
Catheter Ablation for Channelopathies: When Is Less More?
 , ,
, ,
Abstract
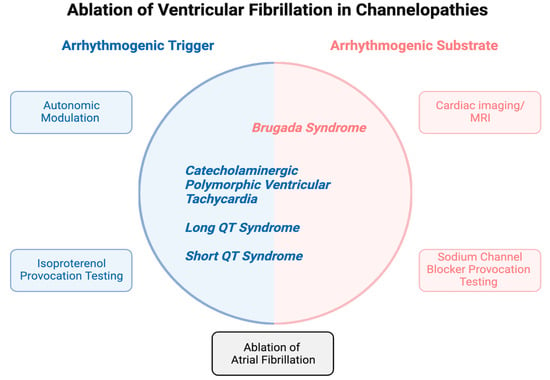
1. Introduction
2. Mechanistic Insights into the Substrate and Triggers of VF
3. Brugada Syndrome
4. Congenital Long QT Syndrome
5. Catecholaminergic Polymorphic Ventricular Tachycardia
6. Short QT Syndrome
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bagnall, R.D.; Weintraub, R.G.; Ingles, J.; Duflou, J.; Yeates, L.; Lam, L.; Davis, A.M.; Thompson, T.; Connell, V.; Wallace, J.; et al. A Prospective Study of Sudden Cardiac Death among Children and Young Adults. N. Engl. J. Med. 2016, 374, 2441–2452. [Google Scholar] [CrossRef] [PubMed]
- Girotra, S.; Nallamothu, B.K.; Spertus, J.A.; Li, Y.; Krumholz, H.M.; Chan, P.S. Trends in Survival after In-Hospital Cardiac Arrest. N. Engl. J. Med. 2012, 367, 1912–1920. [Google Scholar] [CrossRef] [PubMed]
- Mariani, M.V.; Pierucci, N.; Fanisio, F.; Laviola, D.; Silvetti, G.; Piro, A.; La Fazia, V.M.; Chimenti, C.; Rebecchi, M.; Drago, F.; et al. Inherited Arrhythmias in the Pediatric Population: An Updated Overview. Medicina 2024, 60, 94. [Google Scholar] [CrossRef]
- Priori, S.G.; Wilde, A.A.; Horie, M.; Cho, Y.; Behr, E.R.; Berul, C.; Blom, N.; Brugada, J.; Chiang, C.E.; Huikuri, H.; et al. HRS/EHRA/APHRS Expert Consensus Statement on the Diagnosis and Management of Patients with Inherited Primary Arrhythmia Syndromes: Document endorsed by HRS, EHRA, and APHRS in May 2013 and by ACCF, AHA, PACES, and AEPC in June 2013. Heart Rhythm. 2013, 10, 1932–1963. [Google Scholar] [CrossRef] [PubMed]
- Bohnen, M.S.; Peng, G.; Robey, S.H.; Terrenoire, C.; Iyer, V.; Sampson, K.J.; Kass, R.S. Molecular Pathophysiology of Congenital Long QT Syndrome. Physiol. Rev. 2017, 97, 89–134. [Google Scholar] [CrossRef] [PubMed]
- Weiss, J.N.; Garfinkel, A.; Karagueuzian, H.S.; Chen, P.S.; Qu, Z. Early afterdepolarizations and cardiac arrhythmias. Heart Rhythm. 2010, 7, 1891–1899. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Curran, M.E.; Splawski, I.; Burn, T.C.; Millholland, J.M.; VanRaay, T.J.; Shen, J.; Timothy, K.W.; Vincent, G.M.; De Jager, T.; et al. Positional cloning of a novel potassium channel gene: KVLQT1 mutations cause cardiac arrhythmias. Nat. Genet. 1996, 12, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Curran, M.E.; Splawski, I.; Timothy, K.W.; Vincent, G.M.; Green, E.D.; Keating, M.T. A molecular basis for cardiac arrhythmia: HERG mutations cause long QT syndrome. Cell 1995, 80, 795–803. [Google Scholar] [CrossRef]
- Wang, Q.; Shen, J.; Splawski, I.; Atkinson, D.; Li, Z.; Robinson, J.L.; Moss, A.J.; Towbin, J.A.; Keating, M.T. SCN5A mutations associated with an inherited cardiac arrhythmia, long QT syndrome. Cell 1995, 80, 805–811. [Google Scholar] [CrossRef]
- Litovsky, S.H.; Antzelevitch, C. Transient outward current prominent in canine ventricular epicardium but not endocardium. Circ. Res. 1988, 62, 116–126. [Google Scholar] [CrossRef]
- Zaza, A.; Malfatto, G.; Rosen, M.R. Electrophysiologic effects of ketanserin on canine Purkinje fibers, ventricular myocardium and the intact heart. J. Pharmacol. Exp. Ther. 1989, 250, 397–405. [Google Scholar] [PubMed]
- Dun, W.; Boyden, P.A. The Purkinje cell; 2008 style. J. Mol. Cell. Cardiol. 2008, 45, 617–624. [Google Scholar] [CrossRef]
