

Targeted Complement Treatments in Glomerulopathies: A Comprehensive Review

Abstract
:1. Introduction
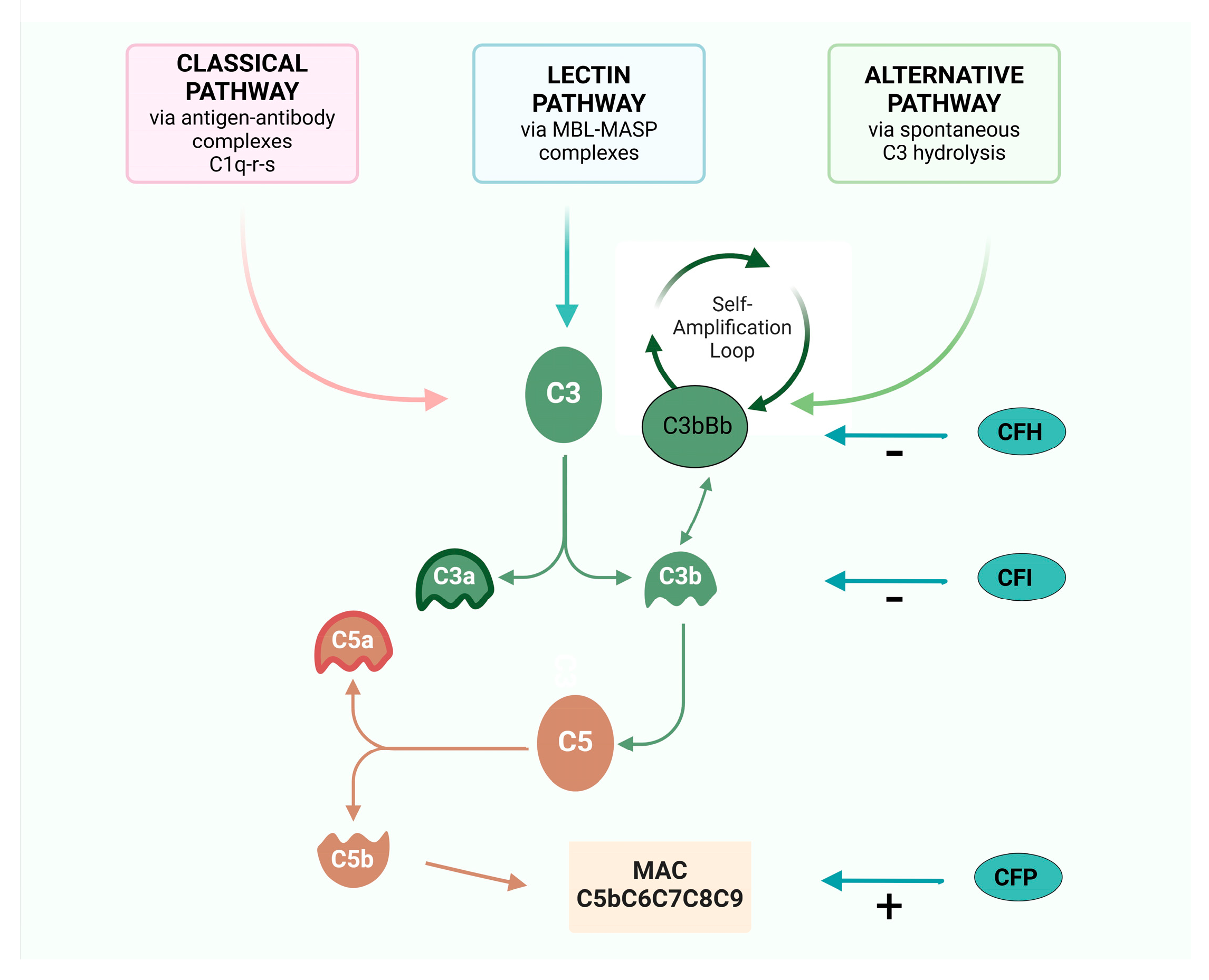
2. The Complement Cascade
- -
- it binds to cell surfaces in complex with factor B, triggering a positive feedback loop (called an “amplification loop”) with the generation of even more C3a and C3b, leading to rapid amplification of the AP [14];
- -
- it promotes the formation of the C5Conv, leading to the formation of the MAC and C5a;
- -
- it operates as an opsonin, coating foreign particles and facilitating the recognition and phagocytosis of pathogens by immune cells such as macrophages and neutrophils [14].
