
Tuberous Sclerosis Complex: A Case Series from a Romanian Genetics Center and a Review of the Literature


 ,
,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Inclusion Criteria
- Confirmed diagnosis of tuberous sclerosis according to the Schwartz 2007 clinical criteria.
- Age: Patients of all ages (children and adults) included in the BRCMG records between 1984 and 2024.
- Availability of complete clinical data: Comprehensive medical information, including family history, clinical examination, and imaging investigations (CT/MRI), and at least one follow-up evaluation after diagnosis to monitor the progression.
- Multidisciplinary evaluation: Performed by a geneticist, pediatrician, neurologist, psychiatrist, and psychologist.
2.2. Exclusion Criteria
- Incomplete data: Patients without sufficient medical data or clinical investigations to confirm the diagnosis of tuberous sclerosis.
- Incorrect diagnoses: Erroneous or insufficiently documented diagnoses based on the Schwartz 2007 criteria.
- Patients who could not be contacted for re-evaluation or long-term follow-up.
2.3. Data Collection Methods
- Detailed clinical history of each patient, including family and personal history.
- Imaging investigations: Imaging examinations (CT, MRI) to identify brain lesions and other involved organs (kidneys, heart, lungs).
- Psychological and neuropsychiatric evaluation, as well as pediatric neuropsychiatry, including information from psychological and psychiatric evaluations to determine the degree of cognitive and neurological impairment.
- Genetic evaluation. In the case of patients for whom genetic testing could be performed, the identified genetic mutational variants as well as deletions/duplications were reported.
2.4. Evaluation Procedure
- Geneticist: Performed genetic evaluation to identify clinical signs consistent with TSC and detect mutations in the TSC1 or TSC2 genes. Assessment included family history and genetic counseling.
- Pediatrician: Conducted general health assessments and monitored growth and development, identifying systemic manifestations of TSC.
- Neurologist/Pediatric Neurologist: Evaluated for neurological manifestations such as cortical tubers, subependymal nodules, subependymal giant cell astrocytomas (SEGAs), and epilepsy/seizures.
- Psychiatrist/Pediatric Psychiatrist: Assessed for TAND (TSC-associated neuropsychiatric disorders), including autism spectrum disorder (ASD), attention-deficit/hyperactivity disorder (ADHD), anxiety, depression, and aggressive behaviors.
- Dermatologist: Identified characteristic skin lesions associated with TSC, including facial angiofibromas, hypomelanotic macules, shagreen patches, and ungual fibromas.
- Cardiologist/Pediatric Cardiologist: Screened for cardiac rhabdomyomas, which are common in infants and may cause arrhythmias or obstructive symptoms.
- Ophthalmologist (preferably with expertise in pediatric or retinal disorders): Evaluated for retinal hamartomas and other ocular manifestations such as astrocytic retinal lesions.
- Psychologist: Conducted cognitive and behavioral assessments to identify developmental delays, learning difficulties, and intellectual disability, which are frequently associated with TSC.
2.5. Data Analysis
2.6. Ethical Considerations
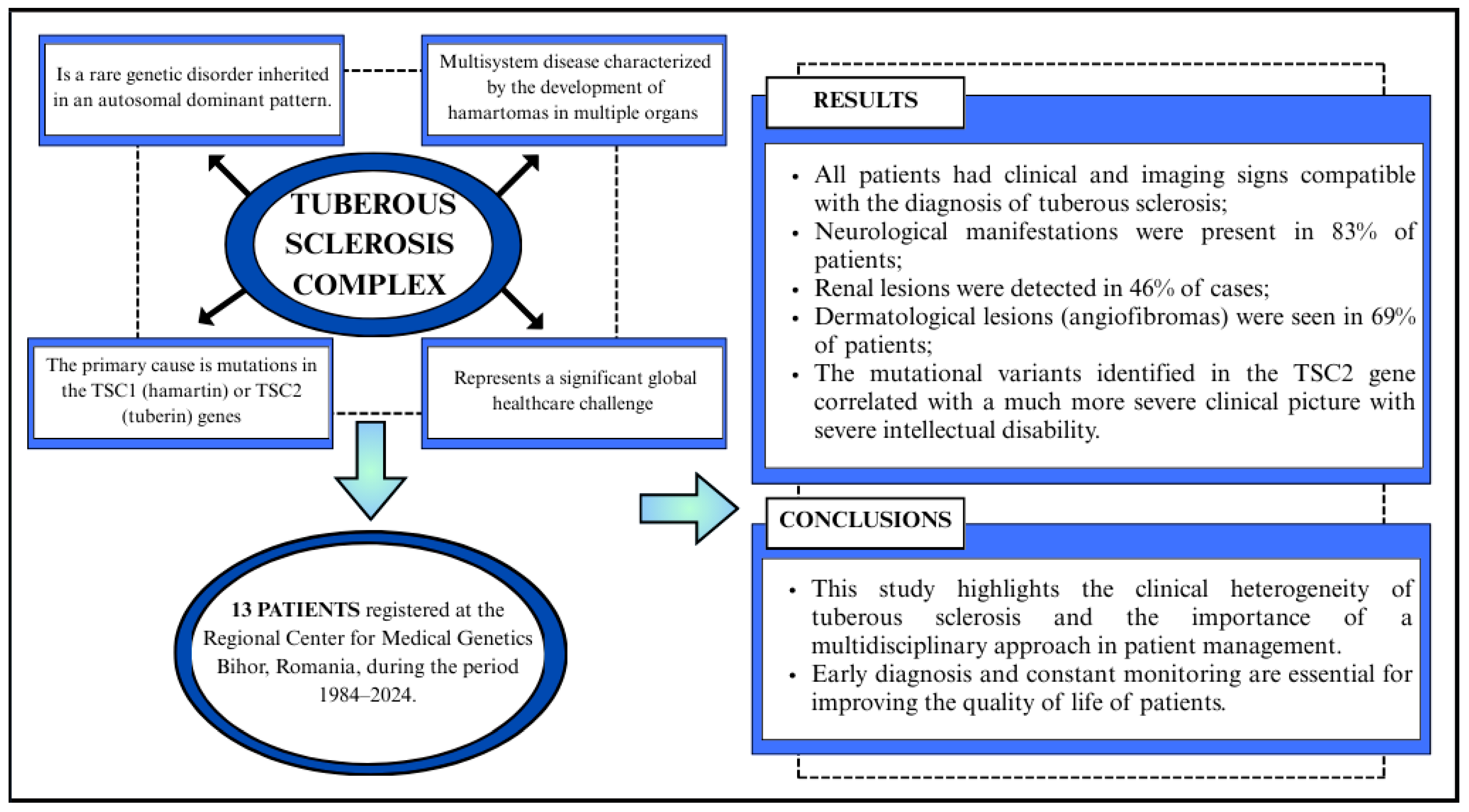
3. Results
3.1. Demographic Characteristics
3.2. Clinical Features
3.2.1. Cardiac, Renal, Bone, and Ophthalmological Ultrasound Clinical Aspects
3.2.2. Neuropsychiatric Aspects
3.2.3. Imaging Characteristics
3.2.4. Molecular Diagnosis
3.2.5. Social, Functional, and Integration Aspects of Patient Evolution
4. Discussion
4.1. Dermatological Changes
4.2. Cardiac Changes
4.3. Renal Changes
