
Castleman Disease—Still More Questions than Answers: A Case Report and Review of the Literature

 ,
,  , , , and
, , , and
Abstract
1. Introduction
2. Case Description
2.1. Initial Presentation (Age 14)
2.2. Early Diagnostic Workup and Management (Age 14–15)
2.3. Tertiary Center Evaluation and First Surgical Intervention (Age 15)
2.4. Initial Treatment Attempts (Age 15–16)
2.5. Disease Progression and Second Surgery (Age 16)
2.6. Further Treatment Attempts (Age 16–17)
2.7. Genetic Testing and Dose Escalation (Age 17)
2.8. Final Diagnosis and Effective Treatment (Age 17–18)
2.9. Transition to Adult Care, Treatment Outcome and Follow-Up (Age 18–20)
3. Discussion
3.1. Epidemiology of Castleman Disease
3.2. Castleman Disease in Differential Diagnosis of FUO
3.3. Classification of Castleman Disease
3.4. Clinical Manifestations of Castleman Disease
3.5. Diagnosis of Castleman Disease
3.5.1. Imaging Tests
3.5.2. Histopathology
3.5.3. Genetic Tests
3.6. Pathophysiology of Castleman Disease
3.7. Treatment of Castleman Disease
3.8. The Importance of Multidisciplinary Approach in Diagnosing Castleman Disease
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CD | Castleman disease |
| UCD | unicentric Castleman disease |
| MCD | multicentric Castleman disease |
| IL | interleukin |
| FUO | Fever of unknown origin |
| iMCD | idiopathic multicentric Castleman disease |
| MRI | magnetic resonance imaging |
| WES | whole-exome sequencing |
| FDG-PET/CT | fluorodeoxyglucose-positron emission tomography/computed tomography |
| CRP | C-reactive protein |
| HHV-8 | human herpes virus-8 |
| POEMS | polyneuropathy, organomegaly, endocrinopathy, monoclonal gammopathy, and skin changes |
| TAFRO | thrombocytopenia, ascites, reticulin fibrosis, renal dysfunction, organomegaly |
| NOS | not otherwise specified |
| CECT | contrast-enhanced computed tomography |
| SUV | standardized uptake value |
| HV-CD | hyaline vascular Castleman disease |
| PC-CD | plasma cell infiltration pattern Castleman disease |
| HUVEC | human umbilical vein endothelial cells |
| TNF | tumor necrosis factor |
| VEGF | vascular endothelial growth factor |
| FDA | Food and Drug Administration |
| EMA | European Medicines Agency |
| HIV | human immunodeficiency virus |
| SAA | serum amyloid A |
References
- Castleman, B.; Towne, V.W. Case records of the Massachusetts General Hospital: Case No. 40231. N. Engl. J. Med. 1954, 250, 1001–1005. [Google Scholar] [CrossRef]
- Carbone, A.; Borok, M.; Damania, B.; Gloghini, A.; Polizzotto, M.N.; Jayanthan, R.K.; Fajgenbaum, D.C.; Bower, M. Castleman disease. Nat. Rev. Dis. Primers 2021, 7, 84. [Google Scholar] [CrossRef]
- Nishimura, M.F.; Nishimura, Y.; Nishikori, A.; Yoshino, T.; Sato, Y. Historical and pathological overview of Castleman disease. J. Clin. Exp. Hematop. 2022, 62, 60–72. [Google Scholar] [CrossRef]
- Kishimoto, T.; Kang, S. IL-6 Revisited: From Rheumatoid Arthritis to CAR T Cell Therapy and COVID-19. Annu. Rev. Immunol. 2022, 40, 323–348. [Google Scholar] [CrossRef]