- Haïssaguerre, M.; Extramiana, F.; Hocini, M.; Cauchemez, B.; Jaïs, P.; Cabrera, J.A.; Farre, G.; Leenhardt, A.; Sanders, P.; Scavée, C.; et al. Mapping and Ablation of Ventricular Fibrillation Associated with Long-QT and Brugada Syndromes. Circulation 2003, 108, 925–928. [Google Scholar] [CrossRef]
- Pappone, C.; Ciconte, G.; Anastasia, L.; Gaita, F.; Grant, E.; Micaglio, E.; Locati, E.T.; Calovic, Z.; Vicedomini, G.; Santinelli, V. Right ventricular epicardial arrhythmogenic substrate in long-QT syndrome patients at risk of sudden death. Europace 2023, 25, 948–955. [Google Scholar] [CrossRef]
- Chaumont, C.; Suffee, N.; Gandjbakhch, E.; Balse, E.; Anselme, F.; Hatem, S.N. Epicardial origin of cardiac arrhythmias: Clinical evidences and pathophysiology. Cardiovasc. Res. 2021, 118, 1693–1702. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Sun, Q.; Zeng, Z.; Li, Q.; Zhou, S.; Zhou, M.; Xue, Y.; Cheng, X.; Xia, Y.; Wang, Q.; et al. Regulation of SCN3B/scn3b by Interleukin 2 (IL-2): IL-2 modulates SCN3B/scn3b transcript expression and increases sodium current in myocardial cells. BMC Cardiovasc. Disord. 2016, 16, 1. [Google Scholar] [CrossRef]
- Diedrich, A.; Jordan, J.; Shannon, J.R.; Robertson, D.; Biaggioni, I. Modulation of QT Interval During Autonomic Nervous System Blockade in Humans. Circulation 2002, 106, 2238–2243. [Google Scholar] [CrossRef]
- Ter Bekke, R.M.; Moers, A.M.; de Jong, M.M.; Johnson, D.M.; Schwartz, P.J.; Vanoli, E.; Volders, P.G. Proarrhythmic proclivity of left-stellate ganglion stimulation in a canine model of drug-induced long-QT syndrome type 1. Int. J. Cardiol. 2019, 286, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Shah, C.; Jiwani, S.; Limbu, B.; Weinberg, S.; Deo, M. Delayed afterdepolarization-induced triggered activity in cardiac purkinje cells mediated through cytosolic calcium diffusion waves. Physiol. Rep. 2019, 7, e14296. [Google Scholar] [CrossRef] [PubMed]
- Lukas, A.; Antzelevitch, C. Differences in the electrophysiological response of canine ventricular epicardium and endocardium to ischemia. Role Transient Outw. Curr. Circ. 1993, 88, 2903–2915. [Google Scholar]
- Antzelevitch, C.; Belardinelli, L. The role of sodium channel current in modulating transmural dispersion of repolarization and arrhythmogenesis. J. Cardiovasc. Electrophysiol. 2006, 17 (Suppl. 1), S79–S85. [Google Scholar] [CrossRef] [PubMed]
- Nademanee, K.; Raju, H.; de Noronha, S.V.; Papadakis, M.; Robinson, L.; Rothery, S.; Makita, N.; Kowase, S.; Boonmee, N.; Vitayakritsirikul, V.; et al. Fibrosis, Connexin-43, and Conduction Abnormalities in the Brugada Syndrome. J. Am. Coll. Cardiol. 2015, 66, 1976–1986. [Google Scholar] [CrossRef] [PubMed]
- Brugada, P.; Brugada, J. Right bundle branch block, persistent ST segment elevation and sudden cardiac death: A distinct clinical and electrocardiographic syndrome. A multicenter report. J. Am. Coll. Cardiol. 1992, 20, 1391–1396. [Google Scholar] [CrossRef] [PubMed]
- Atarashi, H.; Ogawa, S.; Harumi, K.; Sugimoto, T.; Inoue, H.; Murayama, M.; Toyama, J.; Hayakawa, H. Three-year follow-up of patients with right bundle branch block and ST segment elevation in the right precordial leads: Japanese Registry of Brugada Syndrome. Idiopathic Ventricular Fibrillation Investigators. J. Am. Coll. Cardiol. 2001, 37, 1916–1920. [Google Scholar] [PubMed]
