Vitelliform Macular Dystrophy. Investig. Ophthalmol. Vis. Sci. 2019, 60, 2012–2022. [Google Scholar] [CrossRef]
- Bianco, L.; Arrigo, A.; Antropoli, A.; Berni, A.; Saladino, A.; Vilela, M.A.; Mansour, A.M.; Bandello, F.; Parodi, M.B. Multimodal imaging in Best Vitelliform Macular Dystrophy: Literature review and novel insights. Eur. J. Ophthalmol. 2024, 34, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Querques, G.; Regenbogen, M.; Quijano, C.; Delphin, N.; Soubrane, G.; Souied, E.H. High-definition optical coherence tomography features in vitelliform macular dystrophy. Am. J. Ophthalmol. 2008, 146, 501–507. [Google Scholar] [CrossRef]
- Querques, G.; Forte, R.; Querques, L.; Massamba, N.; Souied, E.H. Natural course of adult-onset foveomacular vitelliform dystrophy: A spectral-domain optical coherence tomography analysis. Am. J. Ophthalmol. 2011, 152, 304–313. [Google Scholar] [CrossRef] [PubMed]
- Querques, G.; Zerbib, J.; Santacroce, R.; Margaglione, M.; Delphin, N.; Querques, L.; Rozet, J.-M.; Kaplan, J.; Souied, E.H. The spectrum of subclinical Best vitelliform macular dystrophy in subjects with mutations in BEST1 gene. Investig. Ophthalmol. Vis. Sci. 2011, 52, 4678–4684. [Google Scholar] [CrossRef] [PubMed]
- Romano, F.; Arrigo, A.; Leone, P.P.; Bandello, F.; Battaglia Parodi, M. SHORT-TERM MODIFICATIONS OF ELLIPSOID ZONE IN BEST VITELLIFORM MACULAR DYSTROPHY. Retina 2021, 41, 1010–1017. [Google Scholar] [CrossRef] [PubMed]
- Parodi, M.B.; Romano, F.; Sacconi, R.; Casati, S.; Marchini, G.; Bandello, F.; Iacono, P. Intraretinal hyperreflective foci in best vitelliform macular dystrophy. Retina 2018, 38, 2379–2386. [Google Scholar] [CrossRef] [PubMed]
- Querques, G.; Bocco, M.C.A.; Soubrane, G.; Souied, E.H. Intravitreal ranibizumab (Lucentis) for choroidal neovascularization associated with vitelliform macular dystrophy. Acta Ophthalmol. 2008, 86, 694–695. [Google Scholar] [CrossRef] [PubMed]
- Romano, F.; Arrigo, A.; Leone, P.P.; Saladino, A.; Bandello, F.; Battaglia Parodi, M. Altered ellipsoid zone reflectivity and deep capillary plexus rarefaction correlate with progression in Best disease. Br. J. Ophthalmol. 2020, 104, 461–465. [Google Scholar] [CrossRef]
- Parodi, M.B.; Arrigo, A.; Calamuneri, A.; Aragona, E.; Bandello, F. Multimodal imaging in subclinical best vitelliform macular dystrophy. Br. J. Ophthalmol. 2022, 106, 564–567. [Google Scholar] [CrossRef]
- Jauregui, R.; Parmann, R.; Nuzbrokh, Y.; Tsang, S.H.; Sparrow, J.R. Stage-dependent choriocapillaris impairment in Best vitelliform macular dystrophy characterized by optical coherence tomography angiography. Sci. Rep. 2021, 11, 14300. [Google Scholar] [CrossRef]
- Kay, D.B.; Land, M.E.; Cooper, R.F.; Dubis, A.M.; Godara, P.; Dubra, A.; Carroll, J.; Stepien, K.E. Outer retinal structure in best vitelliform macular dystrophy. JAMA Ophthalmol. 2013, 131, 1207–1215. [Google Scholar] [CrossRef] [PubMed]
- Querques, G.; Bux, A.V.; Prato, R.; Iaculli, C.; Souied, E.H.; Delle Noci, N. Correlation of visual function impairment and optical coherence tomography findings in patients with adult-onset foveomacular vitelliform macular dystrophy. Am. J. Ophthalmol. 2008, 146, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Battaglia Parodi, M.; Bianco, L.; Arrigo, A.; Saladino, A.; Antropoli, A.; Pina, A.; Marchese, A.; Aragona, E.; Rashid, H.F.; Bandello, F. Clinical Correlation Between Optical Coherence Tomography Biomarkers and Retinal Sensitivity in Best Vitelliform Macular Dystrophy. Transl. Vis. Sci. Technol. 2022, 11, 24. [Google Scholar] [CrossRef] [PubMed]