3. Regulator Proteins
4. The Role of Complement in Kidney Disease
4.1. Atypical Hemolytic Uremic Syndrome
4.2. Membranoproliferative Glomerulonephritis
4.3. Secondary IC-MPGN (Lupus Nephritis)
4.4. IgA Nephropathy
4.5. ANCA-Associated Vasculitis
4.6. Membranous Nephropathy
5. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mathern, D.R.; Heeger, P.S. Molecules Great and Small: The Complement System. Clin. J. Am. Soc. Nephrol. 2015, 10, 1636–1650. [Google Scholar] [CrossRef] [PubMed]
- Vivarelli, M.; Barratt, J.; Beck, L.H., Jr.; Fakhouri, F.; Gale, D.P.; de Jorge, E.G.; Mosca, M.; Noris, M.; Pickering, M.C.; Susztak, K.; et al. The role of complement in kidney disease: Conclusions from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney Int. 2024, 106, 369–391. [Google Scholar] [CrossRef] [PubMed]
- Thurman, J.M. Complement and the Kidney: An Overview. Adv. Chronic Kidney Dis. 2020, 27, 86–94. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Anwar, I.J.; DeLaura, I.; Ladowski, J.; Gao, Q.; Knechtle, S.J.; Kwun, J. Complement-targeted therapies in kidney transplantation—Insights from preclinical studies. Front. Immunol. 2022, 13, 984090. [Google Scholar] [CrossRef]
- Noris, M.; Remuzzi, G. Overview of complement activation and regulation. Semin. Nephrol. 2013, 33, 479–492. [Google Scholar] [CrossRef]
- de la Calle, G.M.; Fernández-Ruiz, M.; López-Medrano, F.; Polanco, N.; González, E.; Juan, R.S.; Ruiz-Merlo, T.; Origüen, J.; Paz-Artal, E.; Andrés, A.; et al. Post-transplant hypocomplementemia: A novel marker of cardiovascular risk in kidney transplant recipients? Atherosclerosis 2018, 269, 204–210. [Google Scholar] [CrossRef] [PubMed]
- Speidl, W.S.; Kastl, S.P.; Huber, K.; Wojta, J. Complement in atherosclerosis: Friend or foe? J. Thromb. Haemost. 2011, 9, 428–440. [Google Scholar] [CrossRef]
- Marchio, P.; Guerra-Ojeda, S.; Vila, J.M.; Aldasoro, M.; Victor, V.M.; Mauricio, M.D. Targeting Early Atherosclerosis: A Focus on Oxidative Stress and Inflammation. Oxid. Med. Cell. Longev. 2019, 2019, 8563845. [Google Scholar] [CrossRef]
- Noris, M.; Remuzzi, G. Genetics of Immune-Mediated Glomerular Diseases: Focus on Complement. Semin. Nephrol. 2017, 37, 447–463. [Google Scholar] [CrossRef] [PubMed]
- Noris, M.; Galbusera, M. The complement alternative pathway and hemostasis. Immunol. Rev. 2023, 313, 139–161. [Google Scholar] [CrossRef]
- Bajic, G.; Degn, S.E.; Thiel, S.; Andersen, G.R. Complement activation, regulation, and molecular basis for complement-related diseases. EMBO J. 2015, 34, 2735–2757. [Google Scholar] [CrossRef]
- Lachmann, P.J. The Amplification Loop of the Complement Pathways. In Advances in Immunology; Elsevier: Amsterdam, The Netherlands, 2009; Volume 104, pp. 115–149. [Google Scholar] [CrossRef]
- Noris, M.; Remuzzi, G. Glomerular Diseases Dependent on Complement Activation, Including Atypical Hemolytic Uremic Syndrome, Membranoproliferative Glomerulonephritis, and C3 Glomerulopathy: Core Curriculum 2015. Am. J. Kidney Dis. 2015, 66, 359–375. [Google Scholar] [CrossRef] [PubMed]
- Guo, R.-F.; Ward, P.A. Role of C5a in Inflammatory Responses. Annu. Rev. Immunol. 2005, 23, 821–852. [Google Scholar] [CrossRef] [PubMed]
- Pangburn, M.K. Cutting Edge: Localization of the Host Recognition Functions of Complement Factor H at the Carboxyl-Terminal: Implications for Hemolytic Uremic Syndrome. J. Immunol. 2002, 169, 4702–4706. [Google Scholar] [CrossRef]
- Heurich, M.; McCluskey, G. Complement and coagulation crosstalk—Factor H in the spotlight. Immunobiology 2023, 228, 152707. [Google Scholar] [CrossRef] [PubMed]
- Van Den Bos, R.M.; Pearce, N.M.; Granneman, J.; Brondijk, T.H.C.; Gros, P. Insights into Enhanced Complement Activation by Structures of Properdin and Its Complex with the C-Terminal Domain of C3b. Front. Immunol. 2019, 10, 2097. [Google Scholar] [CrossRef] [PubMed]