- Matsuo, K.; Akahoshi, M.; Nakashima, E.; Suyama, A.; Seto, S.; Hayano, M.; Yano, K. The prevalence, incidence and prognostic value of the Brugada-type electrocardiogram: A population-based study of four decades. J. Am. Coll. Cardiol. 2001, 38, 765–770. [Google Scholar] [CrossRef] [PubMed]
- Junttila, M.J.; Raatikainen, M.J.; Karjalainen, J.; Kauma, H.; Kesäniemi, Y.A.; Huikuri, H.V. Prevalence and prognosis of subjects with Brugada-type ECG pattern in a young and middle-aged Finnish population. Eur. Heart J. 2004, 25, 874–878. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.S.; Anees, S.; Ferrick, K.J. Prevalence of a Brugada pattern electrocardiogram in an urban population in the United States. Pacing Clin. Electrophysiol. 2009, 32, 704–708. [Google Scholar] [CrossRef] [PubMed]
- Vutthikraivit, W.; Rattanawong, P.; Putthapiban, P.; Sukhumthammarat, W.; Vathesatogkit, P.; Ngarmukos, T.; Thakkinstian, A. Worldwide Prevalence of Brugada Syndrome: A Systematic Review and Meta-Analysis. Acta Cardiol. Sin. 2018, 34, 267–277. [Google Scholar]
- Alings, M.; Wilde, A. “Brugada” Syndrome. Circulation 1999, 99, 666–673. [Google Scholar] [CrossRef]
- Sieira, J.; Conte, G.; Ciconte, G.; de Asmundis, C.; Chierchia, G.B.; Baltogiannis, G.; Di Giovanni, G.; Saitoh, Y.; Irfan, G.; Casado-Arroyo, R.; et al. Clinical characterisation and long-term prognosis of women with Brugada syndrome. Heart 2016, 102, 452–458. [Google Scholar] [CrossRef]
- Rodríguez-Mañero, M.; Jordá, P.; Hernandez, J.; Muñoz, C.; Grima, E.Z.; García-Fernández, A.; Cañadas-Godoy, M.V.; Jiménez-Ramos, V.; Oloriz, T.; Basterra, N.; et al. Long-term prognosis of women with Brugada syndrome and electrophysiological study. Heart Rhythm. 2021, 18, 664–671. [Google Scholar] [CrossRef] [PubMed]
- Benito, B.; Sarkozy, A.; Mont, L.; Henkens, S.; Berruezo, A.; Tamborero, D.; Arzamendi, D.; Berne, P.; Brugada, R.; Brugada, P.; et al. Gender differences in clinical manifestations of Brugada syndrome. J. Am. Coll. Cardiol. 2008, 52, 1567–1573. [Google Scholar] [CrossRef] [PubMed]
- Antzelevitch, C.; Brugada, P.; Borggrefe, M.; Brugada, J.; Brugada, R.; Corrado, D.; Gussak, I.; LeMarec, H.; Nademanee, K.; Riera, A.R.P.; et al. Brugada syndrome: Report of the second consensus conference. Heart Rhythm. 2005, 2, 429–440. [Google Scholar] [CrossRef] [PubMed]
- Antzelevitch, C.; Yan, G.X.; Ackerman, M.J.; Borggrefe, M.; Corrado, D.; Guo, J.; Gussak, I.; Hasdemir, C.; Horie, M.; Huikuri, H.; et al. J-Wave syndromes expert consensus conference report: Emerging concepts and gaps in knowledge. Europace 2017, 19, 665–694. [Google Scholar]
- Brugada, J.; Brugada, R.; Brugada, P. Determinants of sudden cardiac death in individuals with the electrocardiographic pattern of Brugada syndrome and no previous cardiac arrest. Circulation 2003, 108, 3092–3096. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Kirsch, G.E.; Zhang, D.; Brugada, R.; Brugada, J.; Brugada, P.; Potenza, D.; Moya, A.; Borggrefe, M.; Breithardt, G.; et al. Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nature 1998, 392, 293–296. [Google Scholar] [CrossRef]
- Brugada, J.; Campuzano, O.; Arbelo, E.; Sarquella-Brugada, G.; Brugada, R. Present Status of Brugada Syndrome: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2018, 72, 1046–1059. [Google Scholar] [CrossRef]
- Campuzano, O.; Sarquella-Brugada, G.; Cesar, S.; Arbelo, E.; Brugada, J.; Brugada, R. Update on Genetic Basis of Brugada Syndrome: Monogenic, Polygenic or Oligogenic? Int. J. Mol. Sci. 2020, 21, 7155. [Google Scholar] [CrossRef]