- Dispenzieri, A.; Fajgenbaum, D.C. Overview of Castleman disease. Blood 2020, 135, 1353–1364. [Google Scholar] [CrossRef]
- Fajgenbaum, D.C.; Pierson, S.K.; Kanhai, K.; Bagg, A.; Alapat, D.; Lim, M.S.; Lechowicz, M.J.; Srkalovic, G.; Uldrick, T.S.; van Rhee, F.; et al. The disease course of Castleman disease patients with fatal outcomes in the ACCELERATE registry. Br. J. Haematol. 2022, 198, 307–316. [Google Scholar] [CrossRef]
- Pierson, S.K.; Lim, M.S.; Srkalovic, G.; Brandstadter, J.D.; Sarmiento Bustamante, M.; Shyamsundar, S.; Mango, N.; Lavery, C.; Austin, B.; Alapat, D.; et al. Treatment consistent with idiopathic multicentric Castleman disease guidelines is associated with improved outcomes. Blood Adv. 2023, 7, 6652–6664. [Google Scholar] [CrossRef]
- Robinson, D., Jr.; Reynolds, M.; Casper, C.; Dispenzieri, A.; Vermeulen, J.; Payne, K.; Schramm, J.; Ristow, K.; Desrosiers, M.P.; Yeomans, K.; et al. Clinical epidemiology and treatment patterns of patients with multicentric Castleman disease: Results from two US treatment centres. Br. J. Haematol. 2014, 165, 39–48. [Google Scholar] [CrossRef]
- Hu, C.; Zou, Y.; Pan, J.; Yang, J.; Yang, T.; Tan, T.; Li, J. Analysis of Clinical Characteristics, Pathological Changes and Changes of Interleukin-6 (IL-6) and C-Reactive Protein (CRP) in Children with Castleman’s Disease. Med. Sci. Monit. 2020, 26, e924783. [Google Scholar] [CrossRef]
- Parez, N.; Bader-Meunier, B.; Roy, C.C.; Dommergues, J.P. Paediatric Castleman disease: Report of seven cases and review of the literature. Eur. J. Pediatr. 1999, 158, 631–637. [Google Scholar] [CrossRef]
- Trapani, S.; Fiordelisi, A.; Stinco, M.; Resti, M. Update on Fever of Unknown Origin in Children: Focus on Etiologies and Clinical Approach. Children 2023, 11, 20. [Google Scholar] [CrossRef]
- Cunha, B.A. Fever of unknown origin: Clinical overview of classic and current concepts. Infect. Dis. Clin. N. Am. 2007, 21, 867–915. [Google Scholar] [CrossRef]
- Haidar, G.; Singh, N. Fever of Unknown Origin. N. Engl. J. Med. 2022, 386, 463–477. [Google Scholar] [CrossRef]
- Wright, W.F.; Yenokyan, G.; Simner, P.J.; Carroll, K.C.; Auwaerter, P.G. Geographic Variation of Infectious Disease Diagnoses Among Patients With Fever of Unknown Origin: A Systematic Review and Meta-analysis. Open Forum Infect. Dis. 2022, 9, ofac151. [Google Scholar] [CrossRef]
- Nowroozizadeh, B.; Haghighi Mehmandari, N.; Gallegos, N.; Perez-Rosendahl, M.; Lu, D. Q Fever Presented as a Large Retroperitoneal Pseudotumoral Mass. Case Rep. Pathol. 2017, 2017, 4076159. [Google Scholar] [CrossRef]
- Foggo, V.; Cavenagh, J. Malignant causes of fever of unknown origin. Clin. Med. 2015, 15, 292–294. [Google Scholar] [CrossRef]
- Sogaard, K.K.; Farkas, D.K.; Leisner, M.Z.; Schmidt, S.A.J.; Lash, T.L.; Sorensen, H.T. Fever of Unknown Origin and Incidence of Cancer. Clin. Infect. Dis. 2022, 75, 968–974. [Google Scholar] [CrossRef]
