Genetic Testing in the Diagnosis of Primary Ciliary Dyskinesia: State-of-the-Art and Future Perspectives

Abstract
:1. Introduction


2. Diagnosis of PCD
3. The Need for Early Diagnostics
4. Genetics of PCD

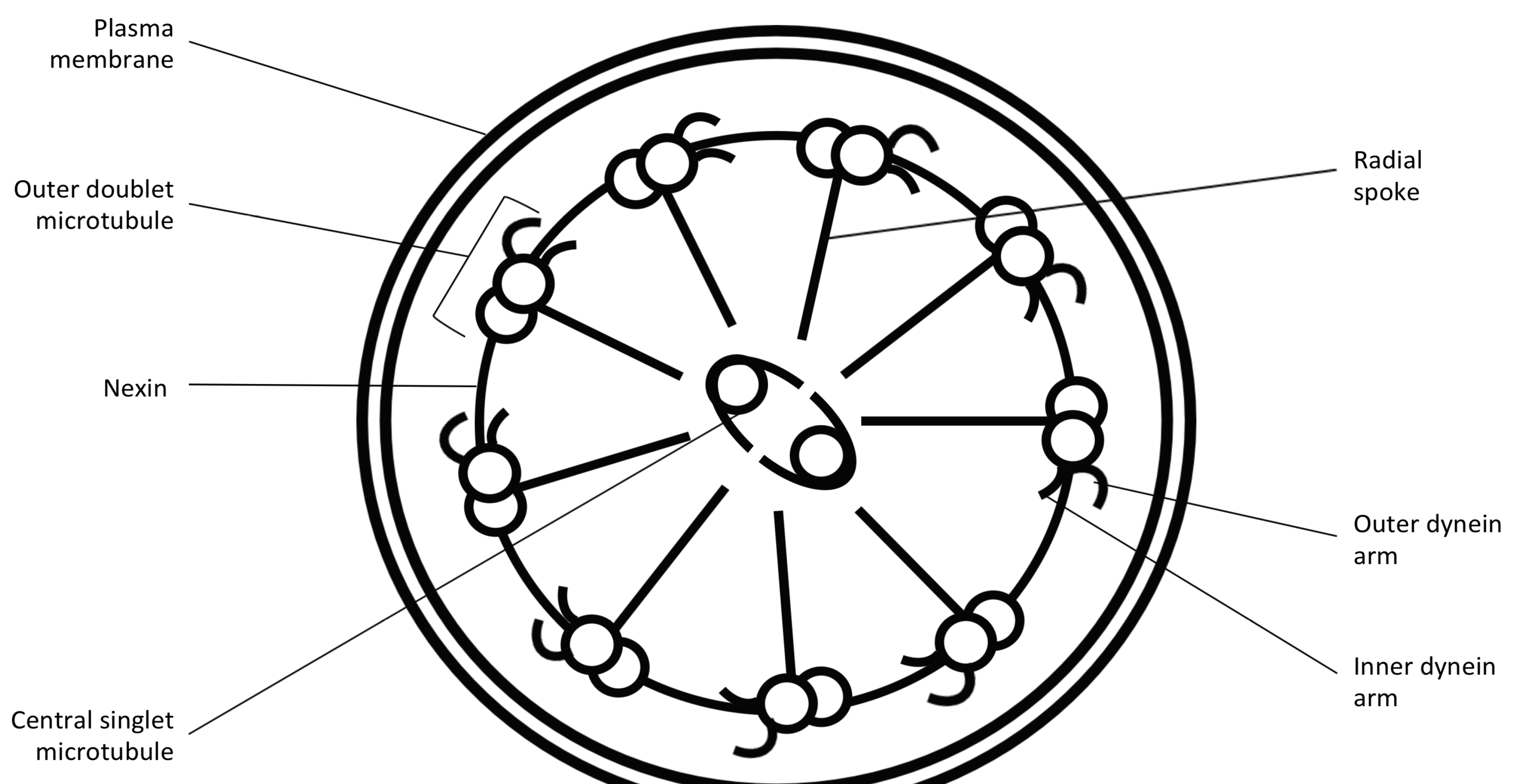{kind=link}
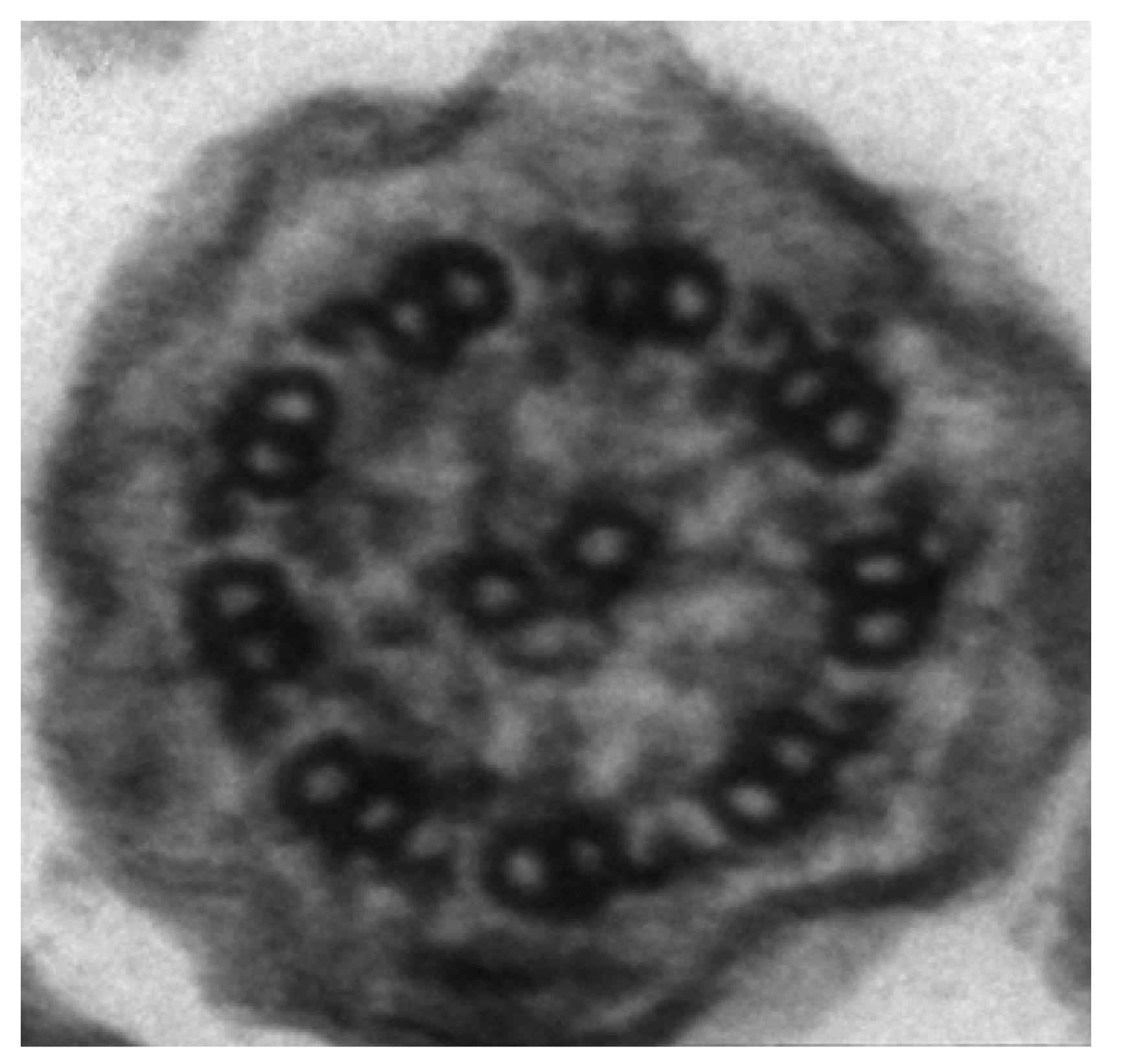{kind=link}
| Gene | Structural Defect |
|---|---|
| Abnormalities in dynein proteins | |
| DNAI1 | ODA defect (+/− IDA) |
| DNAH5 | ODA defect (+/− IDA) |
| DNAH11 | Beat abnormalities (normal structure) |
| DNAI2 | ODA defect |
| DNALI1 | ODA defect |
| TXNDC3 | ODA defect |
| ARMC4 | ODA defect |
| Genes coding for proteins responsible for assembly or transport of axonemal proteins | |
| KTU | ODA and IDA defects |
| LRRC50 | ODA and IDA defects |
| DNAAF3 | ODA and IDA defects |
| CCDC39 | ODA and IDA defects |
| CCDC40 | Axone disorganisation and IDA defect |
| CCDC103 | ODA and IDA defects |
| CCDC114 | ODA defect |
| HEATR2 | Absent ODA |
| CCDC65 | Cilial vibration, normal structure |
| ZMYND10 | Absent ODA + IDA |
| SPAG1 | Absent ODA + IDA |
| C21orf59 | Absent ODA + IDA |
| Central pair abnormalities | |
| RSPH9 | Central pair defects |
| RSPH4A | Central pair defects |
| RSPH1 | Central pair defects |
| HYDIN | Central pair defects |
| Nexin-dynein complex defects | |
| DRC CCDC164 | Nexin link missing |
| CCDC65 | Beat abnormalities |
| Genes causing PCD with associated syndromes | |
| OFD1 | Unknown |
| RPGR | Variable |
5. Comparison of Primary Ciliary Dyskinesia and Cystic Fibrosis
6. Conclusions
Acknowledgements
Author Contributions
Conflicts of Interest
References
- Bush, A.; Chodhari, R.; Collins, N.; Copeland, F.; Hall, P.; Harcourt, J.; Hariri, M.; Hogg, C.; Lucas, J.; Mitchison, H.M.; et al. Primary ciliary dyskinesia: Current state of the art. Arch. Dis. Child. 2007, 92, 1136–1140. [Google Scholar] [CrossRef]
- Kuehni, C.E.; Frischer, T.; Strippoli, M.-P.F.; Maurer, E.; Bush, A.; Nielsen, K.G.; Escribano, A.; Lucas, J.S.A.; Yiallouros, P.; Omran, H.; et al. Factors influencing age at diagnosis of primary ciliary dyskinesia in European children. Eur. Respir. J. 2010, 36, 1248–1258. [Google Scholar] [CrossRef]