4.4. Other Manifestations
4.4.1. Neurological Manifestations
Seizures
4.4.2. Cortical and Subcortical Tubers
4.4.3. Tuberous Sclerosis-Associated Neuropsychiatric Disorders (TANDs)
4.5. Molecular Changes
4.5.1. Comparation Between Mutations in TSC1, TSC2 Gene and Exon Deletions
4.5.2. Exon Deletions
4.5.3. Genotype–Phenotype Correlation
4.6. Treatment
4.7. Social and Functional Evolution of Patients with Tuberous Sclerosis: Monitoring, Survival, and Transition to Adulthood
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Northrup, H.; Aronow, M.E.; Bebin, E.M.; Bissler, J.; Darling, T.N.; de Vries, P.J.; Frost, M.D.; Fuchs, Z.; Gosnell, E.S.; Gupta, N.; et al. Updated international tuberous sclerosis complex diagnostic criteria and surveillance and management recommendations. Pediatr Neurol. 2021, 123, 50–66. [Google Scholar] [CrossRef] [PubMed]
- Devkota, S.; Bhatta, O.P.; Kalikote, A.; Gyawali, P.; Lamichhane, S. Atypical Case of Tuberous Sclerosis with Isolated Neurologic Findings: A Case Report. Clin. Case Rep. 2024, 12, e9379. [Google Scholar] [CrossRef]
- Randle, S.C. Tuberous Sclerosis Complex: A Review. Pediatr. Ann. 2017, 46, e166–e171. [Google Scholar] [CrossRef] [PubMed]
- Northrup, H.; Krueger, D.A.; International Tuberous Sclerosis Complex Consensus Group. Tuberous Sclerosis Complex Diagnostic Criteria Update: Recommendations of the 2012 Iinternational Tuberous Sclerosis Complex Consensus Conference. Pediatr. Neurol. 2013, 49, 243–254. [Google Scholar] [CrossRef]
- Jurca, C.M.; Kozma, K.; Petchesi, C.D.; Zaha, D.C.; Magyar, I.; Munteanu, M.; Faur, L.; Jurca, A.; Bembea, D.; Severin, E.; et al. Tuberous Sclerosis, Type II Diabetes Mellitus and the PI3K/AKT/mTOR Signaling Pathways—Case Report and Literature Review. Genes 2023, 14, 433. [Google Scholar] [CrossRef] [PubMed]
- CNAS. National Program for Rare Diseases. Available online: https://cnas.ro/wp-content/uploads/2022/04/Programul-national-de-tratament-pentru-boli-rare.pdf (accessed on 18 January 2025).
- Kohrman, M.H. Emerging Treatments in the Management of Tuberous Sclerosis Complex. Pediatr. Neurol. 2012, 46, 267–275. [Google Scholar] [CrossRef]
- Plank, T.L.; Yeung, R.S.; Henske, E.P. Hamartin, the Product of the Tuberous Sclerosis 1 (TSC1) Gene, Interacts with Tuberin and Appears to Be Localized to Cytoplasmic Vesicles. Cancer Res. 1998, 58, 4766–4770. [Google Scholar]
- Jones, A.C.; Snell, R.G.; Tachataki, M. SA Molecular Genetic and Phenotypic Analysis Reveals Differences between TSC1 and TSC2 Associated Familial and Sporadic Tuberous Sclerosis. Hum. Mol. Genet. 1997, 6, 2155–2161. [Google Scholar] [CrossRef]
- Schwartz, R.A.; Fernández, G.; Kotulska, K.; Jóźwiak, S. Tuberous Sclerosis Complex: Advances in Diagnosis, Genetics, and Management. J. Am. Acad. Dermatol. 2007, 57, 189–202. [Google Scholar] [CrossRef]
- Teng, J.M.C.; Cowen, E.W.; Wataya-Kaneda, M.; Gosnell, E.S.; Witman, P.M.; Hebert, A.A.; Mlynarczyk, G.; Soltani, K.; Darling, T.N. Dermatologic and Dental Aspects of the 2012 International Tuberous Sclerosis Complex Consensus Statements. JAMA Dermatol. 2014, 150, 1095–1101. [Google Scholar] [CrossRef]
- Jóźwiak, S.; Schwartz, R.A.; Janniger, C.K.; Michałowicz, R.; Chmielik, J. Skin Lesions in Children with Tuberous Sclerosis Complex: Their Prevalence, Natural Course, and Diagnostic Significance: Skin Lesions in Tuberous Sclerosis Complex. Int. J. Dermatol. 1998, 37, 911–917. [Google Scholar] [CrossRef]
- Webb, D.W.; Clarke, A.; Fryer, A.; Osborne, J.P. The Cutaneous Features of Tuberous Sclerosis: A Population Study. Br. J. Dermatol. 1996, 135, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Mceneaney, L.J. AR Finding a Cure for Tuberous Sclerosis Complex: From Genetics through to Targeted Drug Therapies. Adv. Genet. 2019, 103, 91–118. [Google Scholar] [CrossRef] [PubMed]