- Milchert, M.; Makowska, J.; Brzezinska, O.; Brzosko, M.; Wiesik-Szewczyk, E. Monogenic autoinflammatory diseases in adults—A challenge to rheumatologic practice at the onset of the Polish national programme of interleukin 1 inhibitor treatment. Reumatologia 2019, 57, 326–335. [Google Scholar] [CrossRef]
- Fajgenbaum, D.C.; Uldrick, T.S.; Bagg, A.; Frank, D.; Wu, D.; Srkalovic, G.; Simpson, D.; Liu, A.Y.; Menke, D.; Chandrakasan, S.; et al. International, evidence-based consensus diagnostic criteria for HHV-8-negative/idiopathic multicentric Castleman disease. Blood 2017, 129, 1646–1657. [Google Scholar] [CrossRef]
- Molacek, J.; Treska, V.; Skalicky, T.; Vodicka, J.; Ferda, J.; Ferdova, E.; Baxa, J.; Mach, C.; Jungova, A.; Michal, M. Unicentric form of Castleman s disease, pitfalls of diagnosis and surgical treatment. Front. Oncol. 2023, 13, 1057683. [Google Scholar] [CrossRef]
- Kaniewska, M.; Wasielica-Berger, J.; Wereszczyńska-Siemiątkowska, U.; Augustynowicz, A.; Cepowicz, D.; Kędra, B.; Dąbrowski, A. Case report Castleman’s disease imitating pancreatic tumour—Diagnostic difficulties. Gastroenterol. Rev./Prz. Gastroenterol. 2007, 2, 199–204. [Google Scholar]
- Li, D.; Tang, D.; Sun, F. Clinical Analysis of Castleman’s Disease of the Lacrimal Gland. J. Ophthalmol. 2020, 2020, 3718305. [Google Scholar] [CrossRef]
- Eward, W.C.; DeWitt, S.B.; Brigman, B.E.; Kontogeorgakos, V.; Lagoo, A.S. Extranodal Castleman disease of the extremities: A case report and review of the literature. Skelet. Radiol. 2014, 43, 1627–1631. [Google Scholar] [CrossRef]
- Muhammad, T.; Alkheder, A.; Mazloum, A.; Almooay, A.; Naziha, L.; Shaheen, M. Unicentric Castleman disease: A case report of an atypical presentation and successful management. Int. J. Surg. Case Rep. 2024, 118, 109688. [Google Scholar] [CrossRef]
- Zambrana Garcia, J.L.; Torres Serrano, F.; Jansen Chaparro, S.; Lopez Rubio, F.; Jimenez-Pereperez, J.A.; Perez Jimenez, F. Fever of unknown origin as initial manifestation of Castleman’s disease: Apropos of 2 cases. An. Med. Interna 1996, 13, 500–501. [Google Scholar]
- Roca, B.; Torres, V. Castleman’s disease presenting as fever of unknown origin: Diagnostic value of fluorodeoxyglucose-positron emission tomography/computed tomography. Am. J. Med. Sci. 2009, 337, 295–296. [Google Scholar] [CrossRef]
- Nishimura, Y.; Hanayama, Y.; Fujii, N.; Kondo, E.; Otsuka, F. Comparison of the clinical characteristics of TAFRO syndrome and idiopathic multicentric Castleman disease in general internal medicine: A 6-year retrospective study. Intern. Med. J. 2020, 50, 184–191. [Google Scholar] [CrossRef]
- Bustamante, M.S.; Pierson, S.K.; Ren, Y.; Bagg, A.; Brandstadter, J.D.; Srkalovic, G.; Mango, N.; Alapat, D.; Lechowicz, M.J.; Li, H.; et al. Longitudinal, natural history study reveals the disease burden of idiopathic multicentric Castleman disease. Haematologica 2024, 109, 2196–2206. [Google Scholar] [CrossRef]