- O’Callaghan, C.; Chetcuti, P.; Moya, E. High prevalence of primary ciliary dyskinesia in a British Asian population. Arch. Dis. Child. 2010, 95, 51–52. [Google Scholar] [CrossRef]
- Kennedy, M.P.; Omran, H.; Leigh, M.W.; Dell, S.; Morgan, L.; Molina, P.L.; Robinson, B.V.; Minnix, S.L.; Olbrich, H.; Severin, T.; et al. Congenital heart disease and other heterotaxic defects in a large cohort of patients with primary ciliary dyskinesia. Circulation 2007, 115, 2814–2821. [Google Scholar] [CrossRef]
- Coren, M.E.; Meeks, M.; Morrison, I.; Buchdahl, R.M.; Bush, A. Primary ciliary dyskinesia: Age at diagnosis and symptom history. Acta Paediatr. 2002, 91, 667–669. [Google Scholar] [CrossRef]
- Ellerman, A.; Bisgaard, H. Longitudinal study of lung function in a cohort of primary ciliary dyskinesia. Eur. Respir. J. 1997, 10, 2376–2379. [Google Scholar] [CrossRef]
- Farrell, P.M.; Kosorok, M.R.; Rock, M.J.; Laxova, A.; Zeng, L.; Lai, H.C.; Hoffman, G.; Laessig, R.H.; Splaingard, M.L. Early diagnosis of cystic fibrosis through neonatal screening prevents severe malnutrition and improves long-term growth. Wisconsin Cystic Fibrosis Neonatal Screening Study Group. Pediatrics 2001, 107, 1–13. [Google Scholar] [CrossRef]
- Afzelius, B.A. A human syndrome caused by immotile cilia. Science 1976, 193, 317–319. [Google Scholar]
- Omran, H.; Häffner, K.; Völkel, A.; Kuehr, J.; Ketelsen, U.P.; Ross, U.H.; Konietzko, N.; Wienker, T.; Brandis, M.; Hildebrandt, F. Homozygosity mapping of a gene locus for primary ciliary dyskinesia on chromosome 5p and identification of the heavy dynein chain DNAH5 as a candidate gene. Am. J. Respir. Cell Mol. Biol. 2000, 23, 696–702. [Google Scholar] [CrossRef]
- Olbrich, H.; Häffner, K.; Kispert, A.; Völkel, A.; Volz, A.; Sasmaz, G.; Reinhardt, R.; Hennig, S.; Lehrach, H.; Konietzko, N.; et al. Mutations in DNAH5 cause primary ciliary dyskinesia and randomization of left-right asymmetry. Nat. Genet. 2002, 30, 143–144. [Google Scholar]
- Zhu, L.; Belmont, J.W.; Ware, S.M. Genetics of human heterotaxias. Eur. J. Hum. Genet. 2006, 14, 17–25. [Google Scholar]