- Burgess, R.; Millar, I.D.; Leroy, B.P.; Urquhart, J.E.; Fearon, I.M.; De Baere, E.; Brown, P.D.; Robson, A.G.; Wright, G.A.; Kestelyn, P.; et al. Biallelic mutation of BEST1 causes a distinct retinopathy in humans. Am. J. Hum. Genet. 2008, 82, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Boon, C.J.F.; Klevering, B.J.; Leroy, B.P.; Hoyng, C.B.; Keunen, J.E.E.; den Hollander, A.I. The spectrum of ocular phenotypes caused by mutations in the BEST1 gene. Prog. Retin. Eye Res. 2009, 28, 187–205. [Google Scholar] [CrossRef] [PubMed]
- Zerbib, J.; Querques, G.; Massamba, N.; Puche, N.; Tilleul, J.; Lalloum, F.; Srour, M.; Coscas, G.; Souied, E.H. Reticular pattern dystrophy of the retina: A spectral-domain optical coherence tomography analysis. Am. J. Ophthalmol. 2013, 156, 1228–1237. [Google Scholar] [CrossRef] [PubMed]
- Wijesuriya, S.D.; Evans, K.; Jay, M.R.; Davison, C.; Weber, B.H.; Bird, A.C.; Bhattacharya, S.S.; Gregory, C.Y. Sorsby’s fundus dystrophy in the British Isles: Demonstration of a striking founder effect by microsatellite-generated haplotypes. Genome Res. 1996, 6, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Sorsby, A.; Mason, M.E.J. A fundus dystrophy with unusual features. Br. J. Ophthalmol. 1949, 33, 67–97. [Google Scholar] [CrossRef] [PubMed]
- Tsokolas, G. Sorsby fundus dystrophy (SFD): A narrative review. Medicine 2022, 101, e30595. [Google Scholar] [CrossRef]
- Small, K.W.; DeLuca, A.P.; Whitmore, S.S.; Rosenberg, T.; Silva-Garcia, R.; Udar, N.; Puech, B.; Garcia, C.A.; Rice, T.A.; Fishman, G.A.; et al. North Carolina Macular Dystrophy Is Caused by Dysregulation of the Retinal Transcription Factor PRDM13. Ophthalmology 2016, 123, 9–18. [Google Scholar] [CrossRef]
- Rabb, M.F.; Mullen, L.; Yelchits, S.; Udar, N.; Small, K.W. A North Carolina macular dystrophy phenotype in a Belizean family maps to the MCDR1 locus. Am. J. Ophthalmol. 1998, 125, 502–508. [Google Scholar] [CrossRef] [PubMed]
- Boon, C.J.F.; Klevering, B.J.; Cremers, F.P.M.; Zonneveld-Vrieling, M.N.; Theelen, T.; Den Hollander, A.I.; Hoyng, C.B. Central areolar choroidal dystrophy. Ophthalmology 2009, 116, 771–782, 782.e1. [Google Scholar] [CrossRef] [PubMed]
- Carr, R.E. Central areolar choroidal dystrophy. Arch. Ophthalmol. 1965, 73, 32–35. [Google Scholar] [CrossRef] [PubMed]
- Romano, F.; Cozzi, E.; Boon, C.J.F.; Staurenghi, G.; Salvetti, A.P. Multimodal retinal imaging reveals new pathogenic insights in central areolar choroidal dystrophy: A case series. Retin. Cases Brief. Rep. 2024, 18, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Korte, G.E.; Reppucci, V.; Henkind, P. RPE destruction causes choriocapillary atrophy. Investig. Ophthalmol. Vis. Sci. 1984, 25, 1135–1145. [Google Scholar]
- Fujinami, K.; Yang, L.; Joo, K.; Tsunoda, K.; Kameya, S.; Hanazono, G.; Fujinami-Yokokawa, Y.; Arno, G.; Kondo, M.; Nakamura, N.; et al. Clinical and Genetic Characteristics of East Asian Patients with Occult Macular Dystrophy (Miyake Disease): East Asia Occult Macular Dystrophy Studies Report Number 1. Ophthalmology 2019, 126, 1432–1444. [Google Scholar] [CrossRef] [PubMed]
- Miyake, Y.; Ichikawa, K.; Shiose, Y.; Kawase, Y. Hereditary macular dystrophy without visible fundus abnormality. Am. J. Ophthalmol. 1989, 108, 292–299. [Google Scholar] [CrossRef] [PubMed]
