
Gene Expression Analysis of Autophagy Markers in Primary and Secondary Myelofibrosis

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients and the Methodology
2.2. Statistical Methods
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef]
- Lim, S.M.; Mohamad Hanif, E.A.; Chin, S.F. Is targeting autophagy mechanism in cancer a good approach? The possible double-edge sword effect. Cell Biosci. 2021, 11, 56. [Google Scholar] [CrossRef]
- Kroemer, G.; Mariño, G.; Levine, B. Autophagy and the integrated stress response. Mol. Cell 2010, 40, 280–293. [Google Scholar] [CrossRef] [PubMed]
- Bellot, G.; Garcia-Medina, R.; Gounon, P.; Chiche, J.; Roux, D.; Pouysségur, J.; Mazure, N.M. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol. Cell Biol. 2009, 29, 2570–2581. [Google Scholar] [CrossRef]
- Harding, H.P.; Zhang, Y.; Zeng, H.; Novoa, I.; Lu, P.D.; Calfon, M.; Sadri, N.; Yun, C.; Popko, B.; Paules, R.; et al. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol. Cell 2003, 11, 619–633. [Google Scholar] [CrossRef] [PubMed]
- Bhadauriya, P.; Onkar, A.; Nagarajan, K.; Karuppusamy, K.A.; Ganesh, S.; Agarwal, S. Glycogen synthase is required for heat shock-mediated autophagy induction in neuronal cells. Biol. Open 2025, 14, BIO061605. [Google Scholar] [CrossRef]
- Ryter, S.W.; Choi, A.M. Autophagy: An Integral Component of the Mammalian Stress Response. J. Biochem. Pharmacol. Res. 2013, 1, 176–188. [Google Scholar]
- Pietrocola, F.; Izzo, V.; Niso-Santano, M.; Vacchelli, E.; Galluzzi, L.; Maiuri, M.C.; Kroemer, G. Regulation of autophagy by stress-responsive transcription factors. Semin. Cancer Biol. 2013, 23, 310–322. [Google Scholar] [CrossRef]
- Jo, E.K.; Yuk, J.M.; Shin, D.M.; Sasakawa, C. Roles of autophagy in elimination of intracellular bacterial pathogens. Front. Immunol. 2013, 4, 97. [Google Scholar] [CrossRef]
- Di-Luoffo, M.; Schmitter, C.; Barrere, E.C.; Therville, N.; Chaouki, M.; D’Angelo, R.; Arcucci, S.; Thibault, B.; Delarue, M.; Guillermet-Guibert, J. Mechanical compressive forces increase PI3K output signaling in breast and pancreatic cancer cells. Life Sci. Alliance 2025, 8, e202402854. [Google Scholar] [CrossRef]