- Knowles, M.R.; Daniels, L.A.; Davis, S.D.; Zariwala, M.A.; Leigh, M.W. Primary ciliary dyskinesia. Recent advances in diagnostics, genetics, and characterization of clinical disease. Am. J. Respir. Crit. Care Med. 2013, 188, 913–922. [Google Scholar]
- O’Callaghan, C.; Chilvers, M.; Hogg, C.; Bush, A.; Lucas, J. Diagnosing primary ciliary dyskinesia. Thorax 2007, 62, 656–657. [Google Scholar] [CrossRef]
- Barbato, A.; Frischer, T.; Kuehni, C.E.; Snijders, D.; Azevedo, I.; Baktai, G.; Bartoloni, L.; Eber, E.; Escribano, A.; Haarman, E.; et al. Primary ciliary dyskinesia: A consensus statement on diagnostic and treatment approaches in children. Eur. Respir. J. 2009, 34, 1264–1276. [Google Scholar] [CrossRef]
- Stannard, W.A.; Chilvers, M.A.; Rutman, A.R.; Williams, C.D.; O’Callaghan, C. Diagnostic testing of patients suspected of primary ciliary dyskinesia. Am. J. Respir. Crit. Care Med. 2010, 181, 307–314. [Google Scholar] [CrossRef]
- Jorissen, M.; Willems, T.; van der Schueren, B.; Verbeken, E.; de Boeck, K. Ultrastructural expression of primary ciliary dyskinesia after ciliogenesis in culture. Acta Otorhinolaryngol. Belg. 2000, 54, 343–356. [Google Scholar]
- Shoemark, A.; Dixon, M.; Corrin, B.; Dewar, A. Twenty-year review of quantitative transmission electron microscopy for the diagnosis of primary ciliary dyskinesia. J. Clin. Pathol. 2012, 65, 267–271. [Google Scholar] [CrossRef]
- Chilvers, M.A.; O’Callaghan, C. Analysis of ciliary beat pattern and beat frequency using digital high speed imaging: Comparison with the photomultiplier and photodiode methods. Thorax 2000, 55, 314–317. [Google Scholar] [CrossRef]
- Chilvers, M.A.; Rutman, A.; O’Callaghan, C. Ciliary beat pattern is associated with specific ultrastructural defects in primary ciliary dyskinesia. J. Allergy Clin. Immunol. 2003, 112, 518–524. [Google Scholar] [CrossRef]
- Olbrich, H.; Schmidts, M.; Werner, C.; Onoufriadis, A.; Loges, N.T.; Raidt, J.; Banki, N.F.; Shoemark, A.; Burgoyne, T.; Al Turki, S.; et al. Recessive HYDIN mutations cause primary ciliary dyskinesia without randomization of left-right body asymmetry. Am. J. Hum. Genet. 2012, 91, 672–684. [Google Scholar] [CrossRef]
