

A Misdiagnosed Familiar Brooke–Spiegler Syndrome: Case Report and Review of the Literature

 ,
,  ,
,  , and
, and 
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Review of the Literature
2.1. Database Research
2.2. The Diagnostic Challenge
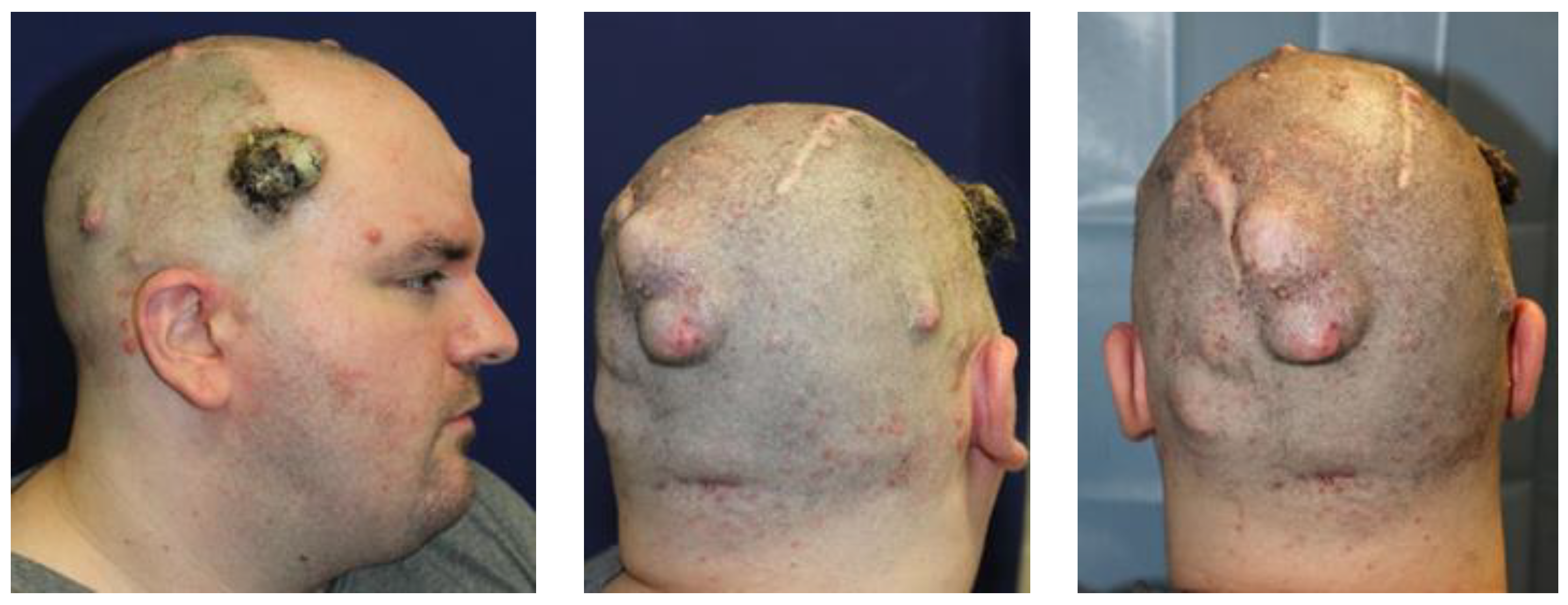
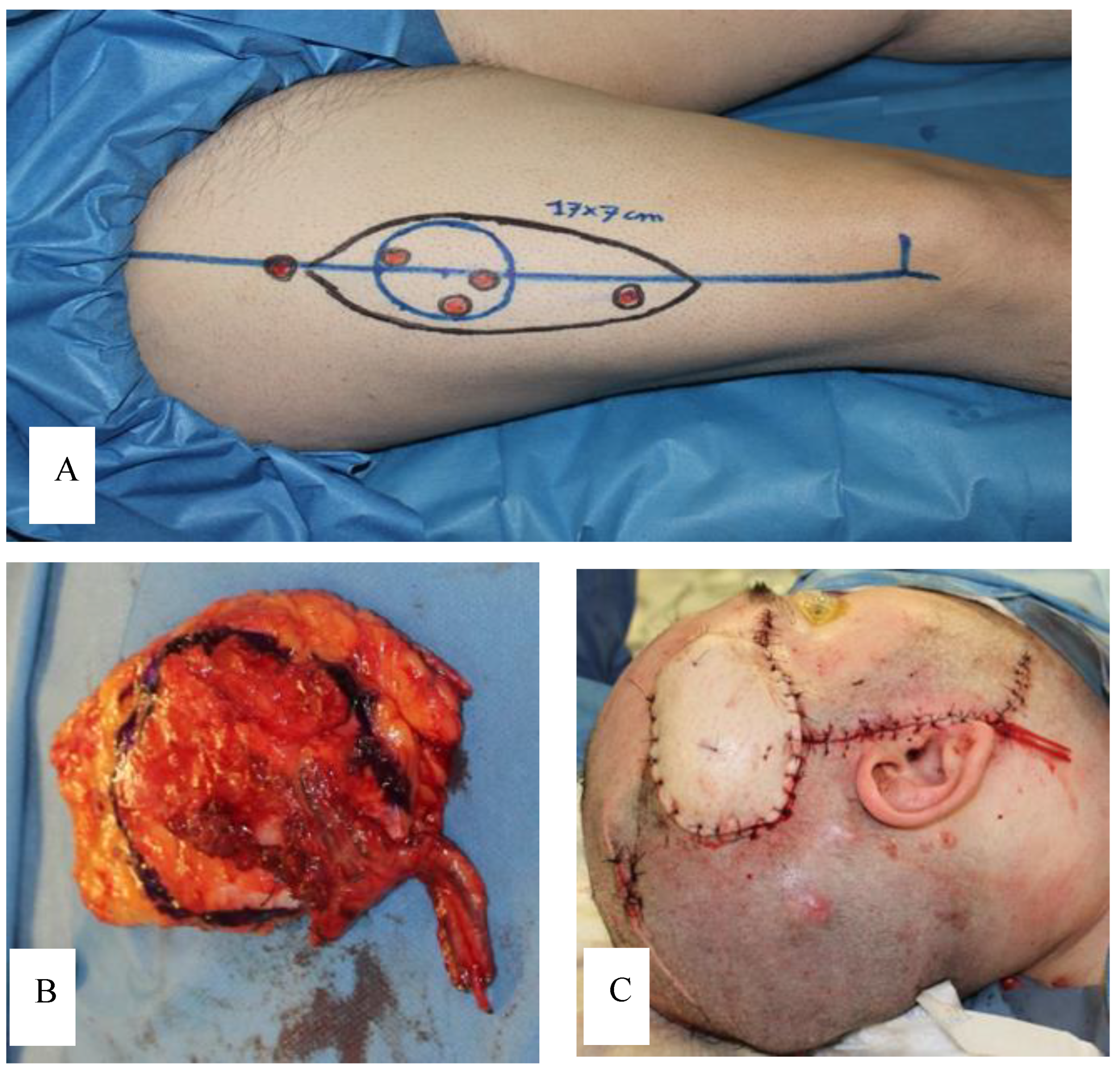
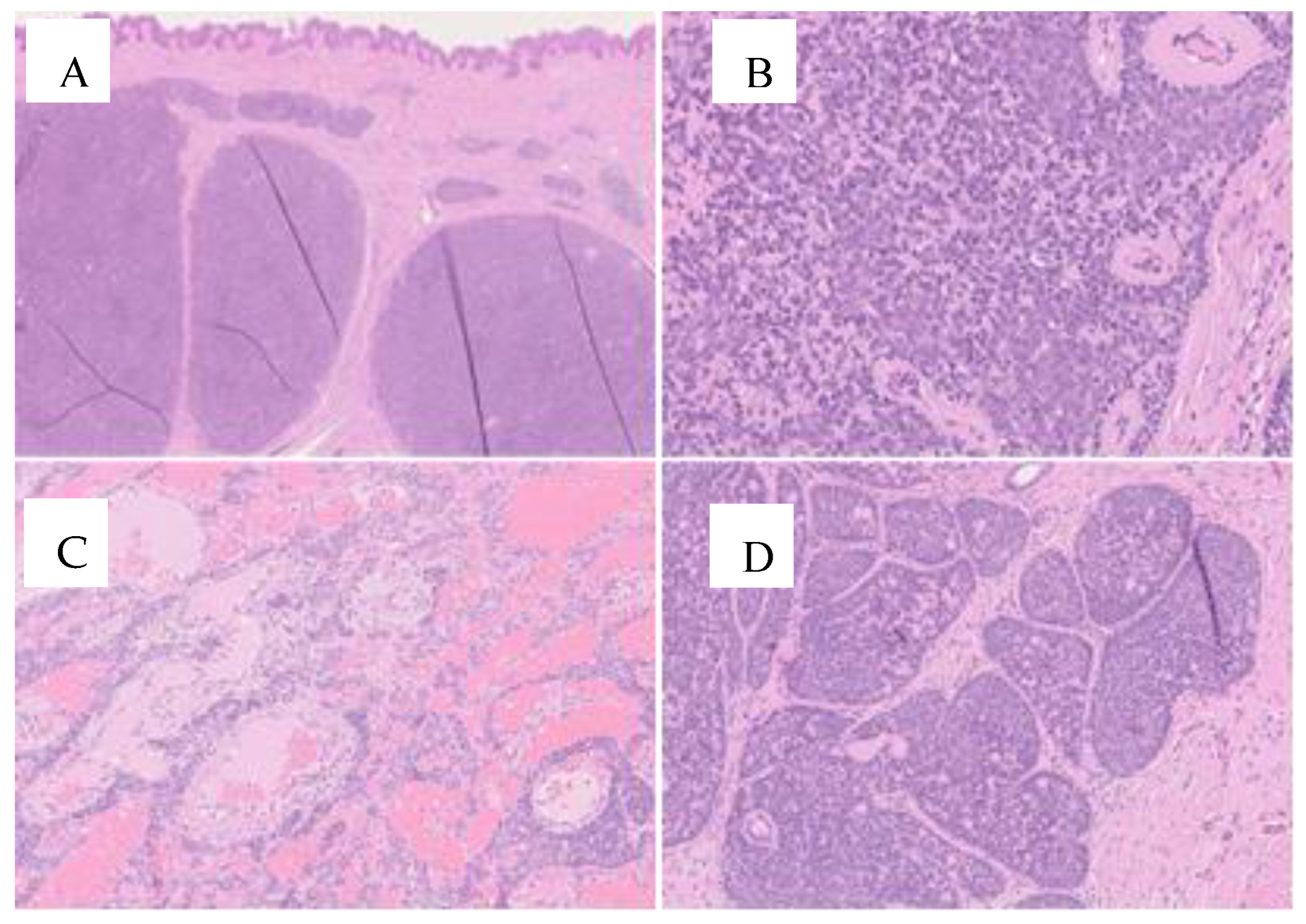
3. Case Report
4. Discussion
5. Conclusions
6. Limitations and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mohiuddin, W.; Laun, J.; Cruse, W. Brooke-Spiegler Syndrome. Eplasty 2018, 18, ic14. [Google Scholar] [PubMed]
- Cranston, A.; Stocken, D.D.; Stamp, E.; Roblin, D.; Hamlin, J.; Langtry, J.; Plummer, R.; Ashworth, A.; Burn, J.; Rajan, N. Tropomyosin Receptor Antagonism in Cylindromatosis (TRAC), an early phase trial of a topical tropomyosin kinase inhibitor as a treatment for inherited CYLD defective skin tumours: Study protocol for a randomised controlled trial. Trials 2017, 18, 111. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Kovich, O.I.; Dosik, J. Brooke-Spiegler syndrome. Dermatol. Online J. 2007, 13, 10. [Google Scholar] [CrossRef]
- Kazakov, D.V. Brooke-Spiegler Syndrome and Phenotypic Variants: An Update. Head Neck Pathol. 2016, 10, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Kazakov, D.V.; Schaller, J.; Vanecek, T.; Kacerovska, D.; Michal, M. Brooke-Spiegler syndrome: Report of a case with a novel mutation in the CYLD gene and different types of somatic mutations in benign and malignant tumors. J. Cutan. Pathol. 2010, 37, 886–890. [Google Scholar] [CrossRef] [PubMed]