- Roach, E.S. Applying the Lessons of Tuberous Sclerosis: The 2015 Hower Award Lecture. Pediatr. Neurol. 2016, 63, 6–22. [Google Scholar] [CrossRef]
- Luo, C.; Ye, W.-R.; Shi, W.; Yin, P.; Chen, C.; He, Y.-B.; Chen, M.-F.; Zu, X.-B.; Cai, Y. Perfect Match: mTOR Inhibitors and Tuberous Sclerosis Complex. Orphanet J. Rare Dis. 2022, 17, 106. [Google Scholar] [CrossRef] [PubMed]
- Hinton, R.B.; Prakash, A.; Romp, R.L.; Krueger, D.A.; Knilans, T.K.; International Tuberous Sclerosis Consensus Group. Cardiovascular Manifestations of Tuberous Sclerosis Complex and Summary of the Revised Diagnostic Criteria and Surveillance and Management Recommendations from the International Tuberous Sclerosis Consensus Group. J. Am. Heart Assoc. 2014, 3, e001493. [Google Scholar] [CrossRef]
- Joźwiak S Kotulska K Kasprzyk-Obara, J.; Domanska-Pakiela, D. Drabik M Clinical and Genotype Studies of Cardiac Tumors in 154 Patients with Tuberous Sclerosis Complex. Pediatrics 2006, 118, e1146–e1151. [Google Scholar] [CrossRef]
- Staley, B.A.; Vail, E.A.; Thiele, E.A. Tuberous Sclerosis Complex: Diagnostic Challenges, Presenting Symptoms, and Commonly Missed Signs. Pediatrics 2011, 127, e117–e125. [Google Scholar] [CrossRef]
- Rout, P.; Zamora, E.A.; Aeddula, N.R. Tuberous Sclerosis. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2025. [Google Scholar]
- Smythe, J.F.; Dyck, J.D.; Smallhorn, J.F.; Freedom, R.M. Natural History of Cardiac Rhabdomyoma in Infancy and Childhood. Am. J. Cardiol. 1990, 66, 1247–1249. [Google Scholar] [CrossRef]
- Nair, N.; Chakraborty, R.; Mahajan, Z.; Sharma, A.; Sethi, S.K.; Raina, R. Renal Manifestations of Tuberous Sclerosis Complex. J. Kidney Cancer VHL 2020, 7, 5–19. [Google Scholar] [CrossRef]
- Curatolo, P.; Maria, B.L. Tuberous Sclerosis. Handb. Clin. Neurol. 2013, 111, 323–331. [Google Scholar] [CrossRef]
- O’Callaghan, F.J.; Noakes, M.J.; Martyn, C.N.; Osborne, J.P. An Epidemiological Study of Renal Pathology in Tuberous Sclerosis Complex. BJU Int. 2004, 94, 853–857. [Google Scholar] [CrossRef] [PubMed]
- Cox, R.L.; Calderon de Anda, F.; Mangoubi, T.; Yoshii, A. Multiple Critical Periods for Rapamycin Treatment to Correct Structural Defects in Tsc-1-Suppressed Brain. Front. Mol. Neurosci. 2018, 11, 409. [Google Scholar] [CrossRef]
- Dhakal, M.; Dhakal, O.P.; Bhandari, D. Polycystic Kidney Disease and Chronic Renal Failure in Tuberous Sclerosis. BMJ Case Rep. 2013, 2013, bcr2013200711. [Google Scholar] [CrossRef] [PubMed]
- Kingswood, J.C.; Belousova, E.; Benedik, M.P.; Carter, T.; Cottin, V.; Curatolo, P.; Dahlin, M.; D’Amato, L.; Beaure d’Augères, G.; de Vries, P.J.; et al. Renal Manifestations of TuberOus SClerosis Complex: Key Findings from the Final Analysis of the TOSCA Study Focussing Mainly on Renal Angiomyolipomas. Front. Neurol. 2020, 11, 972. [Google Scholar] [CrossRef]
- Yates, J.R.W. Tuberous Sclerosis. Eur. J. Hum. Genet. 2006, 14, 1065–1073. [Google Scholar] [CrossRef] [PubMed]
- Sparling, J.D.; Hong, C.-H.; Brahim, J.S.; Moss, J.; Darling, T.N. Oral Findings in 58 Adults with Tuberous Sclerosis Complex. J. Am. Acad. Dermatol. 2007, 56, 786–790. [Google Scholar] [CrossRef]
- Nabbout, R.; Belousova, E.; Benedik, M.P.; Carter, T.; Cottin, V.; Curatolo, P.; Dahlin, M.; D Amato, L.; d’Augères, G.B.; de Vries, P.J.; et al. Epilepsy in Tuberous Sclerosis Complex: Findings from the TOSCA Study. Epilepsia Open 2019, 4, 73–84. [Google Scholar] [CrossRef]
- Jansen, A.C.; TOSCA consortium and TOSCA investigators. TuberOus SClerosis Registry to Increase Disease Awareness (TOSCA)—Baseline Data on 2093 Patients. Orphanet J. Rare Dis. 2017, 12, 2. [Google Scholar] [CrossRef]