- London, B.; Michalec, M.; Mehdi, H.; Zhu, X.; Kerchner, L.; Sanyal, S.; Viswanathan, P.C.; Pfahnl, A.E.; Shang, L.L.; Madhusudanan, M.; et al. Mutation in glycerol-3-phosphate dehydrogenase 1 like gene (GPD1-L) decreases cardiac Na+ current and causes inherited arrhythmias. Circulation 2007, 116, 2260–2268. [Google Scholar] [CrossRef]
- Makarawate, P.; Glinge, C.; Khongphatthanayothin, A.; Walsh, R.; Mauleekoonphairoj, J.; Amnueypol, M.; Prechawat, S.; Wongcharoen, W.; Krittayaphong, R.; Anannab, A.; et al. Common and rare susceptibility genetic variants predisposing to Brugada syndrome in Thailand. Heart Rhythm. 2020, 17, 2145–2153. [Google Scholar] [CrossRef]
- Bezzina, C.R.; Barc, J.; Mizusawa, Y.; Remme, C.A.; Gourraud, J.B.; Simonet, F.; Verkerk, A.O.; Schwartz, P.J.; Crotti, L.; Dagradi, F.; et al. Common variants at SCN5A-SCN10A and HEY2 are associated with Brugada syndrome, a rare disease with high risk of sudden cardiac death. Nat. Genet. 2013, 45, 1044–1049. [Google Scholar] [CrossRef]
- Miyazaki, T.; Mitamura, H.; Miyoshi, S.; Soejima, K.; Aizawa, Y.; Ogawa, S. Autonomic and antiarrhythmic drug modulation of ST segment elevation in patients with Brugada syndrome. J. Am. Coll. Cardiol. 1996, 27, 1061–1070. [Google Scholar] [CrossRef] [PubMed]
- Mizumaki, K.; Fujiki, A.; Tsuneda, T.; Sakabe, M.; Nishida, K.; Sugao, M.; Inoue, H. Vagal activity modulates spontaneous augmentation of ST elevation in the daily life of patients with Brugada syndrome. J. Cardiovasc. Electrophysiol. 2004, 15, 667–673. [Google Scholar] [CrossRef]
- Chung, C.T.; Bazoukis, G.; Radford, D.; Coakley-Youngs, E.; Rajan, R.; Matusik, P.T.; Liu, T.; Letsas, K.P.; Lee, S.; Tse, G. Predictive risk models for forecasting arrhythmic outcomes in Brugada syndrome: A focused review. J. Electrocardiol. 2022, 72, 28–34. [Google Scholar] [CrossRef]
- Catalano, O.; Antonaci, S.; Moro, G.; Mussida, M.; Frascaroli, M.; Baldi, M.; Cobelli, F.; Baiardi, P.; Nastoli, J.; Bloise, R.; et al. Magnetic resonance investigations in Brugada syndrome reveal unexpectedly high rate of structural abnormalities. Eur. Heart J. 2009, 30, 2241–2248. [Google Scholar] [CrossRef]
- Rodríguez-Mañero, M.; Sacher, F.; de Asmundis, C.; Maury, P.; Lambiase, P.D.; Sarkozy, A.; Probst, V.; Gandjbakhch, E.; Castro-Hevia, J.; Saenen, J.; et al. Monomorphic ventricular tachycardia in patients with Brugada syndrome: A multicenter retrospective study. Heart Rhythm. 2016, 13, 669–682. [Google Scholar] [CrossRef] [PubMed]
- Tokioka, S.; Fukamizu, S.; Kitamura, T.; Miyazawa, S.; Kawamura, I.; Hojo, R.; Sakurada, H.; Hiraoka, M. Catheter ablation for monomorphic ventricular tachycardia in Brugada syndrome patients: Detailed characteristics and long-term follow-up. J. Interv. Card. Electrophysiol. 2020, 57, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Szél, T.; Antzelevitch, C. Abnormal Repolarization as the Basis for Late Potentials and Fractionated Electrograms Recorded From Epicardium in Experimental Models of Brugada Syndrome. J. Am. Coll. Cardiol. 2014, 63, 2037–2045. [Google Scholar] [CrossRef]
- Morita, H.; Zipes, D.P.; Morita, S.T.; Lopshire, J.C.; Wu, J. Epicardial ablation eliminates ventricular arrhythmias in an experimental model of Brugada syndrome. Heart Rhythm. 2009, 6, 665–671. [Google Scholar] [CrossRef]