- Puri, D.; Subramanyam, D. Stress—(self) eating: Epigenetic regulation of autophagy in response to psychological stress. FEBS J. 2019, 286, 2447–2460. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.; Zhu, Y.; Xu, X.; Zhu, Y.; Song, Y.; Pang, L.; Chen, Z. The anti-tumor effects of dual PI3K/mTOR inhibitor BEZ235 and histone deacetylase inhibitor Trichostatin A on inducing autophagy in esophageal squamous cell carcinoma. J. Cancer 2018, 9, 987–997. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Chen, Y.; Tooze, S.A. Autophagy pathway: Cellular and molecular mechanisms. Autophagy 2018, 14, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N. Autophagy: Process and function. Genes. Dev. 2007, 21, 2861–2873. [Google Scholar] [CrossRef]
- Phadwal, K.; Watson, A.S.; Simon, A.K. Tightrope act: Autophagy in stem cell renewal, differentiation, proliferation, and aging. Cell Mol. Life Sci. 2013, 70, 89–103. [Google Scholar] [CrossRef]
- Ho, T.T.; Warr, M.R.; Adelman, E.R.; Lansinger, O.M.; Flach, J.; Verovskaya, E.V.; Figueroa, M.E.; Passegué, E. Autophagy maintains the metabolism and function of young and old stem cells. Nature 2017, 543, 205–210. [Google Scholar] [CrossRef]
- Yun, C.W.; Lee, S.H. The Roles of Autophagy in Cancer. Int. J. Mol. Sci. 2018, 19, 3466. [Google Scholar] [CrossRef] [PubMed]
- Arber, D.A.; Orazi, A.; Hasserjian, R.P.; Borowitz, M.J.; Calvo, K.R.; Kvasnicka, H.-M.; Wang, S.A.; Bagg, A.; Barbui, T.; Branford, S.; et al. International Consensus Classification of Myeloid Neoplasms and Acute Leukemias: Integrating morphologic, clinical, and genomic data. Blood 2022, 140, 1200–1228. [Google Scholar] [CrossRef]
- Hultcrantz, M.; Kristinsson, S.Y.; Andersson, T.M.; Landgren, O.; Eloranta, S.; Derolf, A.R.; Dickman, P.W.; Bjorkholm, M. Patterns of survival among patients with myeloproliferative neoplasms diagnosed in Sweden from 1973 to 2008: A population-based study. J. Clin. Oncol. 2012, 30, 2995–3001. [Google Scholar] [CrossRef]
- Tremblay, D.; Yacoub, A.; Hoffman, R. Overview of Myeloproliferative Neoplasms: History, Pathogenesis, Diagnostic Criteria, and Complications. Hematol. Oncol. Clin. N. Am. 2021, 35, 159–176. [Google Scholar] [CrossRef]