- Jeong, A.; Wong, M. Systemic Disease Manifestations Associated with Epilepsy in Tuberous Sclerosis Complex. Epilepsia 2016, 57, 1443–1449. [Google Scholar] [CrossRef]
- Seri, S.; Cerquiglini, A.; Pisani, F.; Michel, C.M.; Pascual Marqui, R.D.; Curatolo, P. Frontal Lobe Epilepsy Associated with Tuberous Sclerosis: Electroencephalographic-Magnetic Resonance Image Fusioning. J. Child. Neurol. 1998, 13, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Diallo, S.M.; Diallo, M.; Barry, I.S.; Touré, M.L.; Barry, M.C.; Diallo, M.T.; Barry, S.D.; Aminou, S.Y.; Othon, G.C.; Diallo, B.; et al. Epileptic Seizures Revealing Tuberous Sclerosis in a Tropical Environment: A Study of 12 Case Series. eNeurologicalSc 2024, 36, 100516. [Google Scholar] [CrossRef]
- Ehninger, D.; Sano, Y.; de Vries, P.J.; Dies, K.; Franz, D.; Geschwind, D.H.; Kaur, M.; Lee, Y.-S.; Li, W.; Lowe, J.K.; et al. Gestational Immune Activation and Tsc2 Haploinsufficiency Cooperate to Disrupt Fetal Survival and May Perturb Social Behavior in Adult Mice. Mol. Psychiatr. 2010, 17, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Vries, P.J.; Whittemore, V.H.; Leclezio, L.; Byars, A.W.; Dunn, D. Tuberous Sclerosis Associated Neuropsychiatric Disorders (TAND) and the TAND Checklist. Pediatr. Neurol. 2015, 52, 25–35. [Google Scholar] [CrossRef]
- Jeste, S.S.; Sahin, M.; Bolton, P.; Ploubidis, G.B.; Humphrey, A. Characterization of Autism in Young Children with Tuberous Sclerosis Complex. J. Child. Neurol. 2008, 23, 520–525. [Google Scholar] [CrossRef]
- van Eeghen, A.M.; Black, M.E.; Pulsifer, M.B.; Kwiatkowski, D.J.; Thiele, E.A. Genotype and Cognitive Phenotype of Patients with Tuberous Sclerosis Complex. Eur. J. Hum. Genet. 2012, 20, 510–515. [Google Scholar] [CrossRef]
- van Slegtenhorst, M.; Nellist, M.; Nagelkerken, B.; Cheadle, J.; Snell, R.; van den Ouweland, A.; Reuser, A.; Sampson, J.; Halley, D.; van der Sluijs, P. Interaction between Hamartin and Tuberin, the TSC1 and TSC2 Gene Products. Hum. Mol. Genet. 1998, 7, 1053–1057. [Google Scholar] [CrossRef] [PubMed]
- Au, K.S.; Williams, A.T.; Roach, E.S.; Batchelor, L.; Sparagana, S.P.; Delgado, M.R.; Wheless, J.W.; Baumgartner, J.E.; Roa, B.B.; Wilson, C.M.; et al. Genotype/Phenotype Correlation in 325 Individuals Referred for a Diagnosis of Tuberous Sclerosis Complex in the United States. Genet. Med. 2007, 9, 88–100. [Google Scholar] [CrossRef]
- Garcia, A.B.; Viola, G.D.; Corrêa, B.D.; Fischer, T.D.; Pinho, M.C.; Rodrigues, G.M.; Ashton-Prolla, P.; Rosset, C. An Overview of Actionable and Potentially Actionable TSC1 and TSC2 Germline Variants in an Online Database. Genet. Mol. Biol. 2023, 46 (Suppl. S1), e20230132. [Google Scholar] [CrossRef]
- Rosset, C.; Vairo, F.; Bandeira, I.C.; Correia, R.L.; de Goes, F.V.; da Silva, R.T.B.; Bueno, L.S.M.; de Miranda Gomes, M.C.S.; de Campos Reis Galvão, H.; Neri, J.I.C.F.; et al. Molecular Analysis of TSC1 and TSC2 Genes and Phenotypic Correlations in Brazilian Families with Tuberous Sclerosis. PLoS ONE 2017, 12, e0185713. [Google Scholar] [CrossRef]
- Singh, A.K.; Olsen, M.F.; Lavik, L.A.S.; Vold, T.; Drabløs, F.; Sjursen, W. Detecting Copy Number Variation in next Generation Sequencing Data from Diagnostic Gene Panels. BMC Med. Genom. 2021, 14, 214. [Google Scholar] [CrossRef] [PubMed]
- 2012 International TSC Clinical Consensus Conference Recommendations. Available online: https://www.tscinternational.org/wp-content/uploads/2019/01/TSCi-Consensus-Guidelines.pdf (accessed on 10 January 2025).