- Kondo, M.; Ito, Y.; Ueno, S.; Piao, C.-H.; Terasaki, H.; Miyake, Y. Foveal thickness in occult macular dystrophy. Am. J. Ophthalmol. 2003, 135, 725–728. [Google Scholar] [CrossRef]
- Nakanishi, A.; Ueno, S.; Kawano, K.; Ito, Y.; Kominami, T.; Yasuda, S.; Kondo, M.; Tsunoda, K.; Iwata, T.; Terasaki, H. Pathologic Changes of Cone Photoreceptors in Eyes with Occult Macular Dystrophy. Investig. Ophthalmol. Vis. Sci. 2015, 56, 7243–7249. [Google Scholar] [CrossRef]
- Zobor, D.; Zobor, G.; Hipp, S.; Baumann, B.; Weisschuh, N.; Biskup, S.; Sliesoraityte, I.; Zrenner, E.; Kohl, S. Phenotype Variations Caused by Mutations in the RP1L1 Gene in a Large Mainly German Cohort. Investig. Ophthalmol. Vis. Sci. 2018, 59, 3041–3052. [Google Scholar] [CrossRef]
- Querques, G.; Guigui, B.; Leveziel, N.; Querques, L.; Bandello, F.; Souied, E.H. Multimodal morphological and functional characterization of Malattia Leventinese. Graefes Arch. Clin. Exp. Ophthalmol. 2013, 251, 705–714. [Google Scholar] [CrossRef] [PubMed]
- Riazuddin, S.A.; Shahzadi, A.; Zeitz, C.; Ahmed, Z.M.; Ayyagari, R.; Chavali, V.R.M.; Ponferrada, V.G.; Audo, I.; Michiels, C.; Lancelot, M.-E.; et al. A mutation in SLC24A1 implicated in autosomal-recessive congenital stationary night blindness. Am. J. Hum. Genet. 2010, 87, 523–531. [Google Scholar] [CrossRef] [PubMed]
- Zeitz, C.; Robson, A.G.; Audo, I. Congenital stationary night blindness: An analysis and update of genotype-phenotype correlations and pathogenic mechanisms. Prog. Retin. Eye Res. 2015, 45, 58–110. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, H.; Kishi, S. Shortening of the rod outer segment in Oguchi disease. Graefes Arch. Clin. Exp. Ophthalmol. 2009, 247, 1561–1563. [Google Scholar] [CrossRef] [PubMed]
- Godara, P.; Cooper, R.F.; Sergouniotis, P.I.; Diederichs, M.A.; Streb, M.R.; Genead, M.A.; McAnany, J.J.; Webster, A.R.; Moore, A.T.; Dubis, A.M.; et al. Assessing retinal structure in complete congenital stationary night blindness and Oguchi disease. Am. J. Ophthalmol. 2012, 154, 987–1001.e1. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, J.P.; Sherman, J.; Zweifel, S.A.; Chen, R.W.S.; Duncker, T.; Kohl, S.; Baumann, B.; Wissinger, B.; Yannuzzi, L.A.; Tsang, S.H. Spectral-domain optical coherence tomography staging and autofluorescence imaging in achromatopsia. JAMA Ophthalmol. 2014, 132, 437–445. [Google Scholar] [CrossRef] [PubMed]
- Aboshiha, J.; Dubis, A.M.; Cowing, J.; Fahy, R.T.A.; Sundaram, V.; Bainbridge, J.W.; Ali, R.R.; Dubra, A.; Nardini, M.; Webster, A.R.; et al. A prospective longitudinal study of retinal structure and function in achromatopsia. Investig. Ophthalmol. Vis. Sci. 2014, 55, 5733–5743. [Google Scholar] [CrossRef] [PubMed]
- Carroll, J.; Neitz, M.; Hofer, H.; Neitz, J.; Williams, D.R. Functional photoreceptor loss revealed with adaptive optics: An alternate cause of color blindness. Proc. Natl. Acad. Sci. USA 2004, 101, 8461–8466. [Google Scholar] [CrossRef] [PubMed]
- Pircher, M.; Zawadzki, R.J. Review of adaptive optics OCT (AO-OCT): Principles and applications for retinal imaging. Biomed. Opt. Express 2017, 8, 2536–2562. [Google Scholar] [CrossRef]
- Langlo, C.S.; Patterson, E.J.; Higgins, B.P.; Summerfelt, P.; Razeen, M.M.; Erker, L.R.; Parker, M.; Collison, F.T.; Fishman, G.A.; Kay, C.N.; et al. Residual Foveal Cone Structure in CNGB3-Associated Achromatopsia. Investig. Ophthalmol. Vis. Sci. 2016, 57, 3984–3995. [Google Scholar] [CrossRef]