- Renner, B.; Laskowski, J.; Poppelaars, F.; Ferreira, V.P.; Blaine, J.; Antonioli, A.H.; Hannan, J.P.; Kovacs, J.M.; van Kooten, C.; You, Z.; et al. Factor H related proteins modulate complement activation on kidney cells. Kidney Int. 2022, 102, 1331–1344. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Goodfellow, R.X.; Ghiringhelli Borsa, N.; Dunlop, H.C.; Presti, S.A.; Meyer, N.C.; Shao, D.; Roberts, S.M.; Jones, M.B.; Pitcher, G.R.; et al. Complement Factor I Variants in Complement-Mediated Renal Diseases. Front. Immunol. 2022, 13, 866330. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, D.V.; Gadeberg, T.A.F.; Thomas, C.; Wang, Y.; Joram, N.; Jensen, R.K.; Mazarakis, S.M.M.; Revel, M.; El Sissy, C.; Petersen, S.V.; et al. Structural Basis for Properdin Oligomerization and Convertase Stimulation in the Human Complement System. Front. Immunol. 2019, 10, 2007. [Google Scholar] [CrossRef]
- Hardy, M.P.; Mansour, M.; Rowe, T.; Wymann, S. The Molecular Mechanisms of Complement Receptor 1-It Is Complicated. Biomolecules 2023, 13, 1522. [Google Scholar] [CrossRef]
- Angeletti, A.; Cantarelli, C.; Petrosyan, A.; Andrighetto, S.; Budge, K.; D’agati, V.D.; Hartzell, S.; Malvi, D.; Donadei, C.; Thurman, J.M.; et al. Loss of decay-accelerating factor triggers podocyte injury and glomerulosclerosis. J. Exp. Med. 2020, 217, e20191699. [Google Scholar] [CrossRef] [PubMed]
- West, E.E.; Kolev, M.; Kemper, C. Complement and the Regulation of T Cell Responses. Annu. Rev. Immunol. 2018, 36, 309–338. [Google Scholar] [CrossRef] [PubMed]
- Genest, D.S.; Patriquin, C.J.; Licht, C.; John, R.; Reich, H.N. Renal Thrombotic Microangiopathy: A Review. Am. J. Kidney Dis. 2023, 81, 591–605. [Google Scholar] [CrossRef]
- George, J.N.; Nester, C.M. Syndromes of Thrombotic Microangiopathy. N. Engl. J. Med. 2014, 371, 654–666. [Google Scholar] [CrossRef] [PubMed]
- Brocklebank, V.; Wood, K.M.; Kavanagh, D. Thrombotic Microangiopathy and the Kidney. Clin. J. Am. Soc. Nephrol. 2018, 13, 300–317. [Google Scholar] [CrossRef] [PubMed]
- Goodship, T.H.; Cook, H.T.; Fakhouri, F.; Fervenza, F.C.; Frémeaux-Bacchi, V.; Kavanagh, D.; Nester, C.M.; Noris, M.; Pickering, M.C.; de Córdoba, S.R.; et al. Atypical hemolytic uremic syndrome and C3 glomerulopathy: Conclusions from a “Kidney Disease: Improving Global Outcomes” (KDIGO) Controversies Conference. Kidney Int. 2017, 91, 539–551. [Google Scholar] [CrossRef]
- Noris, M.; Bresin, E.; Mele, C.; Remuzzi, G. Genetic Atypical Hemolytic-Uremic Syndrome. In GeneReviews®; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. Available online: http://www.ncbi.nlm.nih.gov/books/NBK1367/ (accessed on 18 November 2024).
- Frémeaux-Bacchi, V.; Fakhouri, F.; Roumenina, L.; Dragon–Durey, M.A.; Loirat, C. Syndrome hémolytique et urémique lié à des anomalies du complément. La Rev. Médecine Interne 2011, 32, 232–240. [Google Scholar] [CrossRef]
- Legendre, C.M.; Licht, C.; Muus, P.; Greenbaum, L.A.; Babu, S.; Bedrosian, C.; Bingham, C.; Cohen, D.J.; Delmas, Y.; Douglas, K.; et al. Terminal Complement Inhibitor Eculizumab in Atypical Hemolytic–Uremic Syndrome. N. Engl. J. Med. 2013, 368, 2169–2181. [Google Scholar] [CrossRef]
- Licht, C.; Greenbaum, L.A.; Muus, P.; Babu, S.; Bedrosian, C.L.; Cohen, D.J.; Delmas, Y.; Douglas, K.; Furman, R.R.; Gaber, O.A.; et al. Efficacy and safety of eculizumab in atypical hemolytic uremic syndrome from 2-year extensions of phase 2 studies. Kidney Int. 2015, 87, 1061–1073. [Google Scholar] [CrossRef] [PubMed]
- Fakhouri, F.; Delmas, Y.; Provot, F.; Barbet, C.; Karras, A.; Makdassi, R.; Courivaud, C.; Rifard, K.; Servais, A.; Allard, C.; et al. Insights From the Use in Clinical Practice of Eculizumab in Adult Patients with Atypical Hemolytic Uremic Syndrome Affecting the Native Kidneys: An Analysis of 19 Cases. Am. J. Kidney Dis. 2014, 63, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Zuber, J.; Le Quintrec, M.; Krid, S.; Bertoye, C.; Gueutin, V.; Lahoche, A.; Heyne, N.; Ardissino, G.; Chatelet, V.; Noël, L.H.; et al. Eculizumab for Atypical Hemolytic Uremic Syndrome Recurrence in Renal Transplantation. Am. J. Transplant. 2012, 12, 3337–3354. [Google Scholar] [CrossRef]