- Nademanee, K.; Veerakul, G.; Chandanamattha, P.; Chaothawee, L.; Ariyachaipanich, A.; Jirasirirojanakorn, K.; Likittanasombat, K.; Bhuripanyo, K.; Ngarmukos, T. Prevention of Ventricular Fibrillation Episodes in Brugada Syndrome by Catheter Ablation Over the Anterior Right Ventricular Outflow Tract Epicardium. Circulation 2011, 123, 1270–1279. [Google Scholar] [CrossRef]
- Pappone, C.; Brugada, J.; Vicedomini, G.; Ciconte, G.; Manguso, F.; Saviano, M.; Vitale, R.; Cuko, A.; Giannelli, L.; Calovic, Z.; et al. Electrical Substrate Elimination in 135 Consecutive Patients With Brugada Syndrome. Circ. Arrhythm. Electrophysiol. 2017, 10, e005053. [Google Scholar] [CrossRef] [PubMed]
- Nademanee, K.; Chung, F.-P.; Sacher, F.; Nogami, A.; Nakagawa, H.; Jiang, C.; Hocini, M.; Behr, E.; Veerakul, G.; Jan Smit, J.; et al. Long-Term Outcomes of Brugada Substrate Ablation: A Report from BRAVO (Brugada Ablation of VF Substrate Ongoing Multicenter Registry). Circulation 2023, 147, 1568–1578. [Google Scholar] [CrossRef] [PubMed]
- Talib, A.K.; Takagi, M.; Shimane, A.; Nakano, M.; Hayashi, T.; Okajima, K.; Kentaro, M.; Fukada, K.; Kowase, S.; Kurosaki, K.; et al. Efficacy of Endocardial Ablation of Drug-Resistant Ventricular Fibrillation in Brugada Syndrome. Circ. Arrhythmia Electrophysiol. 2018, 11, e005631. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, G.C.; Fernandes, A.; Cardoso, R.; Nasi, G.; Rivera, M.; Mitrani, R.D.; Goldberger, J.J. Ablation strategies for the management of symptomatic Brugada syndrome: A systematic review. Heart Rhythm. 2018, 15, 1140–1147. [Google Scholar] [CrossRef]
- Zeppenfeld, K.; Tfelt-Hansen, J.; de Riva, M.; Winkel, B.G.; Behr, E.R.; Blom, N.A.; Charron, P.; Corrado, D.; Dagres, N.; de Chillou, C.; et al. 2022 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death. Eur. Heart. J. 2022, 43, 3997–4126. [Google Scholar]
- Sunsaneewitayakul, B.; Yao, Y.; Thamaree, S.; Zhang, S. Endocardial Mapping and Catheter Ablation for Ventricular Fibrillation Prevention in Brugada Syndrome. J. Cardiovasc. Electrophysiol. 2012, 23, s10–s16. [Google Scholar] [CrossRef] [PubMed]
- Brugada, J.; Pappone, C.; Berruezo, A.; Vicedomini, G.; Manguso, F.; Ciconte, G.; Giannelli, L.; Santinelli, V. Brugada Syndrome Phenotype Elimination by Epicardial Substrate Ablation. Circ. Arrhythmia Electrophysiol. 2015, 8, 1373–1381. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Tung, R.; Zhang, Z.; Sheng, X.; Liu, Q.; Jiang, R.; Sun, Y.; Chen, S.; Yu, L.; Ye, Y.; et al. Characterization of the epicardial substrate for catheter ablation of Brugada syndrome. Heart Rhythm. 2016, 13, 2151–2158. [Google Scholar] [CrossRef] [PubMed]
- Chung, F.P.; Raharjo, S.B.; Lin, Y.J.; Chang, S.L.; Lo, L.W.; Hu, Y.F.; Tuan, T.C.; Chao, T.F.; Liao, J.N.; Lin, C.Y.; et al. A novel method to enhance phenotype, epicardial functional substrates, and ventricular tachyarrhythmias in Brugada syndrome. Heart Rhythm. 2017, 14, 508–517. [Google Scholar] [CrossRef]
- Shelke, A.; Tachil, A.; Saggu, D.; Jesuraj, M.L.; Yalagudri, S.; Narasimhan, C. Catheter ablation for electrical storm in Brugada syndrome: Results of substrate based ablation. Indian Heart J. 2018, 70, 296–302. [Google Scholar] [CrossRef]