- Georgieva, B.; Koleva, M.; Todorov, T.; Bojinova, V.; Deneva, D.; Glushkova, M.; Aleksandrova, I.; Rodopska, E.; Miteva, A.; Mitev, V.; et al. Molecular-Genetic Characteristics and Genotype-Phenotype Correlations in Bulgarian Patients with Tuberous Sclerosis Complex. Acta Medica Bulg. 2021, 48, 29–36. [Google Scholar] [CrossRef]
- Rosengren, T.; Nanhoe, S.; de Almeida, L.G.D.; Schönewolf-Greulich, B.; Larsen, L.J.; Hey, C.A.B.; Dunø, M.; Ek, J.; Risom, L.; Nellist, M.; et al. Mutational Analysis of TSC1 and TSC2 in Danish Patients with Tuberous Sclerosis Complex. Sci. Rep. 2020, 10, 9909. [Google Scholar] [CrossRef] [PubMed]
- Alsowat, D.; Whitney, R.; Hewson, S.; Jain, P.; Chan, V.; Kabir, N.; Amburgey, K.; Noone, D.; Lemaire, M.; McCoy, B.; et al. The Phenotypic Spectrum of Tuberous Sclerosis Complex: A Canadian Cohort. Child. Neurol. Open 2021, 8, 2329048X211012817. [Google Scholar] [CrossRef]
- Dabora, S.L.; Jozwiak, S.; Franz, D.N.; Roberts, P.S.; Nieto, A.; Chung, J.; Choy, Y.S.; Reeve, M.P.; Thiele, E.; Egelhoff, J.C.; et al. Mutational Analysis in a Cohort of 224 Tuberous Sclerosis Patients Indicates Increased Severity of TSC2, Compared with TSC1, Disease in Multiple Organs. Am. J. Hum. Genet. 2001, 68, 64–80. [Google Scholar] [CrossRef]
- Sancak, O.; Nellist, M.; Goedbloed, M. Mutational Analysis of the TSC1 and TSC2 Genes in a Diagnostic Setting: Genotype-Phenotype Correlations and Comparison of Diagnostic DNA Techniques in Tuberous Sclerosis Complex. Eur. J. Hum. Genet. 2005, 13, 731–741. [Google Scholar] [CrossRef]
- Farach, L.S.; Pearson, D.A.; Woodhouse, J.P.; Schraw, J.M.; Sahin, M.; Krueger, D.A.; Wu, J.Y.; Bebin, E.M.; Lupo, P.J.; Au, K.S.; et al. Tuberous Sclerosis Complex genotypes and developmental phenotype. Pediatr. Neurol. 2019, 96, 58–63. [Google Scholar] [CrossRef]
- Van Eeghen, A.M.; Nellist, M.; Van Eeghen, E.E.; Thiele, E.A. Central TSC2 missense mutations are associated with a reduced risk of infantile spasms. Epilepsy Res. 2013, 103, 83–87. [Google Scholar] [CrossRef]
- Muzykewicz, D.A.; Newberry, P.; Danforth, N.; Halpern, E.F.; Thiele, E.A. Psychiatric comorbid conditions in a clinic population of 241 patients with Tuberous Sclerosis Complex. Epilepsy Behav. 2007, 11, 506–513. [Google Scholar] [CrossRef]
- Oyazato, Y.; Iijima, K.; Emi, M.; Sekine, T.; Kamei, K.; Takanashi, J.; Nakao, H.; Namai, Y.; Nozu, K.; Matsuo, M. Molecular analysis of TSC2/PKD1 contiguous gene deletion syndrome. Kobe J. Med. Sci. 2011, 57, E1–E10. [Google Scholar]
- Boronat, S.; Barber, I.; Pargaonkar, V.; Chang, J.; Thiele, E.A. Sclerotic Bone Lesions at Abdominal Magnetic Resonance Imaging in Children with Tuberous Sclerosis Complex. Pediatr. Radiol. 2016, 46, 689–694. [Google Scholar] [CrossRef] [PubMed]
- Jones, A.C.; Shyamsundar, M.M.; Thomas, M.W.; Maynard, J.; Idziaszczyk, S.; Tomkins, S.; Sampson, J.R.; Cheadle, J.P. Comprehensive Mutation Analysis of TSC1 and TSC2-and Phenotypic Correlations in 150 Families with Tuberous Sclerosis. Am. J. Hum. Genet. 1999, 64, 1305–1315. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulou, A.; Dinopoulos, A.; Koutsodontis, G.; Pons, R.; Vorgia, P.; Koute, V.; Vratimos, A.; Zafeiriou, D. Screening for TSC1 and TSC2 Mutations Using NGS in Greek Children with Tuberous Sclerosis Syndrome. Eur. J. Paediatr. Neurol. 2018, 22, 419–426. [Google Scholar] [CrossRef]
- O’Connor, S.E.; Kwiatkowski, D.J.; Roberts, P.S.; Wollmann, R.L.; Huttenlocher, P.R. A family with seizures and minor features of Tuberous Sclerosis and a novel TSC2 mutation. Neurology 2003, 61, 409–412. [Google Scholar] [CrossRef] [PubMed]
- Stehlíková, K.; Skálová, D.; Zídková, J.; Haberlová, J.; Voháňka, S.; Mazanec, R.; Mrázová, L.; Vondráček, P.; Ošlejšková, H.; Zámečník, J.; et al. Muscular Dystrophies and Myopathies: The Spectrum of Mutated Genes in the Czech Republic. Clin. Genet. 2016, 91, 463–469. [Google Scholar] [CrossRef]