- Cordero, L.; Cavero, T.; Gutiérrez, E.; Trujillo, H.; Sandino, J.; Auñón, P.; Rivero, M.; Morales, E. Rational use of eculizumab in secondary atypical hemolytic uremic syndrome. Front. Immunol. 2024, 14, 1310469. [Google Scholar] [CrossRef]
- Manenti, L.; Gnappi, E.; Vaglio, A.; Allegri, L.; Noris, M.; Bresin, E.; Pilato, F.P.; Valoti, E.; Pasquali, S.; Buzio, C. Atypical haemolytic uraemic syndrome with underlying glomerulopathies. A case series and a review of the literature. Nephrol. Dial. Transplant. 2013, 28, 2246–2259. [Google Scholar] [CrossRef]
- Park, M.H.; Caselman, N.; Ulmer, S.; Weitz, I.C. Complement-mediated thrombotic microangiopathy associated with lupus nephritis. Blood Adv. 2018, 2, 2090–2094. [Google Scholar] [CrossRef] [PubMed]
- Rondeau, E.; Scully, M.; Ariceta, G.; Barbour, T.; Cataland, S.; Heyne, N.; Miyakawa, Y.; Ortiz, S.; Swenson, E.; Vallee, M.; et al. The long-acting C5 inhibitor, Ravulizumab, is effective and safe in adult patients with atypical hemolytic uremic syndrome naïve to complement inhibitor treatment. Kidney Int. 2020, 97, 1287–1296. [Google Scholar] [CrossRef]
- Barbour, T.; Scully, M.; Ariceta, G.; Cataland, S.; Garlo, K.; Heyne, N.; Luque, Y.; Menne, J.; Miyakawa, Y.; Yoon, S.-S.; et al. Long-Term Efficacy and Safety of the Long-Acting Complement C5 Inhibitor Ravulizumab for the Treatment of Atypical Hemolytic Uremic Syndrome in Adults. Kidney Int. Rep. 2021, 6, 1603–1613. [Google Scholar] [CrossRef]
- Benamu, E.; Montoya, J.G. Infections associated with the use of eculizumab: Recommendations for prevention and prophylaxis. Curr. Opin. Infect. Dis. 2016, 29, 319–329. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, X.; Zhang, J.; Zhang, B. Real-world safety profile of eculizumab: An analysis of FDA adverse event reporting system and systematic review of case reports. Expert Opin. Drug Saf. 2024, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Noris, M.; Remuzzi, G. C3G and Ig-MPGN-treatment standard. Nephrol. Dial. Transpl. 2024, 39, 202–214. [Google Scholar] [CrossRef] [PubMed]
- Sethi, S.; Fervenza, F.C. Membranoproliferative Glomerulonephritis—A New Look at an Old Entity. N. Engl. J. Med. 2012, 366, 1119–1131. [Google Scholar] [CrossRef]
- Smith, R.J.H.; Appel, G.B.; Blom, A.M.; Cook, H.T.; D’Agati, V.D.; Fakhouri, F.; Fremeaux-Bacchi, V.; Józsi, M.; Kavanagh, D.; Lambris, J.D.; et al. C3 glomerulopathy—Understanding a rare complement-driven renal disease. Nat. Rev. Nephrol. 2019, 15, 129–143. [Google Scholar] [CrossRef]
- Sethi, S.; Nester, C.M.; Smith, R.J. Membranoproliferative glomerulonephritis and C3 glomerulopathy: Resolving the confusion. Kidney Int. 2012, 81, 434–441. [Google Scholar] [CrossRef] [PubMed]
- Bomback, A.S.; Smith, R.J.; Barile, G.R.; Zhang, Y.; Heher, E.C.; Herlitz, L.; Stokes, M.B.; Markowitz, G.S.; D’agati, V.D.; Canetta, P.A.; et al. Eculizumab for Dense Deposit Disease and C3 Glomerulonephritis. Clin. J. Am. Soc. Nephrol. 2012, 7, 748–756. [Google Scholar] [CrossRef] [PubMed]
- Bomback, A.S.; Kavanagh, D.; Vivarelli, M.; Meier, M.; Wang, Y.; Webb, N.J.; Trapani, A.J.; Smith, R.J. Alternative Complement Pathway Inhibition with Iptacopan for the Treatment of C3 Glomerulopathy-Study Design of the APPEAR-C3G Trial. Kidney Int. Rep. 2022, 7, 2150–2159. [Google Scholar] [CrossRef] [PubMed]
- Bomback, A.; Herlitz, L.; Yue, H.; Kedia, P.; Schall, T.; Bekker, P. POS-112 Effect of Avacopan, a Selective C5A receptor Inhibitor, on Complement 3 Glomerulopathy Histologic Index of Disease Chronicity. Kidney Int. Rep. 2022, 7, S47–S48. [Google Scholar] [CrossRef]
- Bomback, A.S.; Herlitz, L.C.; Kedia, P.P.; Petersen, J.; Yue, H.; Lafayette, R.A. Safety and Efficacy of Avacopan in Patients with C3 Glomerulopathy: Randomized, Double-Blind Clinical Trial. J. Am. Soc. Nephrol. 2024, 10-1681. [Google Scholar] [CrossRef]
- Ayano, M.; Horiuchi, T. Complement as a Biomarker for Systemic Lupus Erythematosus. Biomolecules 2023, 13, 367. [Google Scholar] [CrossRef]