- Vorechovský, I.; Undén, A.B.; Sandstedt, B.; Toftgård, R.; Ståhle-Bäckdahl, M. Trichoepitheliomas contain somatic mutations in the overexpressed PTCH gene: Support for a gatekeeper mechanism in skin tumorigenesis. Cancer Res. 1997, 57, 4677–4681. [Google Scholar] [PubMed]
- Kazakov, D.V.; Vanecek, T.; Zelger, B.; Carlson, J.A.; Spagnolo, D.V.; Schaller, J.; Nemcova, J.; Kacerovska, D.; Vazmitel, M.; Sangüeza, M.; et al. Multiple (familial) trichoepitheliomas: A clinicopathological and molecular biological study, including CYLD and PTCH gene analysis, of a series of 16 patients. Am. J. Dermatopathol. 2011, 33, 251–265, Erratum in Am. J. Dermatopathol. 2011, 33, 74. [Google Scholar] [CrossRef]
- Iwasaki, J.K.; Srivastava, D.; Moy, R.L.; Lin, H.J.; Kouba, D.J. The molecular genetics underlying basal cell carcinoma pathogenesis and links to targeted therapeutics. J. Am. Acad. Dermatol. 2012, 66, e167–e178. [Google Scholar] [CrossRef] [PubMed]
- Lindström, E.; Shimokawa, T.; Toftgård, R.; Zaphiropoulos, P.G. PTCH mutations: Distribution and analyses. Hum. Mutat. 2006, 27, 215–219. [Google Scholar] [CrossRef]
- Thomas, L.W.; Pham, C.T.; Coakley, B.; Lee, P. Treatment of Brooke-Spiegler Syndrome Trichoepitheliomas with Erbium: Yttrium-Aluminum-Garnet Laser: A Case Report and Review of the Literature. J. Clin. Aesthet Dermatol. 2020, 13, 41–44. [Google Scholar]
- Danilenko, M.; Hodgson, K.; Stones, R.; Husain, A.; Zangarini, M.; Veal, G.; Rajan, N. Diverse assays from a single skin punch biopsy to assess topical drug intervention. Br. J. Dermatol. 2019, 180, 937–938. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.D.; Naujoks, C.; Depprich, R.; Schulte, K.-W.; Jankowiak, F.; Kübler, N.R.; Handschel, J. Cylindroma of head and neck: Review of the literature and report of two rare cases. J. Cranio-Maxillofac. Surg. 2013, 41, 516–521. [Google Scholar] [CrossRef] [PubMed]