- Sikkink, S.K.; Biswas, S.; Parry, N.R.A.; Stanga, P.E.; Trump, D. X-linked retinoschisis: An update. J. Med. Genet. 2007, 44, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, T.; Chang, E.; Nudleman, E.; El-Rayes, E.N.; Yonekawa, Y. Typical and atypical clinical presentations of X-Linked retinoschisis: A case series and literature review. Surv. Ophthalmol. 2023, 68, 347–360. [Google Scholar] [CrossRef] [PubMed]
- Fahim, A.T.; Ali, N.; Blachley, T.; Michaelides, M. Peripheral fundus findings in X-linked retinoschisis. Br. J. Ophthalmol. 2017, 101, 1555–1559. [Google Scholar] [CrossRef]
- Gregori, N.Z.; Berrocal, A.M.; Gregori, G.; Murray, T.G.; Knighton, R.W.; Flynn, H.W., Jr.; Dubovy, S.; Puliafito, C.A.; Rosenfeld, P.J. Macular spectral-domain optical coherence tomography in patients with X linked retinoschisis. Br. J. Ophthalmol. 2009, 93, 373–378. [Google Scholar] [CrossRef] [PubMed]
- Yoshida-Uemura, T.; Katagiri, S.; Yokoi, T.; Nishina, S.; Azuma, N. Different foveal schisis patterns in each retinal layer in eyes with hereditary juvenile retinoschisis evaluated by en-face optical coherence tomography. Graefes Arch. Clin. Exp. Ophthalmol. 2017, 255, 719–723. [Google Scholar] [CrossRef] [PubMed]
- Cozzi, M.; Pagliarini, S. Retinoschisis Microstructure Visualization with En Face Spectral Domain Optical Coherence Tomography. Retina 2016, 36, 227–228. [Google Scholar] [CrossRef] [PubMed]
- De Silva, S.R.; Arno, G.; Robson, A.G.; Fakin, A.; Pontikos, N.; Mohamed, M.D.; Bird, A.C.; Moore, A.T.; Michaelides, M.; Webster, A.R.; et al. The X-linked retinopathies: Physiological insights, pathogenic mechanisms, phenotypic features and novel therapies. Prog. Retin. Eye Res. 2021, 82, 100898. [Google Scholar] [CrossRef]
- Pennesi, M.E.; Yang, P.; Birch, D.G.; Weng, C.Y.; Moore, A.T.; Iannaccone, A.; Comander, J.I.; Jayasundera, T.; Chulay, J.; Halliman, D.; et al. Intravitreal Delivery of rAAV2tYF-CB-hRS1 Vector for Gene Augmentation Therapy in Patients with X-Linked Retinoschisis: 1-Year Clinical Results. Ophthalmol. Retin. 2022, 6, 1130–1144. [Google Scholar] [CrossRef]
- Cukras, C.; Wiley, H.E.; Jeffrey, B.G.; Sen, H.N.; Turriff, A.; Zeng, Y.; Vijayasarathy, C.; Marangoni, D.; Ziccardi, L.; Kjellstrom, S.; et al. Retinal AAV8-RS1 Gene Therapy for X-Linked Retinoschisis: Initial Findings from a Phase I/IIa Trial by Intravitreal Delivery. Mol. Ther. 2018, 26, 2282–2294. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Corradetti, G.; Verma, A.; Tojjar, J.; Almidani, L.; Oncel, D.; Emamverdi, M.; Bradley, A.; Lindenberg, S.; Nittala, M.G.; Sadda, S.R. Retinal Imaging Findings in Inherited Retinal Diseases. J. Clin. Med. 2024, 13, 2079. https://doi.org/10.3390/jcm13072079
Corradetti G, Verma A, Tojjar J, Almidani L, Oncel D, Emamverdi M, Bradley A, Lindenberg S, Nittala MG, Sadda SR. Retinal Imaging Findings in Inherited Retinal Diseases. Journal of Clinical Medicine. 2024; 13(7):2079. https://doi.org/10.3390/jcm13072079
Chicago/Turabian StyleCorradetti, Giulia, Aditya Verma, Jasaman Tojjar, Louay Almidani, Deniz Oncel, Mehdi Emamverdi, Alec Bradley, Sophiana Lindenberg, Muneeswar Gupta Nittala, and SriniVas R. Sadda. 2024. "Retinal Imaging Findings in Inherited Retinal Diseases" Journal of Clinical Medicine 13, no. 7: 2079. https://doi.org/10.3390/jcm13072079
APA StyleCorradetti, G., Verma, A., Tojjar, J., Almidani, L., Oncel, D., Emamverdi, M., Bradley, A., Lindenberg, S., Nittala, M. G., & Sadda, S. R. (2024). Retinal Imaging Findings in Inherited Retinal Diseases. Journal of Clinical Medicine, 13(7), 2079. https://doi.org/10.3390/jcm13072079