- Rossi, G.M.; Maggiore, U.; Peyronel, F.; Fenaroli, P.; Delsante, M.; Benigno, G.D.; Gianfreda, D.; Urban, M.L.; Manna, Z.; Arend, L.J.; et al. Persistent Isolated C3 Hypocomplementemia as a Strong Predictor of End-Stage Kidney Disease in Lupus Nephritis. Kidney Int. Rep. 2022, 7, 2647–2656. [Google Scholar] [CrossRef] [PubMed]
- Wright, R.D.; Bannerman, F.; Beresford, M.W.; Oni, L. A systematic review of the role of eculizumab in systemic lupus erythematosus-associated thrombotic microangiopathy. BMC Nephrol. 2020, 21, 245. [Google Scholar] [CrossRef] [PubMed]
- Thakare, S.B.; So, P.N.; Rodriguez, S.; Hassanein, M.; Lerma, E.; Wiegley, N. Novel Therapeutics for Management of Lupus Nephritis: What Is Next? Kidney Med. 2023, 5, 100688. [Google Scholar] [CrossRef]
- Berger, J. IgA glomerular deposits in renal disease. Transpl. Proc. 1969, 1, 939–944. [Google Scholar]
- Rajasekaran, A.; Julian, B.A.; Rizk, D.V. IgA Nephropathy: An Interesting Autoimmune Kidney Disease. Am. J. Med. Sci. 2021, 361, 176–194. [Google Scholar] [CrossRef]
- Pattrapornpisut, P.; Avila-Casado, C.; Reich, H.N. IgA Nephropathy: Core Curriculum 2021. Am. J. Kidney Dis. 2021, 78, 429–441. [Google Scholar] [CrossRef] [PubMed]
- Tortajada, A.; Gutierrez, E.; Pickering, M.C.; Praga Terente, M.; Medjeral-Thomas, N. The role of complement in IgA nephropathy. Mol. Immunol. 2019, 114, 123–132. [Google Scholar] [CrossRef]
- Suzuki, K.; Honda, K.; Tanabe, K.; Toma, H.; Nihei, H.; Yamaguchi, Y. Incidence of latent mesangial IgA deposition in renal allograft donors in Japan. Kidney Int. 2003, 63, 2286–2294. [Google Scholar] [CrossRef]
- Medjeral-Thomas, N.R.; Cook, H.T.; Pickering, M.C. Complement activation in IgA nephropathy. Semin. Immunopathol. 2021, 43, 679–690. [Google Scholar] [CrossRef] [PubMed]
- Rossi, G.M.; Ricco, F.; Pisani, I.; Delsante, M.; Maggiore, U.; Fiaccadori, E.; Manenti, L. C3 Hypocomplementemia Predicts the Progression of CKD Towards End-Stage Kidney Disease in IgA Nephropathy, Irrespective of Histological Evidence of Thrombotic Microangiopathy. J. Clin. Med. 2024, 13, 2594. [Google Scholar] [CrossRef] [PubMed]
- Medjeral-Thomas, N.R.; Troldborg, A.; Constantinou, N.; Lomax-Browne, H.J.; Hansen, A.G.; Willicombe, M.; Pusey, C.D.; Cook, H.T.; Thiel, S.; Pickering, M.C. Progressive IgA Nephropathy Is Associated with Low Circulating Mannan-Binding Lectin–Associated Serine Protease-3 (MASP-3) and Increased Glomerular Factor H–Related Protein-5 (FHR5) Deposition. Kidney Int. Rep. 2018, 3, 426–438. [Google Scholar] [CrossRef] [PubMed]
- Gentile, M.; Sanchez-Russo, L.; Riella, L.V.; Verlato, A.; Manrique, J.; Granata, S.; Fiaccadori, E.; Pesce, F.; Zaza, G.; Cravedi, P. Immune abnormalities in IgA nephropathy. Clin. Kidney J. 2023, 16, 1059–1070. [Google Scholar] [CrossRef]
- Perkovic, V.; Barratt, J.; Rovin, B.; Kashihara, N.; Maes, B.; Zhang, H.; Trimarchi, H.; Kollins, D.; Papachristofi, O.; Jacinto-Sanders, S.; et al. Alternative Complement Pathway Inhibition with Iptacopan in IgA Nephropathy. N. Engl. J. Med. 2024, NEJMoa2410316. [Google Scholar] [CrossRef] [PubMed]
- Lafayette, R.A.; Rovin, B.H.; Reich, H.N.; Tumlin, J.A.; Floege, J.; Barratt, J. Safety, Tolerability and Efficacy of Narsoplimab, a Novel MASP-2 Inhibitor for the Treatment of IgA Nephropathy. Kidney Int. Rep. 2020, 5, 2032–2041. [Google Scholar] [CrossRef] [PubMed]
- Barratt, J.; Lafayette, R.A.; Floege, J. Therapy of IgA nephropathy: Time for a paradigm change. Front. Med. 2024, 11, 1461879. [Google Scholar] [CrossRef] [PubMed]
- Barratt, J.; Liew, A.; Yeo, S.C.; Fernström, A.; Barbour, S.J.; Sperati, C.J.; Villanueva, R.; Wu, M.-J.; Wang, D.; Borodovsky, A.; et al. Phase 2 Trial of Cemdisiran in Adult Patients with IgA Nephropathy: A Randomized Controlled Trial. Clin. J. Am. Soc. Nephrol. 2024, 19, 452–462. [Google Scholar] [CrossRef] [PubMed]
- Ring, T.; Pedersen, B.B.; Salkus, G.; Goodship, T.H.J. Use of eculizumab in crescentic IgA nephropathy: Proof of principle and conundrum? Clin. Kidney J. 2015, 8, 489–491. [Google Scholar] [CrossRef]