- Oosterkamp, H.; Neering, H.; Nijman, S.; Dirac, A.; Mooi, W.; Bernards, R.; Brummelkamp, T. An evaluation of the efficacy of topical application of salicylic acid for the treatment of familial cylindromatosis. Br. J. Dermatol. 2006, 155, 182–185. [Google Scholar] [CrossRef] [PubMed]
- Ponti, G.; Nasti, S.; Losi, L.; Pastorino, L.; Pollio, A.; Benassi, L.; Giudice, S.; Bertazzoni, G.; Veratti, E.; Azzoni, P.; et al. Brooke-Spiegler syndrome: Report of two cases not associated with a mutation in the CYLD and PTCH tumor-suppressor genes. J. Cutan. Pathol. 2012, 39, 366–371. [Google Scholar] [CrossRef] [PubMed]
- Vanecek, T.; Halbhuber, Z.; Kacerovska, D.; Martinek, P.; Sedivcova, M.; Carr, R.A.; Slouka, D.; Michal, M.; Kazakov, D.V. Large germline deletions of the CYLD gene in patients with Brooke-Spiegler syndrome and multiple familial trichoepithelioma. Am. J. Dermatopathol. 2014, 36, 868–874. [Google Scholar] [CrossRef] [PubMed]
- Nagy, N.; Dubois, A.; Szell, M.; Rajan, N. Genetic Testing in CYLD Cutaneous Syndrome: An Update. Appl. Clin. Genet. 2021, 14, 427–444. [Google Scholar] [CrossRef] [PubMed]
- Kazakov, D.V.; Thoma-Uszynski, S.; Vanecek, T.; Kacerovska, D.; Grossmann, P.; Michal, M. A case of Brooke-Spiegler syndrome with a novel germline deep intronic mutation in the CYLD gene leading to intronic exonization, diverse somatic mutations, and unusual histology. Am. J. Dermatopathol. 2009, 31, 664–673. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.C.; Colome-Grimmer, M.; Kelly, E. Multiple trichoepitheliomas in the lines of Blaschko. Pediatr. Dermatol. 2006, 23, 149–151. [Google Scholar] [CrossRef]