- Smith, C.M.; Kulkarni, H.; Radhakrishnan, P.; Rutman, A.; Bankart, M.J.; Williams, G.; Hirst, R.A.; Easton, A.J.; Andrew, P.W.; O’Callaghan, C. Ciliary dyskinesia is an early feature of respiratory syncytial virus infection. Eur. Respir. J. 2013, 43, 485–496. [Google Scholar]
- Hirst, R.A.; Rutman, A.; Williams, G.; O’Callaghan, C. Ciliated air-liquid cultures as an aid to diagnostic testing of primary ciliary dyskinesia. Chest 2010, 138, 1441–1447. [Google Scholar]
- Omran, H.; Loges, N.T. Immunofluorescence staining of ciliated respiratory epithelial cells. Methods Cell Biol. 2009, 91, 123–133. [Google Scholar] [CrossRef]
- Marthin, J.K.; Mortensen, J.; Pressler, T.; Nielsen, K.G. Pulmonary radioaerosol mucociliary clearance in diagnosis of primary ciliary dyskinesia. Chest 2007, 132, 966–976. [Google Scholar] [CrossRef]
- Walker, W.T.; Jackson, C.L.; Lackie, P.M.; Hogg, C.; Lucas, J.S. Nitric oxide in primary ciliary dyskinesia. Eur. Respir. J. 2012, 40, 1024–1032. [Google Scholar] [CrossRef]
- Lucas, J.S.; Walker, W.T. Nasal nitric oxide is an important test in the diagnostic pathway for primary ciliary dyskinesia. Ann. Am. Thorac. Soc. 2013, 10, 645–647. [Google Scholar] [CrossRef]
- Leigh, M.W.; Hazucha, M.J.; Chawla, K.K.; Baker, B.R.; Shapiro, A.J.; Brown, D.E.; Lavange, L.M.; Horton, B.J.; Qaqish, B.; Carson, J.L.; et al. Standardizing nasal nitric oxide measurement as a test for primary ciliary dyskinesia. Ann. Am. Thorac. Soc. 2013, 10, 574–581. [Google Scholar] [CrossRef]
- American Thoracic Society; European Respiratory Society. ATS/ERS recommendations for standardized procedures for the online and offline measurement of exhaled lower respiratory nitric oxide and nasal nitric oxide, 2005. Am. J. Respir. Crit. Care Med. 2005, 171, 912–930. [Google Scholar] [CrossRef]
- Marthin, J.K.; Nielsen, K.G. Hand-held tidal breathing nasal nitric oxide measurement—A promising targeted case-finding tool for the diagnosis of primary ciliary dyskinesia. PLoS One 2013, 8, e57262. [Google Scholar] [CrossRef]