- Kamakura, T.; Cochet, H.; Juhoor, M.; Nakatani, Y.; Ramirez, F.D.; André, C.; Nakashima, T.; Krisai, P.; Takagi, T.; Tixier, R.; et al. Role of endocardial ablation in eliminating an epicardial arrhythmogenic substrate in patients with Brugada syndrome. Heart Rhythm. 2021, 18, 1673–1681. [Google Scholar] [CrossRef] [PubMed]
- Mamiya, K.; Inden, Y.; Yanagisawa, S.; Fujii, A.; Tomomatsu, T.; Okamoto, H.; Riku, S.; Suga, K.; Furui, K.; Nakagomi, T.; et al. Dynamic Changes in Electrocardiogram Parameters After Epicardial Substrate Catheter Ablation of Brugada Syndrome. Circ. J. 2021, 85, 1283–1293. [Google Scholar] [CrossRef] [PubMed]
- Haissaguerre, M.; Cheniti, G.; Hocini, M.; Sacher, F.; Ramirez, F.D.; Cochet, H.; Bear, L.; Tixier, R.; Duchateau, J.; Walton, R.; et al. Purkinje network and myocardial substrate at the onset of human ventricular fibrillation: Implications for catheter ablation. Eur. Heart J. 2022, 43, 1234–1247. [Google Scholar] [CrossRef] [PubMed]
- Krahn, A.D.; Laksman, Z.; Sy, R.W.; Postema, P.G.; Ackerman, M.J.; Wilde, A.A.M.; Han, H.C. Congenital Long QT Syndrome. JACC Clin. Electrophysiol. 2022, 8, 687–706. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, P.J.; Stramba-Badiale, M.; Crotti, L.; Pedrazzini, M.; Besana, A.; Bosi, G.; Gabbarini, F.; Goulene, K.; Insolia, R.; Mannarino, S.; et al. Prevalence of the congenital long-QT syndrome. Circulation 2009, 120, 1761–1767. [Google Scholar] [CrossRef] [PubMed]
- Abu-Zeitone, A.; Peterson, D.R.; Polonsky, B.; McNitt, S.; Moss, A.J. Efficacy of Different Beta-Blockers in the Treatment of Long QT Syndrome. J. Am. Coll. Cardiol. 2014, 64, 1352–1358. [Google Scholar] [CrossRef]
- Ruan, Y.; Liu, N.; Bloise, R.; Napolitano, C.; Priori, S.G. Gating properties of SCN5A mutations and the response to mexiletine in long-QT syndrome type 3 patients. Circulation 2007, 116, 1137–1144. [Google Scholar] [CrossRef] [PubMed]
- Wilde, A.A.M.; Ackerman, M.J. Counterpoint: Ablation in long QT syndrome. Heart Rhythm. 2023, 20, 1785–1786. [Google Scholar] [CrossRef] [PubMed]
- Dinov, B. Radiofrequency catheter ablation in congenital long QT syndrome: An anatomical approach to a supposedly primary electrical disease. Europace 2023, 25, 253–254. [Google Scholar] [CrossRef]
- Lieve, K.V.; van der Werf, C.; Wilde, A.A. Catecholaminergic Polymorphic Ventricular Tachycardia. Circ. J. 2016, 80, 1285–1291. [Google Scholar] [CrossRef]
- Priori, S.G.; Napolitano, C.; Memmi, M.; Colombi, B.; Drago, F.; Gasparini, M.; DeSimone, L.; Coltorti, F.; Bloise, R.; Keegan, R.; et al. Clinical and molecular characterization of patients with catecholaminergic polymorphic ventricular tachycardia. Circulation 2002, 106, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Walsh, R.; Adler, A.; Amin, A.S.; Abiusi, E.; Care, M.; Bikker, H.; Amenta, S.; Feilotter, H.; Nannenberg, E.A.; Mazzarotto, F.; et al. Evaluation of gene validity for CPVT and short QT syndrome in sudden arrhythmic death. Eur. Heart J. 2022, 43, 1500–1510. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Colombi, B.; Memmi, M.; Zissimopoulos, S.; Rizzi, N.; Negri, S.; Imbriani, M.; Napolitano, C.; Lai, F.A.; Priori, S.G. Arrhythmogenesis in Catecholaminergic Polymorphic Ventricular Tachycardia. Circ. Res. 2006, 99, 292–298. [Google Scholar] [CrossRef] [PubMed]
- Napolitano, C.; Priori, S.G. Diagnosis and treatment of catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm. 2007, 4, 675–678. [Google Scholar] [CrossRef]