- Tsai, P.T.; Rudolph S Guo, C.; Ellegood, J.; Gibson, M. Sensitive Periods for Cerebellar-Mediated Autistic-like Behaviors. Cell Rep. 2018, 25, 357–367. [Google Scholar] [CrossRef] [PubMed]
- Peron, A.; Canevini, M.P.; Ghelma, F.; Di Marco, F.; Vignoli, A. Healthcare Transition from Childhood to Adulthood in Tuberous Sclerosis Complex. Am. J. Med. Genet. C Semin. Med. Genet. 2018, 178, 355–364. [Google Scholar] [CrossRef]
- Yoshinaga, H.; Hayashibara, N.; Inoue, M.; Endoh, F.; Adachi, K.; Nanba, E. Difficulty of Transition to Adult Care in a Patient with Tuberous Sclerosis: A Case Report. Epilepsy Seizure 2021, 13, 36–44. [Google Scholar] [CrossRef]
- Bar, C.; Ghobeira, R.; Azzi, R.; Ville, D.; Riquet, A.; Touraine, R.; Chemaly, N.; Nabbout, R. Experience of Follow-up, Quality of Life, and Transition from Pediatric to Adult Healthcare of Patients with Tuberous Sclerosis Complex. Epilepsy Behav. 2019, 96, 23–27. [Google Scholar] [CrossRef]
- Jansen, A.C.; Vanclooster, S.; de Vries, P.J.; Fladrowski, C.; Beaure d’Augères, G.; Carter, T.; Belousova, E.; Benedik, M.P.; Cottin, V.; Curatolo, P.; et al. Burden of Illness and Quality of Life in TuberOus SClerosis Complex: Findings from the TOSCA Study. Front. Neurol. 2020, 11, 904. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient No. | Sex | Years of Births | Parents’ Age at Birth | Familial = F Sporadic = S | Age at Diagnosis (Months/Years) | Onset Symptoms | OBS | |
|---|---|---|---|---|---|---|---|---|
| Father | Mother | |||||||
| 01 | female | 2010 | 29 | 22 | S | 3 y | Seizures | - |
| 02 | female | 1989 | 40 | 32 | S | 18 y | Seizures | type II diabetes, herniated disc |
| 03 | male | 2021 | 36 | 25 | S | 4 m | cardiac rhabdomyoma | - |
| 04 | female | 2005 | 29 | 27 | F | 5 y | Seizures | - |
| 05 | male | 2010 | 34 | 35 | S | 5 y | Seizures | Left iliac bone cyst |
| 06 | female | 1995 | 22 | 28 | S | 8 y | Seizures | - |
| 07 | male | 2000 | 29 | 32 | S | 3 y | Seizures | Scoliosis, |
| 08 | female | 2001 | 42 | 46 | F | 10 y | Seizures | - |
| 09 | male | 1979 | 36 | 26 | F | 5 y | Seizures | - |
| 10 | female | 1948 | 24 | 21 | S | 35 y | Seizures | - |
| 11 | female | 1976 | 35 | 31 | S | 14 y | Seizures | |
| 12 | male | 2007 | 31 | 28 | S | 11 y | Seizures | - |
| 13 | female | 1997 | 27 | 22 | S | 9–10 y | Seizures | - |
| Patient No. | Facial Angiofibromas | Achromic Spots | Shagreen Patch | Gingival Fibroma | Dental Anomalies | Nail Fibroids |
|---|---|---|---|---|---|---|
| 01 | + | + | - | - | - | - |
| 02 | + | + | + | + | + | + |
| 03 | - | - | - | - | - | - |
| 04 | + | + | + | + | + | - |
| 05 | + | + | + | - | - | - |
| 06 | + | + | + | + | - | + |
| 07 | + | + | - | + | + | + |
| 08 | - | + | + | - | + | - |
| 09 | + | + | - | - | - | - |
| 10 | + | + | + | - | + | - |
| 11 | + | - | + | - | - | - |
| 12 | + | + | + | + | + | + |
| 13 | + | + | + | - | + | + |
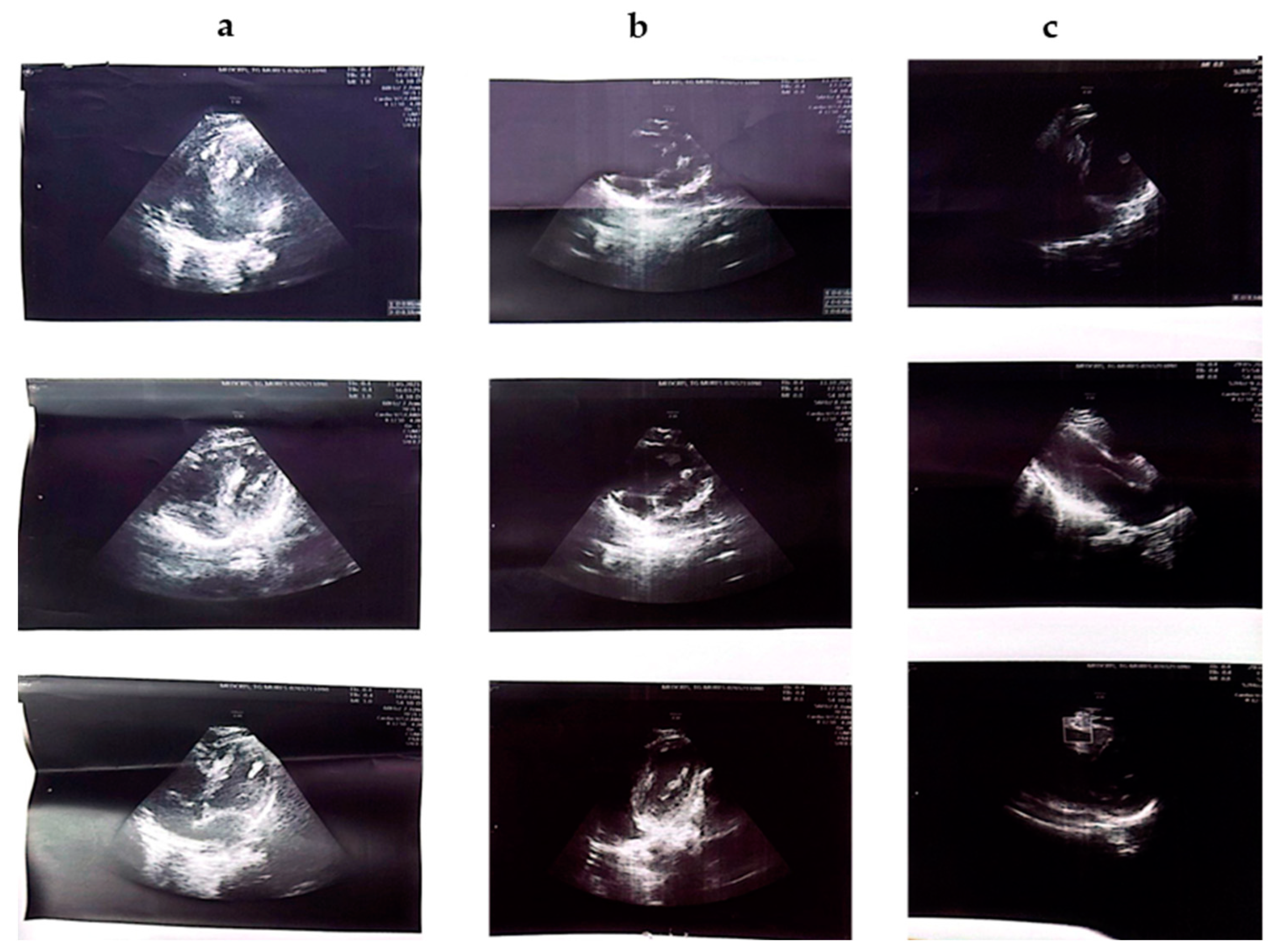
| Patient No. | Cardiac | Renal | Ophthalmology |
|---|---|---|---|
| 01 | Interventricular septal rhabdomyoma | Bilateral renal angiomyolipomas | Tuberous lesions of the right eye, optic nerve, pale, atrophic |
| 02 | Mitral insufficiency | - | - |
| 03 | multiple fetal cardiac rhabdomyomas of the ventricular septum | - | - |
| 04 | - | Bilateral renal angiomyolipomas | - |
| 05 | Hyperechoic SIV, Mitral valve with myxomatous appearance, aberrant chordae | - | - |
| 06 | Mitral valve prolapse, mild mitral regurgitation, nodular formation in the RV wall resembling rhabdomyoma | hypertrophic kidneys, polylobate contour, disappearance of corticomedullary differentiation | - |