- Lust, H.; Gong, S.; Remiker, A.; Rossoff, J. Idiopathic multicentric Castleman disease with TAFRO clinical subtype responsive to IL-6/JAK inhibition: A pediatric case series. Pediatr. Blood Cancer 2021, 68, e29261. [Google Scholar] [CrossRef]
- Oksenhendler, E.; Boutboul, D.; Fajgenbaum, D.; Mirouse, A.; Fieschi, C.; Malphettes, M.; Vercellino, L.; Meignin, V.; Gerard, L.; Galicier, L. The full spectrum of Castleman disease: 273 patients studied over 20 years. Br. J. Haematol. 2018, 180, 206–216. [Google Scholar] [CrossRef]
- Gonzalez Garcia, A.; Fernandez-Martin, J.; Robles Marhuenda, A. Idiopathic multicentric Castleman disease and associated autoimmune and autoinflammatory conditions: Practical guidance for diagnosis. Rheumatology 2023, 62, 1426–1435. [Google Scholar] [CrossRef]
- Soudet, S.; Fajgenbaum, D.; Delattre, C.; Forestier, A.; Hachulla, E.; Hatron, P.Y.; Launay, D.; Terriou, L. Schnitzler syndrome co-occurring with idiopathic multicentric Castleman disease that responds to anti-IL-1 therapy: A case report and clue to pathophysiology. Curr. Res. Transl. Med. 2018, 66, 83–86. [Google Scholar] [CrossRef]
- Din, F.; Mellor, F.; Millard, T.; Pace, E.; Khan, N.; Attygalle, A.D.; Cunningham, D.; Zafar, S.; Sharma, B. Radiology of Castleman disease: The pivotal role of imaging in diagnosis, staging, and response assessment of this rare entity. Clin. Radiol. 2022, 77, 399–408. [Google Scholar] [CrossRef]
- Pitot, M.A.; Tahboub Amawi, A.D.; Alexander, L.F.; LeGout, J.D.; Walkoff, L.; Navin, P.J.; Kawashima, A.; Wood, A.J.; Dispenzieri, A.; Venkatesh, S.K. Imaging of Castleman Disease. Radiographics 2023, 43, e220210. [Google Scholar] [CrossRef]
- Jiang, Y.; Hou, G.; Zhu, Z.; Huo, L.; Li, F.; Cheng, W. 18F-FDG PET/CT imaging features of patients with multicentric Castleman disease. Nucl. Med. Commun. 2021, 42, 833–838. [Google Scholar] [CrossRef]
- He, L.; Chen, Y.; Tan, X.; Sun, X.; Zhang, Q.; Luo, H.; Jiang, L. (18)F-FDG PET/CT and contrast-enhanced CT in the diagnosis of Castleman disease. Jpn. J. Radiol. 2023, 41, 98–107. [Google Scholar] [CrossRef]
- Fajgenbaum, D.C.; Wu, D.; Goodman, A.; Wong, R.; Chadburn, A.; Nasta, S.; Srkalovic, G.; Mukherjee, S.; Leitch, H.; Jayanthan, R.; et al. Insufficient evidence exists to use histopathologic subtype to guide treatment of idiopathic multicentric Castleman disease. Am. J. Hematol. 2020, 95, 1553–1561. [Google Scholar] [CrossRef]
- Wu, D.; Lim, M.S.; Jaffe, E.S. Pathology of Castleman Disease. Hematol. Oncol. Clin. N. Am. 2018, 32, 37–52. [Google Scholar] [CrossRef]
- Liu, X.R.; Tian, M. Glucocorticoids combined with tofacitinib in the treatment of Castleman’s disease: A case report. World J. Clin. Cases 2022, 10, 10794–10802. [Google Scholar] [CrossRef]
- Wang, S.; Wang, R.; Shang, P.; Zhu, X.; Chen, X.; Zhang, G.; Wang, M. Whole-Exome Sequencing Reveals the Genomic Profile and IL6ST Variants as a Prognostic Biomarker of Paraneoplastic Pemphigus-Associated Unicentric Castleman Disease. J. Investig. Dermatol. 2024, 144, 585–592. [Google Scholar] [CrossRef]