- Baraldi, E.; Pasquale, M.F.; Cangiotti, A.M.; Zanconato, S.; Zacchello, F. Nasal nitric oxide is low early in life: Case study of two infants with primary ciliary dyskinesia. Eur. Respir. J. 2004, 24, 881–883. [Google Scholar] [CrossRef]
- Stehling, F.; Roll, C.; Ratjen, F.; Grasemann, H. Nasal nitric oxide to diagnose primary ciliary dyskinesia in newborns. Arch. Dis. Child. Fetal Neonatal Ed. 2006, 91. [Google Scholar] [CrossRef]
- Piacentini, G.L.; Bodini, A.; Peroni, D.G.; Sandri, M.; Brunelli, M.; Pigozzi, R.; Boner, A.L. Nasal nitric oxide levels in healthy pre-school children. Pediatr. Allergy Immunol. 2010, 21, 1139–1145. [Google Scholar] [CrossRef]
- Marthin, J.K.; Nielsen, K.G. Choice of nasal nitric oxide technique as first-line test for primary ciliary dyskinesia. Eur. Respir. J. 2011, 37, 559–565. [Google Scholar] [CrossRef]
- Noone, P.G.; Leigh, M.W.; Sannuti, A.; Minnix, S.L.; Carson, J.L.; Hazucha, M.; Zariwala, M.A.; Knowles, M.R. Primary ciliary dyskinesia: Diagnostic and phenotypic features. Am. J. Respir. Crit. Care Med. 2004, 169, 459–467. [Google Scholar] [CrossRef]
- Riordan, J.R.; Rommens, J.M.; Kerem, B.; Alon, N.; Rozmahel, R.; Grzelczak, Z.; Zielenski, J.; Lok, S.; Plavsic, N.; Chou, J.L. Identification of the cystic fibrosis gene: Cloning and characterization of complementary DNA. Science 1989, 245, 1066–1073. [Google Scholar]
- Guichard, C.; Harricane, M.C.; Lafitte, J.J.; Godard, P.; Zaegel, M.; Tack, V.; Lalau, G.; Bouvagnet, P. Axonemal dynein intermediate-chain gene (DNAI1) mutations result in situs inversus and primary ciliary dyskinesia (Kartagener syndrome). Am. J. Hum. Genet. 2001, 68, 1030–1035. [Google Scholar] [CrossRef]
- Hornef, N.; Olbrich, H.; Horvath, J.; Zariwala, M.A.; Fliegauf, M.; Loges, N.T.; Wildhaber, J.; Noone, P.G.; Kennedy, M.; Antonarakis, S.E.; et al. DNAH5 mutations are a common cause of primary ciliary dyskinesia with outer dynein arm defects. Am. J. Respir. Crit. Care Med. 2006, 174, 120–126. [Google Scholar] [CrossRef]