- Hafner, C.; Schmiemann, V.; Ruetten, A.; Coras, B.; Landthaler, M.; Reifenberger, J.; Vogt, T. PTCH mutations are not mainly involved in the pathogenesis of sporadic trichoblastomas. Hum. Pathol. 2007, 38, 1496–1500. [Google Scholar] [CrossRef]
- Kazakov, D.V.; Soukup, R.; Mukensnabl, P.; Boudova, L.; Michal, M. Brooke-Spiegler syndrome: Report of a case with combined lesions containing cylindromatous, spiradenomatous, trichoblastomatous, and sebaceous differentiation. Am. J. Dermatopathol. 2005, 27, 27–33. [Google Scholar] [CrossRef]
- Dubois, A.; Rajan, N. CYLD Cutaneous Syndrome. 2020 Apr 16. In GeneReviews® [Internet]; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2020. [Google Scholar]
- Dubois, A.; Wilson, V.; Bourn, D.; Rajan, N. CYLD Genetic Testing for Brooke-Spiegler Syndrome, Familial Cylindromatosis and Multiple Familial Trichoepitheliomas. PLoS Curr. 2015, 7–8. [Google Scholar] [CrossRef] [PubMed]
- Arefi, M.; Wilson, V.; Muthiah, S.; Zwolinski, S.; Bajwa, D.; Brennan, P.; Blasdale, K.; Bourn, D.; Burn, J.; Santibanez-Koref, M.; et al. Diverse presentations of cutaneous mosaicism occur in CYLD cutaneous syndrome and may result in parent-to-child transmission. J. Am. Acad. Dermatol. 2019, 81, 1300–1307. [Google Scholar] [CrossRef] [PubMed]