- You, L.; Lin, Q.; Zhao, J.; Shi, F.; Young, K.H.; Qian, W. Whole-exome sequencing identifies novel somatic alterations associated with outcomes in idiopathic multicentric Castleman disease. Br. J. Haematol. 2020, 188, e64–e67. [Google Scholar] [CrossRef]
- Nagy, A.; Bhaduri, A.; Shahmarvand, N.; Shahryari, J.; Zehnder, J.L.; Warnke, R.A.; Mughal, T.; Ali, S.; Ohgami, R.S. Next-generation sequencing of idiopathic multicentric and unicentric Castleman disease and follicular dendritic cell sarcomas. Blood Adv. 2018, 2, 481–491. [Google Scholar] [CrossRef]
- Butzmann, A.; Kumar, J.; Sridhar, K.; Gollapudi, S.; Ohgami, R.S. A Review of Genetic Abnormalities in Unicentric and Multicentric Castleman Disease. Biology 2021, 10, 251. [Google Scholar] [CrossRef]
- Tsoi, L.C.; Spain, S.L.; Knight, J.; Ellinghaus, E.; Stuart, P.E.; Capon, F.; Ding, J.; Li, Y.; Tejasvi, T.; Gudjonsson, J.E.; et al. Identification of 15 new psoriasis susceptibility loci highlights the role of innate immunity. Nat. Genet. 2012, 44, 1341–1348. [Google Scholar] [CrossRef]
- Fischer, M.; Weinberger, T.; Schulz, C. The immunomodulatory role of Regnase family RNA-binding proteins. RNA Biol. 2020, 17, 1721–1726. [Google Scholar] [CrossRef]
- Liu, L.; Zhou, Z.; Huang, S.; Guo, Y.; Fan, Y.; Zhang, J.; Zhang, J.; Fu, M.; Chen, Y.E. Zc3h12c inhibits vascular inflammation by repressing NF-kappaB activation and pro-inflammatory gene expression in endothelial cells. Biochem. J. 2013, 451, 55–60. [Google Scholar] [CrossRef]
- Liu, B.; Huang, J.; Ashraf, A.; Rahaman, O.; Lou, J.; Wang, L.; Cai, P.; Wen, J.; Anwaar, S.; Liu, X.; et al. The RNase MCPIP3 promotes skin inflammation by orchestrating myeloid cytokine response. Nat. Commun. 2021, 12, 4105. [Google Scholar] [CrossRef]
- Clayer, E.; Frank, D.; Anderton, H.; Zhang, S.; Kueh, A.; Heim, V.; Nutt, S.L.; Chopin, M.; Bouillet, P. ZC3H12C expression in dendritic cells is necessary to prevent lymphadenopathy of skin-draining lymph nodes. Immunol. Cell Biol. 2022, 100, 160–173. [Google Scholar] [CrossRef]
- Fajgenbaum, D.C.; Shilling, D. Castleman Disease Pathogenesis. Hematol. Oncol. Clin. N. Am. 2018, 32, 11–21. [Google Scholar] [CrossRef]
- Hirano, T. IL-6 in inflammation, autoimmunity and cancer. Int. Immunol. 2021, 33, 127–148. [Google Scholar] [CrossRef]
- Ostrowska, B.; Romejko-Jarosińska, J.; Domańska-Czyż, K.; Walewski, J. Idiopathic multicentric Castleman disease: Pathogenesis, clinical presentation and recommendations for treatment based on the Castleman Disease Collaborative Network (CDCN). Acta Haematol. Pol. 2021, 52, 29–37. [Google Scholar] [CrossRef]
- van Rhee, F.; Voorhees, P.; Dispenzieri, A.; Fossa, A.; Srkalovic, G.; Ide, M.; Munshi, N.; Schey, S.; Streetly, M.; Pierson, S.K.; et al. International, evidence-based consensus treatment guidelines for idiopathic multicentric Castleman disease. Blood 2018, 132, 2115–2124. [Google Scholar] [CrossRef]