- Passamonti, F.; Giorgino, T.; Mora, B.; Guglielmelli, P.; Rumi, E.; Maffioli, M.; Rambaldi, A.; Caramella, M.; Komrokji, R.; Gotlib, J.; et al. A clinical-molecular prognostic model to predict survival in patients with post polycythemia vera and post essential thrombocythemia myelofibrosis. Leukemia 2017, 31, 2726–2731. [Google Scholar] [CrossRef]
- Lucijanic, M.; Krecak, I.; Soric, E.; Sabljic, A.; Galusic, D.; Holik, H.; Perisa, V.; Moric Peric, M.; Zekanovic, I.; Kusec, R. Patients with post polycythemia vera myelofibrosis might experience increased thrombotic risk in comparison to primary and post essential thrombocythemia myelofibrosis. Leuk. Res. 2022, 119, 106905. [Google Scholar] [CrossRef] [PubMed]
- Watson, A.S.; Mortensen, M.; Simon, A.K. Autophagy in the pathogenesis of myelodysplastic syndrome and acute myeloid leukemia. Cell Cycle 2011, 10, 1719–1725. [Google Scholar] [CrossRef]
- Auberger, P.; Puissant, A. Autophagy, a key mechanism of oncogenesis and resistance in leukemia. Blood 2017, 129, 547–552. [Google Scholar] [CrossRef] [PubMed]
- Khoury, J.D.; Solary, E.; Abla, O.; Akkari, Y.; Alaggio, R.; Apperley, J.F.; Bejar, R.; Berti, E.; Busque, L.; Chan, J.K.C.; et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia 2022, 36, 1703–1719. [Google Scholar] [CrossRef]
- Shi, G.; Kodali, S.; Wong, C.; Gotlieb, V.K.; Wang, J.-C. Defective Autophagy Process and Increased ROS Formation in Primary Myelofibrosis. Blood 2018, 132, 3069. [Google Scholar] [CrossRef]
- Courdy, C.; Platteeuw, L.; Ducau, C.; De Araujo, I.; Boet, E.; Sahal, A.; Saland, E.; Edmond, V.; Tavitian, S.; Bertoli, S.; et al. Targeting PP2A-dependent autophagy enhances sensitivity to ruxolitinib in JAK2(V617F) myeloproliferative neoplasms. Blood Cancer J. 2023, 13, 106. [Google Scholar] [CrossRef] [PubMed]
- Jogalekar, M.P.; Veerabathini, A.; Gangadaran, P. Recent developments in autophagy-targeted therapies in cancer. Exp. Biol. Med. 2021, 246, 207–212. [Google Scholar] [CrossRef]