- Plorina, E.V.; Saulus, K.; Rudzitis, A.; Kiss, N.; Medvecz, M.; Linova, T.; Bliznuks, D.; Lihachev, A.; Lihacova, I. Multispectral Imaging Analysis of Skin Lesions in Patients with Neurofibromatosis Type 1. J. Clin. Med. 2023, 12, 6746. [Google Scholar] [CrossRef] [PubMed]
- Karaconji, T.; Whist, E.; Jamieson, R.V.; Flaherty, M.P.; Grigg, J.R.B. Neurofibromatosis Type 1: Review and Update on Emerging Therapies. Asia-Pacific J. Ophthalmol. 2019, 8, 62–72. [Google Scholar] [CrossRef]
- Magro, G.; Broggi, G.; Angelico, G.; Puzzo, L.; Vecchio, G.M.; Virzì, V.; Salvatorelli, L.; Ruggieri, M. Practical Approach to Histological Diagnosis of Peripheral Nerve Sheath Tumors: An Update. Diagnostics 2022, 12, 1463. [Google Scholar] [CrossRef] [PubMed]
- Kehrer-Sawatzki, H.; Cooper, D.N. Challenges in the diagnosis of neurofibromatosis type 1 (NF1) in young children facilitated by means of revised diagnostic criteria including genetic testing for pathogenic NF1 gene variants. Hum. Genet. 2022, 141, 177–191. [Google Scholar] [CrossRef] [PubMed]
- Boyd, K.P.; Korf, B.R.; Theos, A. Neurofibromatosis type 1. J. Am. Acad. Dermatol. 2009, 61, 1–14. [Google Scholar] [CrossRef] [PubMed]
- De Schepper, S.; Boucneau, J.; Lambert, J.; Messiaen, L.; Naeyaert, J. Pigment cell-related manifestations in neurofibromatosis type 1: An overview. Pigment. Cell Res. 2005, 18, 13–24. [Google Scholar] [CrossRef] [PubMed]
- De Souza Tolentino, E.; de Souza Pinto, G.N.; Maciel, L.; Soares, C.T.; Lara, V.S.; Moreschi, A.R.C. Exuberant manifestation of neurofibromatosis type 1 affecting 3 generations: Delayed diagnosis and the importance of the multidisciplinary approach. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. 2019, 128, e108–e112. [Google Scholar] [CrossRef]