- Bruchfeld, A.; Magin, H.; Nachman, P.; Parikh, S.; Lafayette, R.; Potarca, A.; Miao, S.; Bekker, P. C5a receptor inhibitor avacopan in immunoglobulin A nephropathy—An open-label pilot study. Clin. Kidney J. 2022, 15, 922–928. [Google Scholar] [CrossRef] [PubMed]
- Jennette, J.C.; Falk, R.J.; Bacon, P.A.; Basu, N.; Cid, M.C.; Ferrario, F.; Flores-Suarez, L.F.; Gross, W.L.; Guillevin, L.; Hagen, E.C.; et al. 2012 Revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum. 2013, 65, 603–606. [Google Scholar] [CrossRef]
- Geetha, D.; Jefferson, J.A. ANCA-Associated Vasculitis: Core Curriculum 2020. Am. J. Kidney Dis. 2020, 75, 124–137. [Google Scholar] [CrossRef]
- Flossmann, O.; Berden, A.; de Groot, K.; Hagen, C.; Harper, L.; Heijl, C.; Höglund, P.; Jayne, D.; Luqmani, R.; Mahr, A.; et al. Long-term patient survival in ANCA-associated vasculitis. Ann. Rheum. Dis. 2011, 70, 488–494. [Google Scholar] [CrossRef]
- Cornec, D.; Gall, E.C.-L.; Fervenza, F.C.; Specks, U. ANCA-associated vasculitis—Clinical utility of using ANCA specificity to classify patients. Nat. Rev. Rheumatol. 2016, 12, 570–579. [Google Scholar] [CrossRef] [PubMed]
- Mazzariol, M.; Manenti, L.; Vaglio, A. The complement system in antineutrophil cytoplasmic antibody-associated vasculitis: Pathogenic player and therapeutic target. Curr. Opin. Rheumatol. 2023, 35, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Schreiber, A.; Heeringa, P.; Falk, R.J.; Jennette, J.C. Alternative Complement Pathway in the Pathogenesis of Disease Mediated by Anti-Neutrophil Cytoplasmic Autoantibodies. Am. J. Pathol. 2007, 170, 52–64. [Google Scholar] [CrossRef] [PubMed]
- Noone, D.; Hebert, D.; Licht, C. Pathogenesis and treatment of ANCA-associated vasculitis—A role for complement. Pediatr. Nephrol. 2018, 33, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Poppelaars, F.; Thurman, J.M. Complement-mediated kidney diseases. Mol. Immunol. 2020, 128, 175–187. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, D.J.; Jindal, K.K.; Trillo, A.A. Antineutrophil cytoplasmic antibody-positive crescentic glomerulonephritis and thrombotic microangiopathy. Am. J. Kidney Dis. 1995, 26, 385–386. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.-F.; Wang, H.; Huang, Y.-M.; Li, Z.-Y.; Wang, S.-X.; Yu, F.; Zhao, M.-H.; Chen, M. Clinicopathologic Characteristics and Outcomes of Renal Thrombotic Microangiopathy in Anti-Neutrophil Cytoplasmic Autoantibody-Associated Glomerulonephritis. Clin. J. Am. Soc. Nephrol. 2015, 10, 750–758. [Google Scholar] [CrossRef] [PubMed]
- Manenti, L.; Vaglio, A.; Gnappi, E.; Maggiore, U.; Allegri, L.; Allinovi, M.; Urban, M.L.; Delsante, M.; Galetti, M.; Nicastro, M.; et al. Association of Serum C3 Concentration and Histologic Signs of Thrombotic Microangiopathy with Outcomes Among Patients with ANCA-Associated Renal Vasculitis. Clin. J. Am. Soc. Nephrol. 2015, 10, 2143–2151. [Google Scholar] [CrossRef] [PubMed]
- Manenti, L.; Urban, M.L.; Maritati, F.; Galetti, M.; Vaglio, A. Complement blockade in ANCA-associated vasculitis: An index case, current concepts and future perspectives. Intern. Emerg. Med. 2017, 12, 727–731. [Google Scholar] [CrossRef] [PubMed]
- Huizenga, N.; Zonozi, R.; Rosenthal, J.; Laliberte, K.; Niles, J.L.; Cortazar, F.B. Treatment of Aggressive Antineutrophil Cytoplasmic Antibody–Associated Vasculitis with Eculizumab. Kidney Int. Rep. 2020, 5, 542–545. [Google Scholar] [CrossRef] [PubMed]
- Bekker, P.; Dairaghi, D.; Seitz, L.; Leleti, M.; Wang, Y.; Ertl, L.; Baumgart, T.; Shugarts, S.; Lohr, L.; Dang, T.; et al. Characterization of Pharmacologic and Pharmacokinetic Properties of CCX168, a Potent and Selective Orally Administered Complement 5a Receptor Inhibitor, Based on Preclinical Evaluation and Randomized Phase 1 Clinical Study. PLoS ONE 2016, 11, e0164646. [Google Scholar] [CrossRef] [PubMed]
- Jayne, D.R.W.; Bruchfeld, A.N.; Harper, L.; Schaier, M.; Venning, M.C.; Hamilton, P.; Burst, V.; Grundmann, F.; Jadoul, M.; Szombati, I.; et al. Randomized Trial of C5a Receptor Inhibitor Avacopan in ANCA-Associated Vasculitis. J. Am. Soc. Nephrol. 2017, 28, 2756–2767. [Google Scholar] [CrossRef] [PubMed]