- Foth, M.; McMahon, M. Autophagy Inhibition in BRAF-Driven Cancers. Cancers 2021, 13, 3498. [Google Scholar] [CrossRef]
- Kocaturk, N.M.; Akkoc, Y.; Kig, C.; Bayraktar, O.; Gozuacik, D.; Kutlu, O. Autophagy as a molecular target for cancer treatment. Eur. J. Pharm. Sci. 2019, 134, 116–137. [Google Scholar] [CrossRef]
- Levy, J.M.M.; Towers, C.G.; Thorburn, A. Targeting autophagy in cancer. Nat. Rev. Cancer 2017, 17, 528–542. [Google Scholar] [CrossRef] [PubMed]
- Cheong, H.; Lu, C.; Lindsten, T.; Thompson, C.B. Therapeutic targets in cancer cell metabolism and autophagy. Nat. Biotechnol. 2012, 30, 671–678. [Google Scholar] [CrossRef]
- Ning, F.; Yang, Z.; Xu, L.; Sun, Y. Targeted tumor therapy by autophagy of nanoparticles. Future Oncol. 2020, 16, 793–803. [Google Scholar] [CrossRef] [PubMed]
- Nuñez-Olvera, S.I.; Gallardo-Rincón, D.; Puente-Rivera, J.; Salinas-Vera, Y.M.; Marchat, L.A.; Morales-Villegas, R.; López-Camarillo, C. Autophagy Machinery as a Promising Therapeutic Target in Endometrial Cancer. Front. Oncol. 2019, 9, 1326. [Google Scholar] [CrossRef]
- Pan, H.; Chen, L.; Xu, Y.; Han, W.; Lou, F.; Fei, W.; Liu, S.; Jing, Z.; Sui, X. Autophagy-associated immune responses and cancer immunotherapy. Oncotarget 2016, 7, 21235–21246. [Google Scholar] [CrossRef]
- Becker, I.C.; Barrachina, M.N.; Lykins, J.; Camacho, V.; Stone, A.P.; Chua, B.A.; Signer, R.A.J.; Machlus, K.R.; Whiteheart, S.W.; Roweth, H.G.; et al. Inhibition of RhoA-mediated secretory autophagy in megakaryocytes mitigates myelofibrosis in mice. bioRxiv 2024. [Google Scholar] [CrossRef]
- Lucijanic, M.; Livun, A.; Stoos-Veic, T.; Pejsa, V.; Jaksic, O.; Cicic, D.; Lucijanic, J.; Romic, Z.; Orehovec, B.; Aralica, G.; et al. High absolute basophil count is a powerful independent predictor of inferior overall survival in patients with primary myelofibrosis. Hematology 2018, 23, 201–207. [Google Scholar] [CrossRef]
- Elliott, M.A.; Verstovsek, S.; Dingli, D.; Schwager, S.M.; Mesa, R.A.; Li, C.Y.; Tefferi, A. Monocytosis is an adverse prognostic factor for survival in younger patients with primary myelofibrosis. Leuk. Res. 2007, 31, 1503–1509. [Google Scholar] [CrossRef]