- Roston, T.M.; Jones, K.; Hawkins, N.M.; Bos, J.M.; Schwartz, P.J.; Perry, F.; Ackerman, M.J.; Laksman, Z.W.; Kaul, P.; Lieve, K.V.; et al. Implantable cardioverter-defibrillator use in catecholaminergic polymorphic ventricular tachycardia: A systematic review. Heart Rhythm. 2018, 15, 1791–1799. [Google Scholar] [CrossRef]
- Miyake, C.Y.; Webster, G.; Czosek, R.J.; Kantoch, M.J.; Dubin, A.M.; Avasarala, K.; Atallah, J. Efficacy of Implantable Cardioverter Defibrillators in Young Patients with Catecholaminergic Polymorphic Ventricular Tachycardia. Circ. Arrhythmia Electrophysiol. 2013, 6, 579–587. [Google Scholar] [CrossRef]
- Shen, L.; Liu, S.; Hu, F.; Zhang, Z.; Li, J.; Lai, Z.; Zheng, L.; Yao, Y. Electrophysiological Characteristics and Ablation Outcomes in Patients with Catecholaminergic Polymorphic Ventricular Tachycardia. J. Am. Heart Assoc. 2023, 12, e031768. [Google Scholar] [CrossRef]
- Kaneshiro, T.; Nogami, A.; Kato, Y.; Kuroki, K.; Komatsu, Y.; Tada, H.; Sekiguchi, Y.; Horigome, H.; Aonuma, K. Effects of Catheter Ablation Targeting the Trigger Beats in Inherited Catecholaminergic Polymorphic Ventricular Tachycardia. JACC Clin. Electrophysiol. 2017, 3, 1062–1063. [Google Scholar] [CrossRef]
- Kawada, S.; Morita, H.; Watanabe, A.; Ito, H. Radiofrequency catheter ablation for drug-refractory atrial tachyarrhythmias in a patient with catecholaminergic polymorphic ventricular tachycardia: A case report. J. Cardiol. Cases 2019, 19, 36–39. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, Y.; Watanabe, A.; Morita, H.; Nishii, N.; Nakamura, K.; Ito, H. Successful radiofrequency catheter ablation of a premature ventricular contraction triggering ventricular fibrillation in a patient with short QT syndrome. Heart Case Rep. 2019, 5, 262–265. [Google Scholar] [CrossRef]

{kind=link}
| No. | First Author, Year | Study Design | Total Population | Repeat Ablation | Notable Points | |
|---|---|---|---|---|---|---|
| Ablated | Not Ablated | |||||
| 1. | Haïssaguerre, 2003 [13] | Cohort Retrospective | 3 | 0 | 0 | PVC triggers successfully ablated from endocardial RVOT (n = 2) RV and Purkinje fibers (n = 1). |
| 2. | Sunsaneewiyatakul, 2012 [56] | Cohort Retrospective | 4 | 6 | 0 | Endocardial RVOT ablation. No more VF storm at 12–30-month follow-up. |
| 3. | Brugada, 2015 [57] | Cohort Prospective | 14 | 0 | 0 | RV epicardial ablation (anterior RV free wall and RVOT) of low voltage areas identified with mapping before and after flecainide. VA not inducible after 5-month follow-up. |
| 4. | Rodríguez-Mañero, 2015 [46] | Cohort Retrospective | 6 | 828 | 0 | 4.2% of the patients of BrS with ICD experienced monomorphic VT: Successful ablation of RVOT tachycardia (from the endocardium, n = 4 and epicardial and endocardial ablation, n = 1), endocardial lateral mitral annulus, n = 1, and BBRVT, n = 2. |
| 5. | Zhang, 2016 [58] | Cohort Prospective | 11 | 0 | 0 | Despite epicardial RVOT ablation, 27% of the patients experienced recurrence of VA. |
| 6. | Chung, 2017 [59] | Cohort Retrospective | 15 | 0 | 1 | Flecainide provocation test, n = 15, and epicardial warm water instillation (n = 6) led to enhancement of functional epicardial substrates. Successful ablation in 14 of 15 patients; 1 patient required repeat ablation with no further recurrence of VA at 1-year follow-up. |