- Merkel, P.A.; Niles, J.; Jimenez, R.; Spiera, R.F.; Rovin, B.H.; Bomback, A.; Pagnoux, C.; Potarca, A.; Schall, T.J.; Bekker, P.; et al. Adjunctive Treatment with Avacopan, an Oral C5a Receptor Inhibitor, in Patients with Antineutrophil Cytoplasmic Antibody–Associated Vasculitis. ACR Open Rheumatol. 2020, 2, 662–671. [Google Scholar] [CrossRef]
- Jayne, D.R.W.; Merkel, P.A.; Schall, T.J.; Bekker, P. Avacopan for the Treatment of ANCA-Associated Vasculitis. N. Engl. J. Med. 2021, 384, 599–609. [Google Scholar] [CrossRef] [PubMed]
- Dantas, M.; Silva, L.B.B.; Pontes, B.T.M.; Dos Reis, M.A.; de Lima, P.S.N.; Moysés Neto, M. Membranous nephropathy. Braz. J. Nephrol. 2023, 45, 229–243. [Google Scholar] [CrossRef]
- Couser, W.G. Primary Membranous Nephropathy. Clin. J. Am. Soc. Nephrol. 2017, 12, 983–997, Erratum in Clin. J. Am. Soc. Nephrol. 2017, 12, 1528. [Google Scholar] [CrossRef] [PubMed]
- Ronco, P.; Debiec, H. Target antigens and nephritogenic antibodies in membranous nephropathy: Of rats and men. Semin. Immunopathol. 2007, 29, 445–458. [Google Scholar] [CrossRef]
- Cunningham, P.N.; Quigg, R.J. Contrasting Roles of Complement Activation and Its Regulation in Membranous Nephropathy. J. Am. Soc. Nephrol. 2005, 16, 1214–1222. [Google Scholar] [CrossRef]
- Chung, E.Y.M.; Wang, Y.M.; Keung, K.; Hu, M.; McCarthy, H.; Wong, G.; Kairaitis, L.; Bose, B.; Harris, D.C.H.; Alexander, S.I. Membranous nephropathy: Clearer pathology and mechanisms identify potential strategies for treatment. Front. Immunol. 2022, 13, 1036249. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Drug | Disease | Clinical Trial | Primary Aims | Status |
|---|---|---|---|---|
| ECULIZUMAB Target: C5 | aHUS | phase 2—NCT01194973 | Percentage of patients with complete TMA response. | completed |
| aHUS | phase 2—NCT00844545 | 1. Platelet count change from baseline to 26 weeks. 2. Percentage of patients with platelet count normalization. 3. Percentage of patients with hematologic normalization. | completed | |
| RAVULIZUMAB Target: C5 | aHUS | phase 3—NCT03131219 | Percentage of complement inhibitor treatment-naïve participants with complete TMA response at week 26. | completed |
| aHUS | phase 3—NCT02949128 | Percentage of participants with complete TMA response at week 26. | completed | |
| LN–IgAN | phase 2—NCT04564339 | Percentage change in proteinuria from baseline to week 26, assessed using 24-h urine collections. | recruiting | |
| IgAN | phase 3—NCT06291376 | 1. Change from baseline in proteinuria based on 24-h urine protein creatinine ratio at week 34. 2. Glomerular filtration rate over 106 weeks. | recruiting | |
| IPTACOPAN Target: Factor B | aHUS | phase 3—NCT04889430 | 1. Percentage of participants with complete TMA response without the use of plasma exchange and anti-C5 antibody. 2. Long-term safety and efficacy evaluations. | recruiting |
| aHUS | phase 3—NCT05795140 | 1. Number of participants with adverse events and serious adverse events. 2. Number of participants with abnormal safety laboratory parameters, vital signs, and electrocardiography. | recruiting | |
| aHUS | phase 3—NCT05935215 | Percentage of participants free of TMA manifestation. | recruiting | |
| MN | Phase 2—NCT04154787 | Ratio between baseline urine protein creatinine ratio and urine protein creatinine ratio at 24 weeks of treatment. | completed | |
| IgAN | phase 3—NCT04557462 | Number and percentage of participants with adverse event and serious adverse event and with abnormalities in clinical laboratory evaluations. | recruiting | |
| LN | phase 2—NCT05268289 | Proportion of patients achieving complete kidney response at week 24 in the absence of kidney flares. | recruiting | |
| C3G | phase 3—NCT04817618 (APPEAR-G3G) | 1. Log-transformed ratio to baseline in urine protein creatinine ratio. 2. Change from baseline in log-transformed urine protein creatinine ratio at 12 months. | recruiting | |
| AAV | phase 2—NCT06388941 | Sustained remission through week 48 defined as complete remission at week 24 without major relapse up to week 48. | recruiting | |