- Denadai, R.; Stelini, R.F.; Roberto, W.M.; Raposo-Amaral, C.A.; Raposo-Amaral, C.E. Brooke-Spiegler syndrome clinically misdiagnosed as neurofibromatosis type 1. J. Craniofacial Surg. 2015, 26, 323–325. [Google Scholar] [CrossRef]
- Goodman, J.C.; Baskin, D.S. Autosomal dominant familial angiolipomatosis clinically mimicking neurofibromatosis. Neurofibromatosi 1989, 2, 326–331. [Google Scholar]
- Hwang, D.-Y.; Yim, Y.-M.; Kwon, H.; Jung, S.-N. Multiple huge epidermal inclusion cysts mistaken as neurofibromatosis. J. Craniofacial Surg. 2008, 19, 1683–1686. [Google Scholar] [CrossRef] [PubMed]
- Nosé, V.; Lazar, A.J. Update from the 5th Edition of the World Health Organization Classification of Head and Neck Tumors: Familial Tumor Syndromes. Head Neck Pathol. 2022, 16, 143–157. [Google Scholar] [CrossRef] [PubMed]
- Mori, S.; Navarrete-Dechent, C.; Petukhova, T.A.; Lee, E.H.; Rossi, A.M.; Postow, M.A.; Dunn, L.A.; Roman, B.R.; Yin, V.T.; Coit, D.G.; et al. Tumor Board Conferences for Multidisciplinary Skin Cancer Management: A Survey of US Cancer Centers. J. Natl. Compr. Cancer Netw. 2018, 16, 1209–1215. [Google Scholar] [CrossRef] [PubMed]
- National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology, Basal Cell Skin Cancer, Version 1.2018 2018. Available online: https://oncolife.com.ua/doc/nccn/Basal_Cell_Skin_Cancer.pdf (accessed on 3 March 2024).
- National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology, Squamous Cell Skin Cancer, Version 2.2018 2018. Available online: https://oncolife.com.ua/doc/nccn/Squamous_Cell_Skin_Cancer.pdf (accessed on 3 March 2024).