| 7. | Pappone, 2017 [51] | Cohort Prospective | 135 | 0 | 2 | Ajmaline provocation test. Arrhythmogenic electrophysiological substrate commonly present in the RV epicardium. |
| 8. | Shelke, 2017 [60] | Cohort Retrospective | 5 | 0 | 0 | Substrate modification of RVOT was performed endocardially in one patient, both endocardial and epicardial in three, and only epicardially in one patient. One patient had an episode of VF at 24 months after ablation. |
| 9. | Talib, 2023 [53] | Cohort Retrospective | 21 | 102 | 0 | Endocardial ablation of PVCs triggering VF storm from RVOT (n = 18) and RV anterior free wall (n = 2), RV inflow tract (n = 1). Notching of QRS in lead V1 was associated with high VF recurrence. |
| 10. | Tokioka, 2019 [47] | Cohort Retrospective | 7 | 181 | 0 | Ablation in BrS patients with monomorphic VT: RVOT (n = 4), tricuspid annulus (n = 1), LV septum (n = 1), BBRVT (n = 1). One patient had recurrent VT. |
| 11. | Kamakura, 2021 [61] | Cohort Retrospective | 16 | 0 | 0 | Ajmaline provocation. Abnormal epicardial potentials in RVOT in all 16 patients and in the epicardial RV inferior wall in 12/16 patients. Combined endocardial and epicardial ablation was suggested, with endocardial ablation as an alternative to epicardial ablation for ablating areas close to the coronary artery. No patients had recurrent VF storm after ablation. |
| 12. | Mamiya, 2021 [62] | Cohort Retrospective | 11 | 16 | 0 | Pilsicainide provocation test. Epicardial RVOT ablation. Improvements in the ECG parameters were associated with decreased VF recurrence post-procedure. A total of 4/11 patients had recurrent VF after ablation. |
| 13. | Haïssaguerre, 2022 [63] | Cohort Retrospective | 17 | 2 | 3 | Most common triggers were located in the epicardial RVOT and epicardial inferior RV; epicardial ablation in all patients, and combined endo-epi ablation in 2/17. A total of 6/17 patients had recurrent VF. |
| No. | First Author, Year | Study Design | Total Population | Repeat Ablation | Notable Points | |
|---|---|---|---|---|---|---|
| Ablated | Not Ablated | |||||
| 1. | Haïssaguerre, 2003 [13] | Cohort Retrospective | 4 | 0 | 0 | PVC triggers successfully ablated from LV Purkinje fibers (n = 3) and endocardial RVOT (n = 1). |
| 2. | Pappone, 2023 [14] | Cohort Prospective | 11 | 0 | 0 | Endo- and epicardial mapping of RV and LV. Ablation of structural electrophysiological abnormalities in the epicardium of RV prevents recurrences. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mehta, A.; Chandiramani, R.; Ghosh, B.; Asatryan, B.; Hajra, A.; Barth, A.S. Catheter Ablation for Channelopathies: When Is Less More? J. Clin. Med. 2024, 13, 2384. https://doi.org/10.3390/jcm13082384
Mehta A, Chandiramani R, Ghosh B, Asatryan B, Hajra A, Barth AS. Catheter Ablation for Channelopathies: When Is Less More? Journal of Clinical Medicine. 2024; 13(8):2384. https://doi.org/10.3390/jcm13082384
Chicago/Turabian StyleMehta, Adhya, Rishi Chandiramani, Binita Ghosh, Babken Asatryan, Adrija Hajra, and Andreas S. Barth. 2024. "Catheter Ablation for Channelopathies: When Is Less More?" Journal of Clinical Medicine 13, no. 8: 2384. https://doi.org/10.3390/jcm13082384
APA StyleMehta, A., Chandiramani, R., Ghosh, B., Asatryan, B., Hajra, A., & Barth, A. S. (2024). Catheter Ablation for Channelopathies: When Is Less More? Journal of Clinical Medicine, 13(8), 2384. https://doi.org/10.3390/jcm13082384