- Rehman, M.E.U.; Chattaraj, A.; Neupane, K.; Rafae, A.; Saeed, S.; Basit, J.; Ibrahim, A.; Khouri, J.; Mukherjee, S.; Anwer, F. Efficacy and safety of regimens used for the treatment of multicentric Castleman disease: A systematic review. Eur. J. Haematol. 2022, 109, 309–320. [Google Scholar] [CrossRef]
- Kapriniotis, K.; Lampridis, S.; Mitsos, S.; Patrini, D.; Lawrence, D.R.; Panagiotopoulos, N. Biologic Agents in the Treatment of Multicentric Castleman Disease. Turk. Thorac. J. 2018, 19, 220–225. [Google Scholar] [CrossRef]
- Rossini, B.; Cecchi, N.; Clemente, F.; De Paolis, M.R.; Hohaus, S.; Innao, V.; Lucignano, M.; Massaiu, R.; Palumbo, G.; Rigolin, G.M.; et al. Real-practice management and treatment of idiopathic multicentric Castleman disease with siltuximab: A collection of clinical experiences. Drugs Context 2024, 13. [Google Scholar] [CrossRef]
- Lang, E.; Sande, B.; Brodkin, S.; van Rhee, F. Idiopathic multicentric Castleman disease treated with siltuximab for 15 years: A case report. Ther. Adv. Hematol. 2022, 13, 20406207221082552. [Google Scholar] [CrossRef]
- El-Osta, H.; Janku, F.; Kurzrock, R. Successful treatment of Castleman’s disease with interleukin-1 receptor antagonist (Anakinra). Mol. Cancer Ther. 2010, 9, 1485–1488. [Google Scholar] [CrossRef]
- Galeotti, C.; Tran, T.A.; Franchi-Abella, S.; Fabre, M.; Pariente, D.; Kone-Paut, I. IL-1RA agonist (anakinra) in the treatment of multifocal castleman disease: Case report. J. Pediatr. Hematol. Oncol. 2008, 30, 920–924. [Google Scholar] [CrossRef]
- Borocco, C.; Ballot-Schmit, C.; Ackermann, O.; Aladjidi, N.; Delaleu, J.; Giacobbi-Milet, V.; Jannier, S.; Jeziorski, E.; Maurier, F.; Perel, Y.; et al. The French paediatric cohort of Castleman disease: A retrospective report of 23 patients. Orphanet J. Rare Dis. 2020, 15, 95. [Google Scholar] [CrossRef]
- Pelliccia, S.; Rogges, E.; Cardoni, A.; Lopez, G.; Conte, E.; Faccini, A.L.; De Vito, R.; Girardi, K.; Bianchi, A.; Annibali, O.; et al. The application of a multidisciplinary approach in the diagnosis of Castleman disease and Castleman-like lymphadenopathies: A 20-year retrospective analysis of clinical and pathological features. Br. J. Haematol. 2024, 204, 534–547. [Google Scholar] [CrossRef]

{kind=link}
| Time | Clinical Course and Interventions |
|---|---|
| Age 14 | Fever of unknown origin, fatigue, and significant weight loss |
| Age 14–15 | Early diagnostic workup including multiple hospital admissions Laboratory investigations: elevated inflammatory markers, anemia, hypergammaglobulinemia Imaging (chest X-ray, ultrasound, echocardiogram) without significant findings Infectious disease workup (negative) Multiple antibiotic courses—no clinical response |
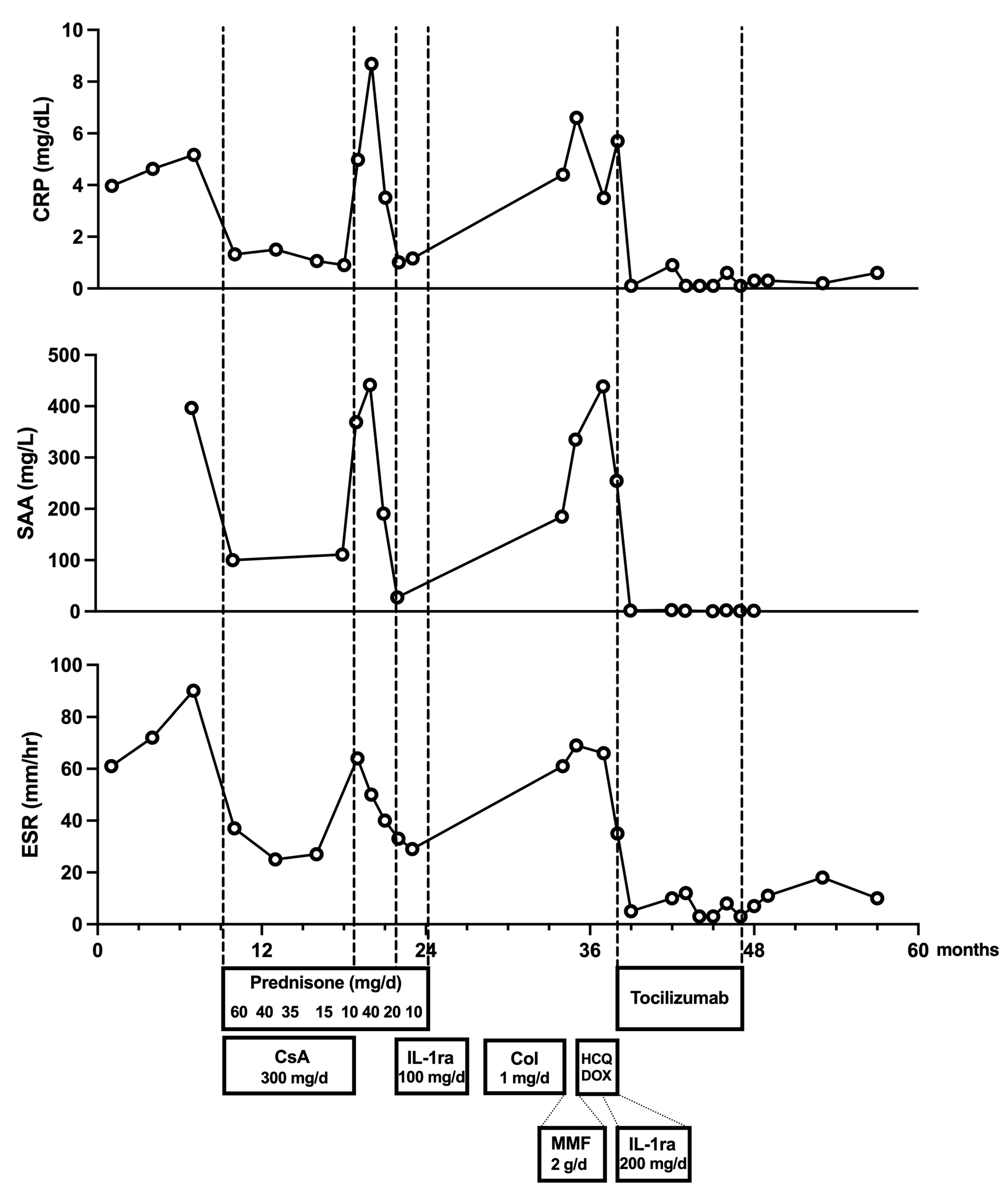
| Age 15 | Tertiary center evaluation at Children’s Memorial Health Institute Abdominal ultrasound and MRI revealing enlarged lymph nodes in the paraaortic region First surgical intervention: partial laparoscopic resection of lymph node cluster Histopathological analysis suggesting an inflammatory pseudotumor |