- Moore, D.J.; Onoufriadis, A.; Shoemark, A.; Simpson, M.A.; zur Lage, P.I.; de Castro, S.C.; Bartoloni, L.; Gallone, G.; Petridi, S.; Woollard, W.J.; et al. Mutations in ZMYND10, a gene essential for proper axonemal assembly of inner and outer dynein arms in humans and flies, cause primary ciliary dyskinesia. Am. J. Hum. Genet. 2013, 93, 346–356. [Google Scholar]
- Tarkar, A.; Loges, N.T.; Slagle, C.E.; Francis, R.; Dougherty, G.W.; Tamayo, J.; et al. DYX1C1 is required for axonemal dynein assembly and ciliary motility. Nat. Genet. 2013, 45, 995–1003. [Google Scholar] [CrossRef]
- Antony, D.; Becker-Heck, A.; Zariwala, M.A.; Schmidts, M.; Onoufriadis, A.; Forouhan, M.; Wilson, R.; Taylor-Cox, T.; Dewar, A.; Jackson, C.; et al. Mutations in CCDC39 and CCDC40 are the major cause of primary ciliary dyskinesia with axonemal disorganization and absent inner dynein arms. Hum. Mutat. 2013, 34, 462–472. [Google Scholar] [CrossRef]
- Schwabe, G.C.; Hoffmann, K.; Loges, N.T.; Birker, D.; Rossier, C.; de Santi, M.M.; Olbrich, H.; Fliegauf, M.; Failly, M.; Liebers, U.; et al. Primary ciliary dyskinesia associated with normal axoneme ultrastructure is caused by DNAH11 mutations. Hum. Mutat. 2008, 29, 289–298. [Google Scholar] [CrossRef]
- Horani, A.; Brody, S.L.; Ferkol, T.W. Picking up speed: Advances in the genetics of primary ciliary dyskinesia. Pediatr. Res. 2014, 75, 158–164. [Google Scholar] [CrossRef]
- Onoufriadis, A.; Shoemark, A.; Munye, M.M.; James, C.T.; Schmidts, M.; Patel, M.; Rosser, E.M.; Bacchelli, C.; Beales, P.L.; Scambler, P.J.; et al. Combined exome and whole-genome sequencing identifies mutations in ARMC4 as a cause of primary ciliary dyskinesia with defects in the outer dynein arm. J. Med. Genet. 2014, 51, 61–67. [Google Scholar] [CrossRef] [Green Version]
- Horani, A.; Druley, T.E.; Zariwala, M.A.; Patel, A.C.; Levinson, B.T.; van Arendonk, L.G.; Thornton, K.C.; Giacalone, J.C.; Albee, A.J.; Wilson, K.S.; et al. Whole-exome capture and sequencing identifies HEATR2 mutation as a cause of primary ciliary dyskinesia. Am. J. Hum. Genet. 2012, 91, 685–693. [Google Scholar] [CrossRef]
- Cystic Fibrosis Mutation Database. Available online: http://www.genet.sickkids.on.ca/cftr/app (accessed on 10 February 2014).