| ANX009 Target: C1q | LN | phase 1B—NCT05780515 | Number of participants with treatment-emergent adverse events. | completed |
| CROVALIMAB Target: C5 | aHUS | phase 3—NCT04958265 | Percentage of participants with complete TMA response (naive cohort only). | recruiting |
| aHUS | phase 3—NCT04861259 | Percentage of participants with complete TMA response. | recruiting | |
| NOMACOPAN Target: C5 | TA-TMA | phase 3—NCT04784455 | RBC transfusion independence or urine protein creatinine ratio ≤ 2 mg/mg maintained over ≥28 days immediately prior to any scheduled clinical visit up to week 24. | terminated |
| PEGCETACOPLAN Target: C3 | TA-TMA | phase 2—NCT05148299 | PK parameter concentration maximum, time of maximum measured serum concentration. | completed |
| IgAN | phase 2—NCT03453619 | Proteinuria reduction from baseline to week 48, based on urinary protein-to-creatinine ratio. | completed | |
| C3G | phase 3—NCT05067127 (VALIANT) | The log-transformed ratio of urine proteine creatinine ratio at week 26 compared to baseline. | active, not recruiting | |
| C3G | phase 3—NCT05809531 (VALE) | Proportion of participants with a reduction in urine protein creatinine ratio of at least 50% from the pretreatment value over time. | active, not recruiting | |
| CEMDISIRAN Target: C5 | IgAN | phase 2—NCT03841448 | Percent change from baseline in urine protein creatinine ratio at week 32. | completed |
| AVACOPAN Target: C5R | AAV | phase 3—NCT02994927 (ADVOCATE) | Percentage of subjects achieving sustained disease remission at week 52. | completed |
| IgAN | phase 2—NCT02384317 | 1. Change in slope of urinary proteine creatinine ratio from the 8-week renin angiotensin aldosteron blocker run-in period to the 12-week treatment period. 2. Number of participants with advers events. | completed | |
| C3G | phase 2 -NCT03301467 (ACCOLADE) | Change from baseline to week 26 in the C3G Histologic Index for disease activity—subjects with elevated C5b-9. | completed | |
| VILOBELIMAB Target: C5a | AAV | phase 2—NCT03712345 | Number and percentage of participants with at least one treatment-emergent adverse event per treatment group. | completed |
| IONIS-FB-LRX Target: Factor B | IgAN | phase 2A—NCT04014335 | Percent reduction in 24-h urine protein excretion. | completed |
| NM8074 Target: Factor B | IgAN | phase 2—NCT06454110 | Change from baseline or percent change from baseline in urine protein-to-creatinine concentration ratio. | Not yet recruiting |
| C3G | phase 1b—NCT05647811 | 1. Monitoring for incidence of adverse events/serious adverse events. 2. Change from baseline or percent change from baseline in urine protein to creatine concentration ratio. | not yet recruiting | |
| NARSOPLIMAB Target: MASPs | IgAN | phase 2—NCT02682407 (ARTEMISAN-IgAN) | 1. Proportion of subjects with treatment-related adverse events. 2. Change from baseline in serum and urine complement component levels. | completed |
| IgAN | phase 3—NCT03608033 | Change from baseline in 24-h urine protein excretion in IgAN assessed at 36 weeks from baseline. | completed | |
| ZALTENIBART Target: MASPs | C3G | Phase 2—NCT06209736 | To assess 5 mg/kg IV administration at 4-week intervals. | recruiting |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gentile, M.; Manenti, L. Targeted Complement Treatments in Glomerulopathies: A Comprehensive Review. J. Clin. Med. 2025, 14, 702. https://doi.org/10.3390/jcm14030702
Gentile M, Manenti L. Targeted Complement Treatments in Glomerulopathies: A Comprehensive Review. Journal of Clinical Medicine. 2025; 14(3):702. https://doi.org/10.3390/jcm14030702
Chicago/Turabian StyleGentile, Micaela, and Lucio Manenti. 2025. "Targeted Complement Treatments in Glomerulopathies: A Comprehensive Review" Journal of Clinical Medicine 14, no. 3: 702. https://doi.org/10.3390/jcm14030702
APA StyleGentile, M., & Manenti, L. (2025). Targeted Complement Treatments in Glomerulopathies: A Comprehensive Review. Journal of Clinical Medicine, 14(3), 702. https://doi.org/10.3390/jcm14030702