| Age 15–16 | Treatment with glucocorticoids (methylprednisolone pulses, then prednisone) combined with ciclosporin—initial improvement followed by recurrence of fever and inflammatory markers upon steroid tapering |
| Age 16 | Disease progression noted on follow-up MRI: enlargement of lymph node clusters Second surgery (laparotomy) with complete resection of remaining lymph node masses Histopathology confirmed inflammatory pseudotumor (no malignancy) |
| Age 16–17 | Further treatment attempts: introduction of anakinra (100 mg/day), followed by colchicine and mycophenolate mofetil—no improvement in clinical or laboratory parameters |
| Age 17 | Genetic testing (WES) revealed heterozygous ZC3H12C gene variant (NM_033390.2: c.1819C>T, p.Pro607Ser)—variant of uncertain significance Dose escalation of anakinra to 200 mg/day—partial improvement (reduced evening fever spikes, partial reduction of inflammatory markers) |
| Age 17–18 | FDG-PET/CT—increased tracer uptake in reactive lymph nodes Reevaluation of biopsy samples from both surgical interventions—Castleman disease-like changes (hypervascular type) Final diagnosis of idiopathic multicentric Castleman disease Initiation of targeted therapy with tocilizumab (8 mg/kg every 2–3 weeks)—rapid clinical improvement, normalization of inflammatory markers |
| Age 18–20 | Transition from pediatric to adult clinical immunology care Resolution of lymphadenopathy, steroid discontinuation Tocilizumab discontinued after nine months of treatment Patient remains asymptomatic with no disease recurrence during two-year follow-up period |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sikora, M.; Dąbrowska-Leonik, N.; Buda, P.; Wolska-Kuśnierz, B.; Jahnz-Różyk, K.; Pac, M.; Więsik-Szewczyk, E. Castleman Disease—Still More Questions than Answers: A Case Report and Review of the Literature. J. Clin. Med. 2025, 14, 2799. https://doi.org/10.3390/jcm14082799
Sikora M, Dąbrowska-Leonik N, Buda P, Wolska-Kuśnierz B, Jahnz-Różyk K, Pac M, Więsik-Szewczyk E. Castleman Disease—Still More Questions than Answers: A Case Report and Review of the Literature. Journal of Clinical Medicine. 2025; 14(8):2799. https://doi.org/10.3390/jcm14082799
Chicago/Turabian StyleSikora, Mariusz, Nel Dąbrowska-Leonik, Piotr Buda, Beata Wolska-Kuśnierz, Karina Jahnz-Różyk, Małgorzata Pac, and Ewa Więsik-Szewczyk. 2025. "Castleman Disease—Still More Questions than Answers: A Case Report and Review of the Literature" Journal of Clinical Medicine 14, no. 8: 2799. https://doi.org/10.3390/jcm14082799
APA StyleSikora, M., Dąbrowska-Leonik, N., Buda, P., Wolska-Kuśnierz, B., Jahnz-Różyk, K., Pac, M., & Więsik-Szewczyk, E. (2025). Castleman Disease—Still More Questions than Answers: A Case Report and Review of the Literature. Journal of Clinical Medicine, 14(8), 2799. https://doi.org/10.3390/jcm14082799