- Lucijanic, M.; Prka, Z.; Pejsa, V.; Stoos-Veic, T.; Lucijanic, J.; Kusec, R. Prognostic implications of low transferrin saturation in patients with primary myelofibrosis. Leuk. Res. 2018, 66, 89–95. [Google Scholar] [CrossRef]
- Lucijanic, M.; Galusic, D.; Krecak, I.; Sedinic, M.; Soric, E.; Holik, H.; Perisa, V.; Moric Peric, M.; Zekanovic, I.; Stoos-Veic, T.; et al. C reactive protein to albumin ratio as prognostic marker in primary and secondary myelofibrosis. Leuk. Lymphoma 2020, 61, 2969–2974. [Google Scholar] [CrossRef]
- Shah, S.; Mudireddy, M.; Hanson, C.A.; Ketterling, R.P.; Gangat, N.; Pardanani, A.; Tefferi, A. Marked elevation of serum lactate dehydrogenase in primary myelofibrosis: Clinical and prognostic correlates. Blood Cancer J. 2017, 7, 657. [Google Scholar] [CrossRef] [PubMed]
- Lucijanic, M.; Pejsa, V.; Jaksic, O.; Mitrovic, Z.; Tomasovic-Loncaric, C.; Stoos-Veic, T.; Prka, Z.; Pirsic, M.; Haris, V.; Vasilj, T.; et al. The Degree of Anisocytosis Predicts Survival in Patients with Primary Myelofibrosis. Acta Haematol. 2016, 136, 98–100. [Google Scholar] [CrossRef]
- Kuykendall, A.T.; Mo, Q.; Sallman, D.A.; Ali, N.A.; Chan, O.; Yun, S.; Sweet, K.L.; Padron, E.; Lancet, J.E.; Komrokji, R.S. Disease-related thrombocytopenia in myelofibrosis is defined by distinct genetic etiologies and is associated with unique prognostic correlates. Cancer 2022, 128, 3495–3501. [Google Scholar] [CrossRef]
- Rabinowitz, J.D.; White, E. Autophagy and metabolism. Science 2010, 330, 1344–1348. [Google Scholar] [CrossRef] [PubMed]
- Schwertz, H.; Middleton, E.A. Autophagy and its consequences for platelet biology. Thromb. Res. 2023, 231, 170–181. [Google Scholar] [CrossRef] [PubMed]
- Bielawska, M.; Warszyńska, M.; Stefańska, M.; Błyszczuk, P. Autophagy in Heart Failure: Insights into Mechanisms and Therapeutic Implications. J. Cardiovasc. Dev. Dis. 2023, 10, 352. [Google Scholar] [CrossRef]
- Shao, B.Z.; Han, B.Z.; Zeng, Y.X.; Su, D.F.; Liu, C. The roles of macrophage autophagy in atherosclerosis. Acta Pharmacol. Sin. 2016, 37, 150–156. [Google Scholar] [CrossRef]
- Saville, D.J. Multiple Comparison Procedures: The Practical Solution. Am. Stat. 1990, 44, 174–180. [Google Scholar] [CrossRef]