- Lucas, J.S.; Adam, E.C.; Goggin, P.M.; Jackson, C.L.; Powles-Glover, N.; Patel, S.H.; Humphreys, J.; Fray, M.D.; Falconnet, E.; Blouin, J.-L.; et al. Static respiratory cilia associated with mutations in DNAHC11/DNAH11: A mouse model of PCD. Hum. Mutat. 2012, 33, 495–503. [Google Scholar] [CrossRef]
- Zariwala, M.A.; Omran, H.; Ferkol, T.W. The emerging genetics of primary ciliary dyskinesia. Proc. Am. Thorac. Soc. 2011, 8, 430–433. [Google Scholar] [CrossRef]
- Palomaki, G.E.; Haddow, J.E.; Bradley, L.A.; FitzSimmons, S.C. Updated assessment of cystic fibrosis mutation frequencies in non-Hispanic Caucasians. Genet. Med. 2002, 4, 90–94. [Google Scholar] [CrossRef]
- Strom, C.M.; Huang, D.; Chen, C.; Buller, A.; Peng, M.; Quan, F.; Redman, J.; Sun, W. Extensive sequencing of the cystic fibrosis transmembrane regulator gene: Assay validation and unexpected benefits of developing a comprehensive test. Genet. Med. 2003, 5, 9–14. [Google Scholar] [CrossRef]
- McCormick, J.; Green, M.W.; Mehta, G.; Culross, F.; Mehta, A. Demographics of the UK cystic fibrosis population: Implications for neonatal screening. Eur. J. Hum. Genet. 2002, 10, 583–590. [Google Scholar] [CrossRef]
- NHS Newborn Blood Spot Screening Programme Home Page. Available online: http://newbornbloodspot.screening.nhs.uk/ (accessed on 15 February 2014).
- Sobczyńska-Tomaszewska, A.; Ołtarzewski, M.; Czerska, K.; Wertheim-Tysarowska, K.; Sands, D.; Walkowiak, J.; Bal, J.; Mazurczak, T. Newborn screening for cystic fibrosis: Polish 4 years’ experience with CFTR sequencing strategy. Eur. J. Hum. Genet. 2013, 21, 391–396. [Google Scholar] [CrossRef]
- Lim, M.; Wallis, C.; Price, J.F.; Carr, S.B.; Chavasse, R.J.; Shankar, A.; Seddon, P.; Balfour-Lynn, I.M. Diagnosis of cystic fibrosis in London and South East England before and after the introduction of newborn screening. Arch. Dis. Child. 2014, 99, 197–202. [Google Scholar] [CrossRef]
- Leigh, M.W.; Pittman, J.E.; Carson, J.L.; Ferkol, T.W.; Dell, S.D.; Davis, S.D.; Knowles, M.R.; Zariwala, M.A. Clinical and genetic aspects of primary ciliary dyskinesia/Kartagener syndrome. Genet. Med. 2009, 11, 473–487. [Google Scholar] [CrossRef]
- Crossley, J.R.; Elliott, R.B.; Smith, P.A. Dried-blood spot screening for cystic fibrosis in the newborn. Lancet 1979, 1, 472–474. [Google Scholar]
- Ramsey, B.W.; Davies, J.; McElvaney, N.G.; Tullis, E.; Bell, S.C.; Dřevínek, P.; Griese, M.; McKone, E.F.; Wainwright, C.E.; Konstan, M.W.; et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N. Engl. J. Med. 2011, 365, 1663–1672. [Google Scholar] [CrossRef] [Green Version]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Collins, S.A.; Walker, W.T.; Lucas, J.S. Genetic Testing in the Diagnosis of Primary Ciliary Dyskinesia: State-of-the-Art and Future Perspectives. J. Clin. Med. 2014, 3, 491-503. https://doi.org/10.3390/jcm3020491
Collins SA, Walker WT, Lucas JS. Genetic Testing in the Diagnosis of Primary Ciliary Dyskinesia: State-of-the-Art and Future Perspectives. Journal of Clinical Medicine. 2014; 3(2):491-503. https://doi.org/10.3390/jcm3020491
Chicago/Turabian StyleCollins, Samuel A., Woolf T. Walker, and Jane S. Lucas. 2014. "Genetic Testing in the Diagnosis of Primary Ciliary Dyskinesia: State-of-the-Art and Future Perspectives" Journal of Clinical Medicine 3, no. 2: 491-503. https://doi.org/10.3390/jcm3020491
APA StyleCollins, S. A., Walker, W. T., & Lucas, J. S. (2014). Genetic Testing in the Diagnosis of Primary Ciliary Dyskinesia: State-of-the-Art and Future Perspectives. Journal of Clinical Medicine, 3(2), 491-503. https://doi.org/10.3390/jcm3020491