| 07 | - | hypertrophic kidneys, disappearance of corticomedullary differentiation | - |
| 08 | - | - | - |
| 09 | - | Bilateral renal angiomyolipomas | - |
| 10 | - | - | - |
| 11 | - | Both without cortical-medullary demarcation | - |
| 12 | - | Right kidney, the disappearance of corticomedullary differentiation and intramedullary microcalcifications | - |
| 13 | - | - | - |
| Patient No. | Seizures | ADHD | Language | NM Development | TSA | Behavior Disorders |
|---|---|---|---|---|---|---|
| 01 | + | - | - | - | - | - |
| 02 | + | - | - | - | - | - |
| 03 | - | - | - | - | - | - |
| 04 | + | - | - | + | - | + |
| 05 | + | - | - | - | - | - |
| 06 | + | - | + | + | + | + |
| 07 | + | - | + | + | - | + |
| 08 | + | - | - | - | - | - |
| 09 | + | - | - | - | - | - |
| 10 | - | - | - | - | - | + |
| 11 | + | + | + | - | - | |
| 12 | + | + | + | + | - | + |
| 13 | + | - | - | - | - | - |
| Patient No. | CT Scan | Brain MRI |
|---|---|---|
| 01 | - | 2023: Inhomogeneous tumor formation in the third ventricle and a calcified formation in the lateral wall of the right lateral ventricle, frontoparietooccipital cortico-subcortical tubers. Secondary hydrocephalus. 2023: cortico-subcortical tubers stabilized compared to the previous examination, calcified subependymal nodules. Parietal narrow tumor formation corresponding to a giant cell astrocytoma (SEGA) |
| 02 | - | 2022: four bilateral hamartomatous subependymal nodules—the largest 3 mm in diameter, partially calcified—associating cortical/subcortical tubers with frontal, temporal and occipital distribution on the right and frontal and parieto-occipital distribution on the left |
| 03 | - | 2021–2022 Subependymal micronodules in the lateral ventricles; 2024. subretentorial cortico-subcortical cortical tubers |
| 04 | Nodular calcifications of the cerebral hemispheres and the left cerebellar hemisphere, diffuse osteosclerosis of the skull bones, diffuse aplasia of the skull bones, aplasia of the frontal sinus | subcortical tubers; subependymal nodules |
| 05 | supratentorial subcortical tubers, subependymal nodules with calcareous inclusions, swelling of the overlying gyrus | |
| 06 | calcified subependymal tubers of the lateral ventricles, left lenticular angioma, cortico-subcortical demyelinating lesions, disseminated cerebral and cerebellar calcifications, | Nodular lesions in the walls of the lateral ventricles, areas of supratentorial paraventricular and supraventricular demyelination |
| 07 | Subependymal calcifications in the lateral ventricles max 9 mm, calcifications in the depth of the Sylvian sulcus of 14 mm, calcifications on the surface of the left cerebellum of 4 mm | Cortical and subcortical tubers, subependymal nodules, lateral ventricles, right temporal arachnoid cyst |
| 08 | Mild asymmetry of the skull, hypo/hyperdense intraparenchymal cerebral infra/supratentorial areas | Bihemispheric cortical tubers cortico-subcortical fronto-temporo-parietal Calcified subependymal and ventrolateral micronodules |
| 09 | Subependymal calcifications of the lateral ventricle | - |
| 10 | Bilateral parietal and temporal cortical tubers | - |
| 11 | - | Fronto-parieto-temporal cortical tubers, perinatal hypoxic leukoencephalopathy with multiple areas of ischemic gliosis |
| 12 | - | Bihemispheric F-P-T cortical tubers Periventricular leukomalacia, corpus callosum hypoplasia, Bilateral occipital polymicrogyria |
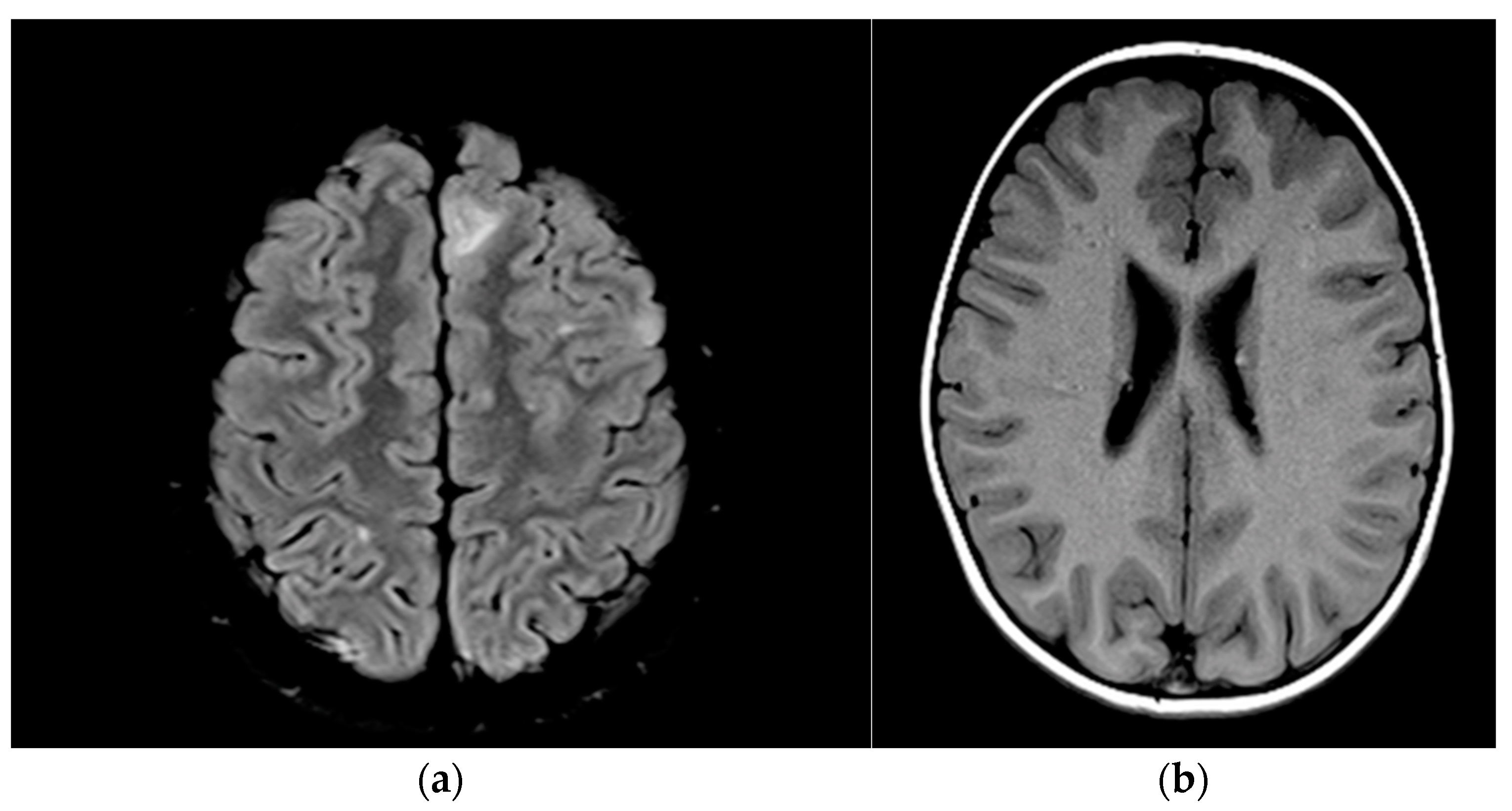
| 13 | - | Subependymal calcareous nodules and subcortical tubers; cortical tubers, photo-calcified nodular lesions, bilateral subependymar and cortical tubers, calcified periventricular and cortical tubers (Figure 3a,b) |