- Lu, G.; Wu, Z.; Shang, J.; Xie, Z.; Chen, C.; Zhang, C. The effects of metformin on autophagy. Biomed. Pharmacother. 2021, 137, 111286. [Google Scholar] [CrossRef]
- Lee, M.H.; Hobbs, G.S. Metformin for MPN: Teaching an old drug new tricks. Blood Adv. 2024, 8, 4476–4477. [Google Scholar] [CrossRef]
- Campos, P.D.M.; Pagnano, K.B.; Mancuso, R.I.; Della Via, F.I.; Tinoco, Â.C.; Assis-Mendonça, G.R.; Freitas, L.L.L.; Traina, F.; Olalla Saad, S.T. Final Results of the Fibromet Trial: An Open Label Phase II Study to Evaluate Metformin Effects on Bone Marrow Fibrosis and Disease Progression in Primary Myelofibrosis Patients. Blood 2021, 138, 2584. [Google Scholar] [CrossRef]
- Hassanpour, M.; Rahbarghazi, R.; Nouri, M.; Aghamohammadzadeh, N.; Safaei, N.; Ahmadi, M. Role of autophagy in atherosclerosis: Foe or friend? J. Inflamm. 2019, 16, 8. [Google Scholar] [CrossRef]
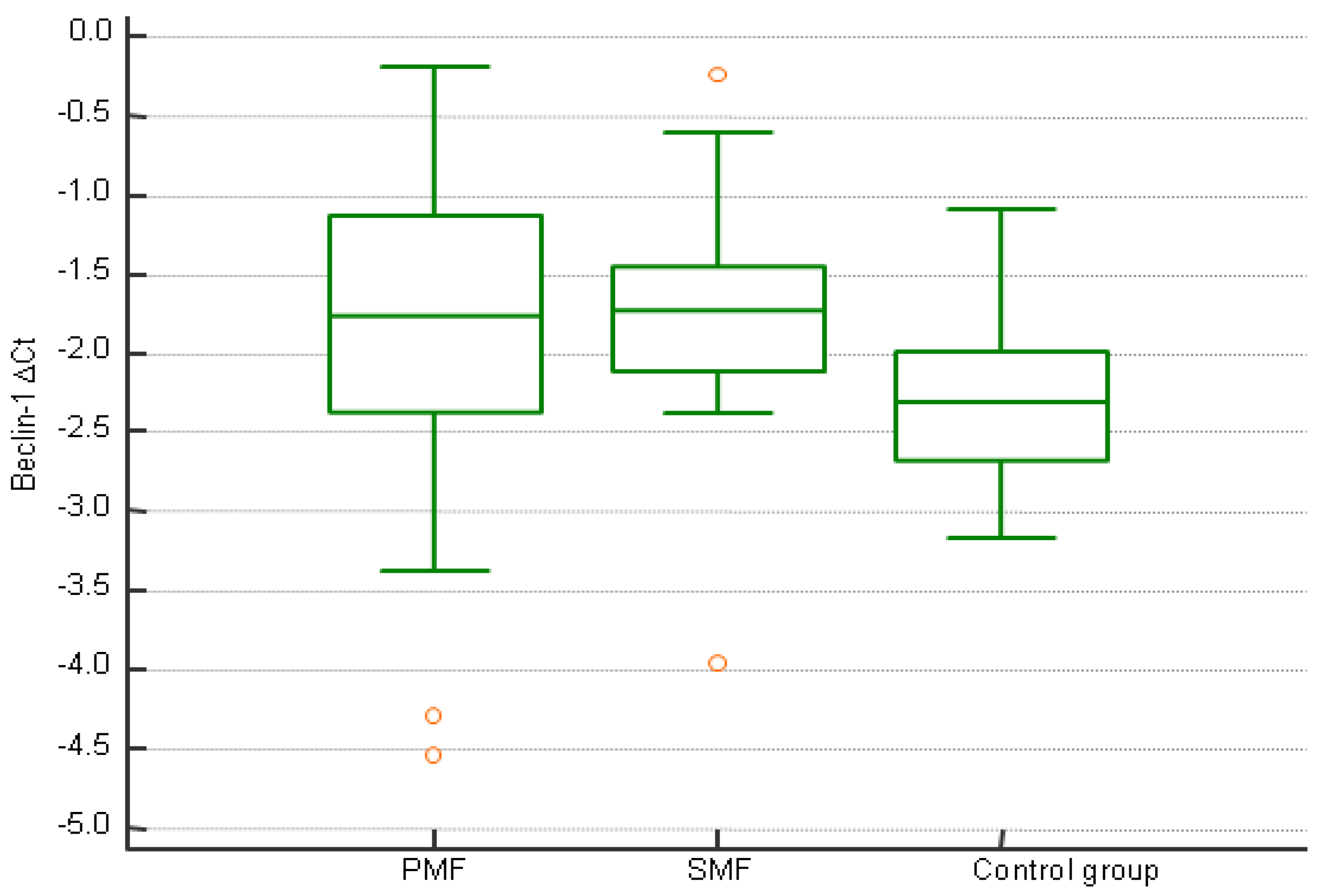

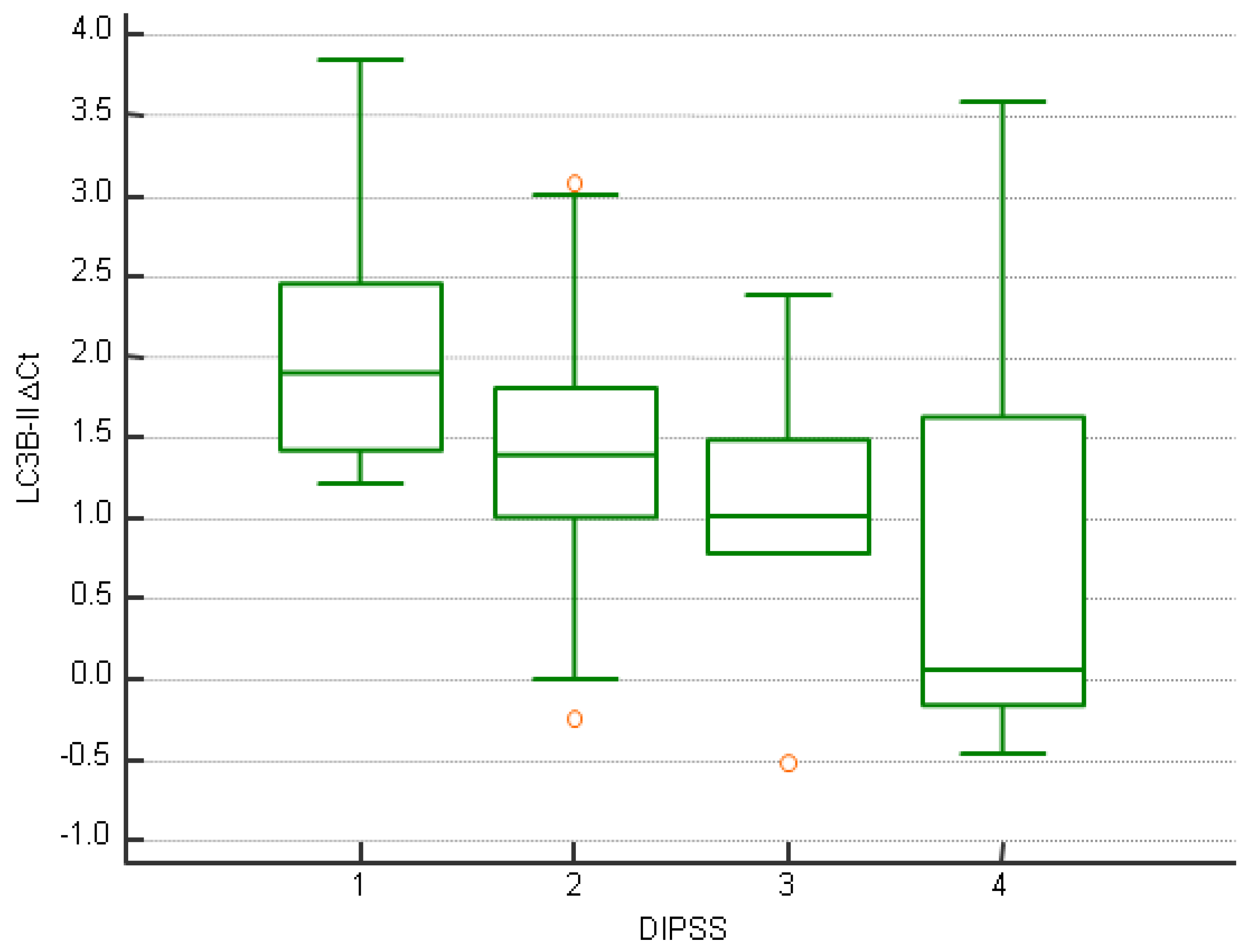
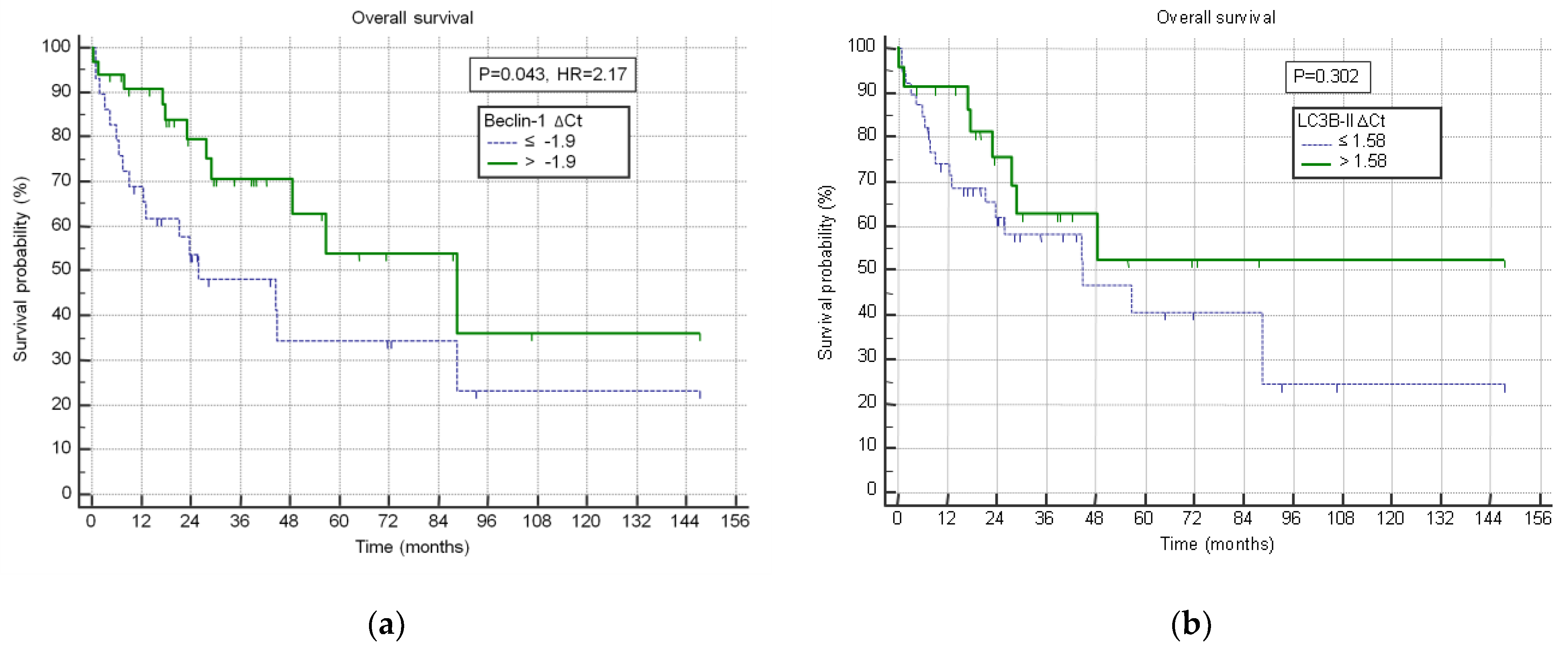

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ∆CtBeclin-1 ≥ −1.76 | ∆CtBeclin-1 < −1.76 | p | |
|---|---|---|---|
| Age (years) | 66 IQR (58–77) | 67 IQR (58–73.25) | 0.659 |
| Sex | |||
| Male | 22/37 (59.5%) | 20/37 (54.1%) | |
| Female | 15/37 (40.5%) | 17/37 (45.9%) | 0.639 |
| Origin of myelofibrosis | |||
| PMF | 27/37 (73%) | 29/37 (78.4%) | |
| Post-PV SMF | 5/37 (13.5%) | 5/37 (13.5%) | |
| Post-ET SMF | 5/37 (13.5%) | 3/37 (8.1%) | 0.751 |
| Grade of fibrosis of bone marrow | |||
| 0-I | 13/29 (44.8%) | 12/33 (36.4%) | |
| II-III | 16/29 (55.2%) | 21/33 (63.6%) | 0.498 |
| DIPSS (PMF) | 0.176 | ||
| Low | 6/21 (28.6%) | 3/27 (11.1%) | |
| Intermediate-1 | 12/21 (57.1%) | 13/27 (48.1%) | |
| Intermediate-2 | 2/21 (9.5%) | 7/27 (25.9%) | |
| High | 1/21 (4.8%) | 4/27 (14.8%) | |
| Mysec-PM (SMF) | 0.771 | ||
| Low | 1/8 (12.5%) | 0/4 (0%) | |
| Intermediate-1 | 3/8 (37.5%) | 1/4 (25%) | |
| Intermediate-2 | 2/8 (25%) | 2/4 (50%) | |
| High | 2/8 (25%) | 1/4 (25%) | |
| JAK2V617F-positive | 22/29 (75.9%) | 19/32 (59.4%) | 0.171 |