- Brambullo, T.; Azzena, G.P.; Toninello, P.; Masciopinto, G.; De Lazzari, A.; Biffoli, B.; Vindigni, V.; Bassetto, F. Current Surgical Therapy of Locally Advanced cSCC: From Patient Selection to Microsurgical Tissue Transplant. Review. Front. Oncol. 2021, 11, 783257. [Google Scholar] [CrossRef] [PubMed]
- Berardi, R.; Morgese, F.; Rinaldi, S.; Torniai, M.; Mentrasti, G.; Scortichini, L.; Giampieri, R. Benefits and Limitations of a Multidisciplinary Approach in Cancer Patient Management. Cancer Manag. Res. 2020, 12, 9363–9374. [Google Scholar] [CrossRef]
- Clarke, J.; Ioffreda, M.; Helm, K.F. Multiple familial trichoepitheliomas: A folliculosebaceous-apocrine genodermatosis. Am. J. Dermatopathol. 2002, 24, 402–405. [Google Scholar] [CrossRef] [PubMed]
- Shang, X.; Lin, X.; Howell, S.B. Claudin-4 controls the receptor tyrosine kinase EphA2 pro-oncogenic switch through β-catenin. Cell Commun. Signal. 2014, 12, 59. [Google Scholar] [CrossRef] [PubMed]
- Shang, X.; Lin, X.; Alvarez, E.; Manorek, G.; Howell, S.B. Tight junction proteins claudin-3 and claudin-4 control tumor growth and metastases. Neoplasia 2012, 14, 974–985. [Google Scholar] [CrossRef]
- Sung, C.O.; Choi, H.; Lee, K.-W.; Kim, S.-H. Sarcomatoid carcinoma represents a complete phenotype with various pathways of epithelial mesenchymal transition. J. Clin. Pathol. 2013, 66, 601–606. [Google Scholar] [CrossRef]
- Goyal, A.; Marghitu, T.; Goyal, N.; Rubin, N.; Patel, K.; Goyal, K.; O’leary, D.; Bohjanen, K.; Maher, I. Surgical management and lymph-node biopsy of rare malignant cutaneous adnexal carcinomas: A population-based analysis of 7591 patients. Arch. Dermatol. Res. 2021, 313, 623–632. [Google Scholar] [CrossRef] [PubMed]
- Wargo, J.J.; Carr, D.R.; Plaza, J.A.; Verschraegen, C.F. Metastatic Spiradenocarcinoma Managed With PD-1 Inhibition. J. Natl. Compr. Cancer Netw. 2022, 20, 318–320. [Google Scholar] [CrossRef] [PubMed]
- Portincasa, A.; Cecchino, L.; Trecca, E.; Lembo, F.; Annacontini, L.; Ciancio, F.; Corsi, F.; Cassano, M.; Parisi, D. A rare case of Brooke-Spiegler syndrome: Integrated surgical treatment of multiple giant eccrine spiradenomas of the head and neck in a young girl. Int. J. Surg. Case Rep. 2018, 51, 277–281. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, S.; Dayal, S. Radiofrequency ablation: A safe and economical modality in treatment of Brooke-Spiegler syndrome. Dermatol. Online J. 2012, 18, 7. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brambullo, T.; De Lazzari, A.; Franchi, A.; Trevisson, E.; Garau, M.L.; Scarmozzino, F.; Vindigni, V.; Bassetto, F. A Misdiagnosed Familiar Brooke–Spiegler Syndrome: Case Report and Review of the Literature. J. Clin. Med. 2024, 13, 2240. https://doi.org/10.3390/jcm13082240
Brambullo T, De Lazzari A, Franchi A, Trevisson E, Garau ML, Scarmozzino F, Vindigni V, Bassetto F. A Misdiagnosed Familiar Brooke–Spiegler Syndrome: Case Report and Review of the Literature. Journal of Clinical Medicine. 2024; 13(8):2240. https://doi.org/10.3390/jcm13082240
Chicago/Turabian StyleBrambullo, Tito, Alberto De Lazzari, Arianna Franchi, Eva Trevisson, Maria Luisa Garau, Federico Scarmozzino, Vincenzo Vindigni, and Franco Bassetto. 2024. "A Misdiagnosed Familiar Brooke–Spiegler Syndrome: Case Report and Review of the Literature" Journal of Clinical Medicine 13, no. 8: 2240. https://doi.org/10.3390/jcm13082240
APA StyleBrambullo, T., De Lazzari, A., Franchi, A., Trevisson, E., Garau, M. L., Scarmozzino, F., Vindigni, V., & Bassetto, F. (2024). A Misdiagnosed Familiar Brooke–Spiegler Syndrome: Case Report and Review of the Literature. Journal of Clinical Medicine, 13(8), 2240. https://doi.org/10.3390/jcm13082240