| No. | Gene | Technique | NM Number | Transcript | Variant | Zygosity | Clasificaton |
|---|---|---|---|---|---|---|---|
| 01 | TSC2 | MLPA | Deletion of exon 41 | structural variant | heterozygous | likely pathogenic | |
| 02 | TSC1 | NGS | NM_000368.4:c.2 | 347C > T | nonsense | heterozygous | pathogenic |
| 03 | TSC1 | NGS | NM_000368.4:c.1 | c.1270A > T (p. Arg424-) | nonsense | heterozygous | pathogenic |
| 04 | TSC2 | NGS | NM_001318831.2 | c.3466del | frameshifts | heterozygous | pathogenic |
| 05 | TSC2 | NGS | Partial Deletion (Exon 40) | structural variant | heterozygous | likely pathogenic | |
| 06 | MLPA | MLPA negative | |||||
| 07 | TSC2 | NM_000548.5 | c.3559dup (p. Val1187Glyfs-47) | frameshift | heterozygous | pathogenic | |
| 08 | TSC1 | NGS | NM_000368.4 | c.5:26dup | frameshift | heterozygous | pathogenic |
| 13 | TSC2 | NGS | NM_000548.5 | c.4005 + 1G > T | splicesite | heterozygous | pathogenic |
| DMD | NGS | NM_004006.3 | c.960 + 2T > G | splice site | unknown | likely pathogenic |
| No. | No Education | Kindergarten | Special Class | Primary School | High School | Faculty | Employee | Age of Death (Years) | |
|---|---|---|---|---|---|---|---|---|---|
| 01 | FR | - | - | - | + | - | - | - | - |
| 02 | VA | - | - | + | - | - | - | - | - |
| 03 | BG | - | + | - | - | - | - | - | - |
| 04 | PO | + | - | - | - | - | - | - | - |
| 05 | BA | - | - | - | + | - | - | - | - |
| 06 | BI | + | - | - | - | - | - | - | - |
| 07 | HR | + | - | - | - | - | - | - | - |
| 08 | MC | - | - | - | - | + | - | + | - |
| 09 | BSt | - | - | - | + | - | - | + | 48 |
| 10 | BO | - | - | + | - | - | - | + | 52 |
| 11 | DN | - | - | - | + | - | - | + | 40 |
| 12 | DR | + | - | - | - | - | - | - | 16 |
| 13 | VL | - | - | - | - | - | + | + | - |
| Major Criteria (11) | Observation |
|---|---|
| Depigmented macules with a 3–5 mm diameter Facial angiofibromas (over 3) Ungual fibromas (over 2) Shagreen patch Retinal hamartomas Cortical dysplasia Astrocytomas and subependymal nodules Cardiac rhabdomyomas Lymphangioleiomyomatosis renal angiomyolipomas | Genetic diagnosis: For a positive diagnosis, it is sufficient to identify a pathogenic variant in one of the two genes |
| Minor criteria (7) | |
| “Confetti” skin lesions Intraoral fibromas Changes in tooth enamel White patches on the retina Renal cysts Other hamartomas Sclerotic bone changes |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jurca, A.A.; Hodisan, R.; Jurca, A.D.; Severin, E.; Jurca, S.; Trandafir, A.; Ilias, T.; Vesa, C.; Jurca, C.M. Tuberous Sclerosis Complex: A Case Series from a Romanian Genetics Center and a Review of the Literature. J. Clin. Med. 2025, 14, 2974. https://doi.org/10.3390/jcm14092974
Jurca AA, Hodisan R, Jurca AD, Severin E, Jurca S, Trandafir A, Ilias T, Vesa C, Jurca CM. Tuberous Sclerosis Complex: A Case Series from a Romanian Genetics Center and a Review of the Literature. Journal of Clinical Medicine. 2025; 14(9):2974. https://doi.org/10.3390/jcm14092974
Chicago/Turabian StyleJurca, Aurora Alexandra, Ramona Hodisan, Alexandru Daniel Jurca, Emilia Severin, Sanziana Jurca, Ana Trandafir, Tiberia Ilias, Cosmin Vesa, and Claudia Maria Jurca. 2025. "Tuberous Sclerosis Complex: A Case Series from a Romanian Genetics Center and a Review of the Literature" Journal of Clinical Medicine 14, no. 9: 2974. https://doi.org/10.3390/jcm14092974
APA StyleJurca, A. A., Hodisan, R., Jurca, A. D., Severin, E., Jurca, S., Trandafir, A., Ilias, T., Vesa, C., & Jurca, C. M. (2025). Tuberous Sclerosis Complex: A Case Series from a Romanian Genetics Center and a Review of the Literature. Journal of Clinical Medicine, 14(9), 2974. https://doi.org/10.3390/jcm14092974