| WBC (×109/L; n.v. 3.4–9.7) | 11.1 IQR (6.9–15.8) | 11.5 IQR (8.3–16) | 0.530 |
| Monocytes (×109/L; n.v. 0.12–0.84) | 0.5 IQR (0.3–0.6) | 0.5 IQR (0.35–0.85) | 0.291 |
| Basophils (×109/L; n.v. 0–0.06) | 0.1 IQR (0.07–0.17) | 0.2 IQR (0.1–0.3) | 0.032 *,** |
| Lymphocytes (×109/L; n.v. 1.19–3.35) | 1.3 IQR (1–1.7) | 1.8 IQR (1.45–2.75) | 0.012 *,** |
| Circulatory blasts (%, n.v. <1) | 0 IQR (0–1) | 2 IQR (0–5) | 0.013 *,** |
| Hemoglobin (g/L, n.v. 119–157) | 125 IQR (108–142) | 111 IQR (97–122) | 0.093 |
| RDW (%, n.v. 9.0–15.0) | 18.5 IQR (15.5–20.8) | 19.6 IQR (17.78–21.7) | 0.050 * |
| Platelets (×109/L; n.v. 158–424) | 574 IQR (297–758) | 342 IQR (181–550) | 0.021 * |
| MPV (fL; n.v. 6.8–10.4) | 8.8 IQR (8.1–9.7) | 9.5 IQR (8.75–10.8) | 0.024 *,** |
| LDH (U/L; n.v. <241) | 359 IQR (234–485) | 546.5 IQR (342.75–887.25) | 0.004 *,** |
| CRP (mg/L; n.v. <5) | 2.6 IQR (1.2–3.6) | 4.3 IQR (2.18–13.4) | 0.032 * |
| ∆CtLC3B-II ≥ 1.43 | ∆CtLC3B-II < 1.43 | p | |
|---|---|---|---|
| Age (years) | 64 IQR (58–77) | 68 IQR (59.5–74.25) | 0.842 |
| Sex | |||
| Male | 20/37 (54.1%) | 22/37 (59.5%) | |
| Female | 17/37 (45.9%) | 15/37 (40.5%) | 0.639 |
| Origin of myelofibrosis | |||
| PMF | 27/37 (73%) | 29/37 (78.4%) | |
| Post-PV SMF | 7/37 (18.9%) | 3/37 (8.1%) | |
| Post-ET SMF | 3/37 (8.1%) | 5/37 (13.5%) | 0.337 |
| Grade of fibrosis of bone marrow | |||
| 0–I | 14/30 (46.7%) | 11/32 (34.4%) | |
| II–III | 16/30 (53.3%) | 21/32 (65.6%) | 0.324 |
| DIPSS (PMF) | 0.068 ** | ||
| Low | 7/22 (31.8%) | 2/26 (7.7%) | |
| Intermediate-1 | 12/22 (54.5%) | 13/26 (50%) | |
| Intermediate-2 | 2/22 (9.1%) | 7/26 (26.9%) | |
| High | 1/22 (4.5%) | 4/26 (15.4%) | |
| Mysec-PM (SMF) | 0.560 | ||
| Low | 0/7 (0%) | 1/5 (20%) | |
| Intermediate-1 | 3/7 (42.9%) | 1/5 (20%) | |
| Intermediate-2 | 2/7 (28.6%) | 2/5 (40%) | |
| High | 2/7 (28.6%) | 1/5 (20%) | |
| JAK2V617F-positive | 23/30 (76.7%) | 18/31 (58.1%) | 0.122 |
| WBC (×109/L; n.v. 3.4–9.7) | 10.3 IQR (7.35–15.05) | 13.1 IQR (8.23–17.85) | 0.130 |
| Monocytes (×109/L; n.v. 0.12–0.84) | 0.4 IQR (0.3–0.6) | 0.6 IQR (0.4–0.9) | 0.021 *,** |
| Basophils (×109/L; n.v. 0–0.06) | 0.1 IQR (0.02–0.2) | 0.2 IQR (0.1–0.25) | 0.060 ** |
| Lymphocytes (×109/L; n.v. 1.19–3.35) | 1.3 IQR (1–1.6) | 1.8 IQR (1.5–2.8) | 0.004 *,** |
| Circulatory blasts (%, n.v. <1) | 0 IQR (0–1.33) | 0 IQR (0–3.65) | 0.320 |
| Hemoglobin (g/L, n.v. 119–157) | 121 IQR (108–132) | 116 IQR (97–127) | 0.223 |
| RDW (%, n.v. 9.0–15.0) | 18.7 IQR (16.93–20.5) | 19.6 IQR (17.15–22.35) | 0.207 |
| Platelets (×109/L; n.v. 158–424) | 546.5 IQR (281.25–683.5) | 351.5 IQR (179.25–570.25) | 0.128 |
| MPV (fL; n.v. 6.8–10.4) | 9.1 IQR (8.15–9.7) | 9.3 IQR (8.43–10.68) | 0.336 |
| LDH (U/L; n.v. <241) | 375.5 IQR (252.75–556.6) | 515 IQR (341.5–744.5) | 0.045 *,** |
| CRP (mg/L; n.v. <5) | 3.2 IQR (1.65–10.3) | 3.5 IQR (1.3–8.55) | 0.960 |
| Fe (μmol/L; n.v. 11–32) | 12 IQR (9.75–14) | 15.9 IQR (11.9–18.4) | 0.016 *,** |
| TIBC (μmol/L; n.v. 49–72) | 55.6 IQR (48.5–58.45) | 50.2 IQR (46.3–53.25) | 0.092 ** |
| TSAT (%, n.v. >20) | 21.7 IQR (13.8–28.38) | 30.2 IQR (24.18–35.85) | 0.013 *,** |
| Ferritin (μg/L; n.v. 10–120) | 83 IQR (18–208) | 232.5 IQR (177.75–352.25) | 0.027 *,** |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Medugorac, M.; Glick, K.M.; Livun, A.; Lucijanic, M.; Galusic, D.; Kusec, R. Gene Expression Analysis of Autophagy Markers in Primary and Secondary Myelofibrosis. J. Clin. Med. 2025, 14, 2333. https://doi.org/10.3390/jcm14072333
Medugorac M, Glick KM, Livun A, Lucijanic M, Galusic D, Kusec R. Gene Expression Analysis of Autophagy Markers in Primary and Secondary Myelofibrosis. Journal of Clinical Medicine. 2025; 14(7):2333. https://doi.org/10.3390/jcm14072333
Chicago/Turabian StyleMedugorac, Marin, Katarina Marija Glick, Ana Livun, Marko Lucijanic, Davor Galusic, and Rajko Kusec. 2025. "Gene Expression Analysis of Autophagy Markers in Primary and Secondary Myelofibrosis" Journal of Clinical Medicine 14, no. 7: 2333. https://doi.org/10.3390/jcm14072333
APA StyleMedugorac, M., Glick, K. M., Livun, A., Lucijanic, M., Galusic, D., & Kusec, R. (2025). Gene Expression Analysis of Autophagy Markers in Primary and Secondary Myelofibrosis. Journal of Clinical Medicine, 14(7), 2333. https://doi.org/10.3390/jcm14072333